
Abstract
Given the role of mitochondria in oxygen consumption, metabolism and cell death regulation, alterations in mitochondrial function or dysregulation of cell death pathways contribute to the genesis and progression of cancer. Cancer cells exhibit an array of metabolic transformations induced by mutations leading to gain-of-function of oncogenes and loss-of-function of tumor suppressor genes that include increased glucose consumption, reduced mitochondrial respiration, increased reactive oxygen species generation and cell death resistance, all of which ensure cancer progression. Cholesterol metabolism is disturbed in cancer cells and supports uncontrolled cell growth. In particular, the accumulation of cholesterol in mitochondria emerges as a molecular component that orchestrates some of these metabolic alterations in cancer cells by impairing mitochondrial function. As a consequence, mitochondrial cholesterol loading in cancer cells may contribute, in part, to the Warburg effect stimulating aerobic glycolysis to meet the energetic demand of proliferating cells, while protecting cancer cells against mitochondrial apoptosis due to changes in mitochondrial membrane dynamics. Further understanding the complexity in the metabolic alterations of cancer cells, mediated largely through alterations in mitochondrial function, may pave the way to identify more efficient strategies for cancer treatment involving the use of small molecules targeting mitochondria, cholesterol homeostasis/trafficking and specific metabolic pathways.
Similar content being viewed by others

Introduction
Cancer cells exhibit critical metabolic transformations induced by mutations leading to gain-of-function of oncogenes and loss-of-function of tumor suppressor genes that result in cell deregulation associated with increased cellular stress. Hanahan and Weinberg identified the six conceptual hallmarks of human cancer: (1) self-sufficient growth signaling, (2) evasion of growth suppressors, (3) cell death resistance, (4) replicative immortalization, (5) angiogenesis and (6) invasion/metastasis [1]. Other common characteristics of cancer cells include enhanced anabolism, avoidance of immune destruction and altered autophagy [2, 3]. Of these characteristic features of cancer cells, mitochondria are directly involved in a number of them. Indeed, mitochondria are critical mediators of apoptosis and the source of reactive oxygen species (ROS) generation and energy production. Consequently, altered mitochondrial function of cancer cells underlies several phenotypes, including: (1) resistance to apoptosis; (2) increased biosynthetic anabolism to support uncontrolled growth and proliferation; (3) increased ROS generation that activates metastatic proteases, tumor-promoting inflammation, genetic instability and DNA mutagenesis; (4) decreased mitochondrial oxidative phosphorylation (OXPHOS), increased aerobic glycolysis and decrease of pH in the extracellular milieu. Furthermore, due to its role as a hub in several signaling pathways [4], mitochondria are central for key metabolic alterations of cancer cells, some of which will be described below.
Experimental evidence indicates that high cell proliferation [5, 6] and tumor growth [7, 8] are closely associated with enhanced cholesterol requirement. Some types of cancers, such as hepatocellular carcinoma (HCC), are dependent on cholesterol for growth [9], and observational studies show a protective association between the use of statins and the risk of developing liver cancer [10], although this trend has been also observed in other cancer types, such as prostate and gastrointestinal cancers [11]. In line with this, genome-scale metabolic models of hepatocellular carcinoma found that among 101 metabolites relevant to HCC development, 30 % of them are related to cholesterol biosynthesis [12]. This protective effect of statins has been attributed to the inhibition of the mevalonate pathway (see below), preventing the posttranslational modification of the oncogenes MYC, RAS and RHO [11, 13, 14]. Moreover, analyses of the Cancer Genome Atlas (TCGA) database revealed a correlation between increased activity of the cholesterol synthesis pathway and decreased survival in patients with sarcoma, acute myeloid leukemia and melanoma [15, 16], supporting the concept that cholesterol promotes carcinogenesis. In this regard, cholesterol trafficking to mitochondria has been reported in tumor cells [17, 18] and may account for the recognized mitochondrial dysfunction and contribute to chemotherapy and apoptosis resistance and metabolic reprogramming of cancer cells, which will be discussed in the following sections.
Mitochondria in cell life and death
Life-sustaining functions
Mitochondria are complex organelles, which differ from the often-held view of isolated, small rounded double-membrane structures. They constitute a dynamic network that continuously undergoes fusion and fission controlled by specific mechanisms [19], and have interactions with other cell structures such as cytoskeleton and endoplasmic reticulum (ER) [20, 21]. Mitochondria contain multiple copies of their own maternally-inherited mitochondrial DNA (mtDNA), with an epigenetic complexity not completely understood [22]. Mitochondrial DNA is a circular molecule of approximately 16.5 kilobases present from hundreds to thousands of copies per cell, which encodes 13 polypeptides of the OXPHOS and respiratory chain, as well as 2 ribosomal RNAs and 22 transfer RNAs necessary for translation of polypeptides inside mitochondria. Most mitochondrial proteins (approximately 1500) are encoded by nuclear DNA, translated in the cytosol and imported into the mitochondria through specific translocator complexes (TIM and TOM) of the mitochondrial inner (MIM) and outer membranes (MOM), respectively. In addition, a disulfide relay molecular device consisting of MIA40 and augmenter of liver regeneration (ALR) are responsible for the import of nuclear encoded sulfur Fe/S cluster proteins to the mitochondrial intermembrane space that are essential for mitochondrial function [23, 24]. Recent data have shown that ALR links mitochondrial function to HCC development [25, 26]. Indeed, mitochondrial proteome has significant cell-type differences, allowing mitochondria to serve in a highly adaptive fashion to the cellular specific functional requirements [27].
Mitochondria are the power plants of the cell, providing the energy for countless cellular functions through OXPHOS. OXPHOS is coordinated by a cascade of redox reactions organized in five protein complexes embedded in the MIM, known as the electron transport chain (ETC), which transfers electrons to oxygen [28, 29]. The fall in electron potential energy through the ETC is used to pump protons out of the mitochondrial matrix to the intermembrane space, generating an electrochemical gradient known as the mitochondrial transmembrane potential (Δψm), which induces a proton motive force used by complex V to regenerate ATP from ADP. Moreover, many additional mitochondrial processes, especially those related to transport of solutes across the MIM [30] are dependent on the electrochemical driving force of the Δψm. Additional metabolic pathways that are located within mitochondria comprise the tricarboxylic acid cycle (TCA or Krebs cycle), β-oxidation of fatty acids, steroidogenesis, metabolism of amino acids, formation of Fe/S clusters, heme biosynthesis as well as reactions involved in lipogenesis, gluconeogenesis, ketogenesis and ammonium detoxification (urea cycle) [31].
Physiologically under aerobic conditions, cells degrade glucose via glycolysis to pyruvate, which is imported into mitochondria. Pyruvate enters the TCA cycle in the form of acetyl-CoA that along with oxaloacetate generates citrate, in a reaction catalyzed by citrate synthase. Citrate is processed in the TCA cycle to generate reducing equivalents that feed the ETC and generate energy with the consumption of oxygen. However, in conditions where macromolecular biosynthesis is active, citrate may be exported to cytosol where is converted to acetyl-CoA by ATP citrate lyase (ACLY), which is used for lipogenesis. Besides their role in metabolism, mitochondria are involved in calcium homeostasis, innate immunity, integration of signaling pathways and autophagy [32, 33]. Moreover, in response to metabolic and genetic stress mitochondria and nucleus engage in bidirectional signaling pathways, which modulate cell function [4, 34, 35].
Electron transport through the ETC can leak the chain and react with oxygen to generate ROS [36, 37]. Complex I and complex III are the major sources of mitochondrial ROS generation [28], although other mitochondrial sites also contribute to ROS production, including complex II [38]. The existence of an efficient antioxidant defense system, of which mitochondrial glutathione (mGSH) is a central component, prevents or repairs oxidative damage generated during normal aerobic metabolism [39]. The primary ROS generated in mitochondria is superoxide [40], which is produced in the mitochondrial matrix and undergoes dismutation to hydrogen peroxide (H2O2) [40], a reaction catalyzed by mitochondrial superoxide dismutases (SOD2). Hydrogen peroxide is further inactivated by the mitochondrial glutathione peroxidase (mGSH/GPX) and peroxiredoxin/thioredoxin (Prx/Trx) antioxidant systems [41]. Both systems use the reducing equivalents of NADPH to regenerate the mitochondrial oxidized glutathione (mGSSG) and Trx back to the reduced forms. The Prx/Trx system is thought to be responsible for scavenging hydrogen peroxide at nanomolar concentrations, while mGSH/GPX system is important for buffering high ROS levels [42, 43]. However, both systems are mutually regulated, as selective depletion of mGSH results in decreased levels of Trx2 and Prx3 [44], highlighting the central role of mGSH in maintaining an adequate hydrogen peroxide homeostasis. Due to its more stable and diffusible nature, hydrogen peroxide acts as a second messenger because of its reactions with specific oxidation-prone protein cysteinyl residues [45], which confers properties to hydrogen peroxide as a mitochondrial signaling molecule [4]. In line with this, mitochondrial hydrogen peroxide bursts have self-sustained circadian oscillations, acting as a redox intracellular pacemaker [46].
Death promoting pathways
Besides their fundamental role in energy generation, mitochondria also play a strategic role in the regulation of several forms of cell death, including apoptosis (both caspase-dependent and independent), necrosis and programmed necrosis [47]. The central mediators of apoptosis include a group of cysteine proteases named caspases, which become activated by a proteolytic processing cascade in response to pro-apoptotic signals. The series of events leading to apoptosis have been categorized in two modes, the extrinsic and intrinsic apoptotic pathways. The extrinsic pathway involves extracellular ligand binding to a transmembrane death receptor, such as TNF receptor or FAS receptor, followed by recruitment of cytosolic adaptor proteins and activation of an initiator caspase (usually caspase-8), which stimulates an effector caspase (such as caspase-3). Conversely, the intrinsic (or mitochondrial) pathway involves the destabilization of the MOM and the release of mitochondrial proteins that activate effector caspases. The BCL-2 family of proteins regulates this pathway with opposing pro-apoptotic effector functions (BAX, BAK), pro-apoptotic BH3-only proteins (BAD, BIM, BID, BIK, Noxa, PUMA, HRK, BMF) and anti-apoptotic functions (BCL-2, BCL-xL, MCL-1, A1, BCL-B, BCL-w) [48]. Activation of the intrinsic pathway of apoptosis by a number of stimuli and stresses, triggers the binding and activation of pro-apoptotic proteins BAX or BAK to the MOM leading to the MOM permeabilization (MOMP) without disruption of the inner membrane and the subsequent release of proteins from the mitochondrial intermembrane space (IMS), such as cytochrome c [49, 50]. Although active BAX or BAK are required to induce MOMP, the underlying mechanism is controversial [51]. While the model of pro-apoptotic activation or neutralization by anti-apoptotic members are still incompletely known, recent findings have shown that BCL-2 ovarian killer (BOK), which displays a high sequence similarity to BAX and BAK, engages the mitochondrial apoptotic pathway independently of BAK/BAX [52]. Although mitochondrial proteins are normally secured in the IMS the rupture of the physical barrier (MOM) constitutes a point-of-no-return in cell death [49, 50]. Pro-apoptotic BH3-only proteins act as stress sentinels that relay the diverse array of apoptotic signals via BAX/BAK activation to induce MOMP. In contrast, anti-apoptotic BCL-2-family proteins prevent MOMP and apoptosis by binding BH3-only proteins, preventing their interaction with BAX/BAK, or by binding activated BAX/BAK [53]. Pro- and anti-apoptotic BCL-2 protein interactions are mediated between BH-3 domains and the BH3 binding cleft in anti-apoptotic BCL-2 proteins.
Once released from the mitochondria into the cytosol through MOMP, cytochrome c binds to the adaptor molecule APAF-1, causing it to oligomerise and form a heptameric structure called apoptosome [54]. This complex recruits pro-caspase 9, which in turn, activates the executioner caspases-3 and -7, triggering the cascade of events that lead to controlled cell death and fragmentation. In addition to cytochrome c, other IMS proteins (Table 1) are also mobilized and released into the cytosol following MOMP where they promote or counteract caspase activation and hence cell death [55–60].
For the execution of mitochondrial apoptosis cytochrome c detaches from the MIM and dissociates from the phospholipid cardiolipin, which binds cytochrome c by an electrostatic bond [61]. Cardiolipin can be oxidized by ROS or by the cardiolipin–cytochrome c complex [62] resulting in oxidized cardiolipin, which exhibits lower affinity for cytochrome c than the reduced form, and therefore contributes to cytochrome c detachment from MIM and its release to cytosol. Since mitochondrial ROS are controlled by antioxidants [63, 64], mGSH arises as an important modulator of apoptotic cell death by indirectly controlling the redox state of cardiolipin [63, 65]. In addition, it has been described that oxidized cardiolipin modulates the biophysical properties of MOM to allow oligomerized BAX to insert and permeabilize the MOM [63, 65, 66].
Integrin-mediated attachment of normal cells to the extracellular matrix elicits anti-apoptotic and pro-survival signaling. The loss of cell–matrix interaction induces anoikis, a specific form of apoptosis [67]. Cell detachment leads to upregulation and activation of several BH3-only pro-apoptotic proteins (BID, BIM and BDF) that, in turn, activate BAX and BAK resulting in MOMP and the apoptotic cascade, resulting in cell death [68]. In addition to MOMP, the generation of mitochondrial ROS in cells undergoing anoikis is required for cell death, as antioxidants treatment suppressed anoikis [69, 70]. Normal cells detached from the matrix undergo dramatic global metabolic changes characterized by decreased mitochondrial respiration and SOD2 induction. Indeed, cells depleted of SOD2 are hypersensitive to cell death by anoikis [71], suggesting the importance of ROS generated in mitochondria in the execution of anoikis.
As opposed to apoptosis, necrosis is a morphologically distinct form of cell death responsible for irreversible tissue destruction due to bioenergetic failure and oxidative damage. Permeabilization of the MIM by the mitochondrial permeability transition (MPT) and secondary rupture of the MOM is a key event of necrosis. MPT is a regulated pore-forming protein complex whose molecular characterization remains elusive [72–74]. Of the MPT components, cyclophillin D is a key constituent, while the role of other putative components, such as voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT) and translocator protein (TSPO, also called benzodiazepine receptor, PBR) is controversial [49, 75, 76]. Mitochondrial ROS regulate MPT by targeting specific cyclophillin D cysteine residues. Necrosis is characterized by mitochondrial swelling, loss of Δψm, and impaired OXPHOS and ATP generation. The fundamental difference with respect to apoptosis is the rapid loss of cellular membrane potential due to energy depletion and ion pump/channel failure, leading to swelling and cytolysis. Concomitantly, water influx causes matrix swelling, rupture of MOM and release of apoptogenic proteins sequestered in IMS. These events, however, block apoptotic cell death due to energetic failure, ATP exhaustion and oxidative stress-mediated caspase inactivation. Moreover, TNFα has been recently shown to induce a caspase-independent form of programmed cell death, named programmed necrosis or necroptosis [77, 78], involving receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 kinases, which interact with the pseudokinase mixed lineage kinase domain-like protein (MLKL). The execution of necroptosis requires mitochondrial ROS generation, which is dependent of MPT and involves cyclophyllin D but it is independent of BAX or BAK [79].
Cholesterol homeostasis and mitochondrial trafficking
Cholesterol synthesis and deregulation in cancer cells
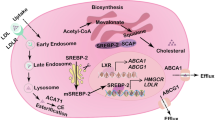
Cholesterol is an essential component of membrane bilayers that plays a key role in their integrity and function. While intake of cholesterol from the diet ends up in different cell membranes, the predominant mechanism that provides the cholesterol needed for cellular functions is its de novo synthesis from acetyl-CoA in the so-called mevalonate pathway, which generates not only cholesterol but also non-sterol components, such as dolichol, ubiquinol and isoprenoids. The hydroxymethylglutaryl-CoA reductase (HMGCoAR) catalyzes the reduction of HMG CoA to mevalonate, the rate-limiting step in the synthesis of cholesterol [80]. Mevalonate is phosphorylated to pyrophosphomevalonate, which is then converted to isopentenyl pyrophosphate (IPP). IPP can be reversibly transformed to dimethylallylpyrophosphate (DMAPP), which can combine with IPP to generate the 10-carbon isoprenoid geranyl pyrophosphate (GPP). The secuential addition of 1 or 2 more IPP units to GPP generates farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), respectively. Isoprenoids generation in the mevalonate pathway is an essential mechanism of posttranslational modification of proteins and these lipid moieties anchor target proteins to cell membranes. FPP is used to prenylate proteins of the Ras family, while GGPP prenylates those of the Rho family [81]. In addition, FPP can be converted into squalene by squalene synthase (SS), which catalyzes the first step in the committed pathway for cholesterol synthesis. Statins, whose chemical structure is similar to that of HMGCoA, compete with and inhibit HMGCoAR, preventing the formation of mevalonate and its downstream product IPP. Therefore, the therapeutic effects of statins can extend beyond cholesterol inhibition and impact in the regulation of a number of proteins due to the blockade of isoprenoids (FPP and GGPP) generation. In contrast to statins, the inhibition of SS results in selective cholesterol downregulation without exerting a major effect in the isoprenylation of proteins [82].
As HMGCoAR is the regulatory enzyme in the mevalonate pathway its feedback and transcriptional control impact in cholesterol and isoprenoids regulation. One mechanism for feedback control involves the rapid degradation of HMGCoAR mediated by ER resident proteins, Insigs. Accumulation of sterols in the ER membrane triggers binding of the membrane domain of HMGCoAR to a subset of Insigs, which carry a membrane-anchored ubiquitin ligase called GP78 which ubiquitinates HMGCoAR, marking it for proteasomal degradation [83]. HMGCoAR is regulated at the transcriptional level by the transcription factor SREBP-2, which resides in the ER is an inactive form. When sterols levels are low, SREBP-2 is transported from the ER to the Golgi to undergo a proteolytic processing by specific proteases, resulting in the mature form of SREBP-2, which translocates to the nuclei to induce HMGCoAR as well as other targets involved in the regulation of cholesterol homeostasis, including the LDL receptor.
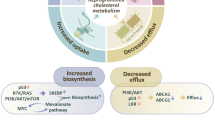
As cholesterol synthesis requires oxygen, which is used for the biotransformation of squalene to cholesterol, an additional mechanism that regulates cholesterol synthesis is oxygen availability. Indeed, the bulk for the oxygen requirement centers on the sequential transformation of lanosterol to cholesterol, involving several redox reactions. Moreover, hypoxia has been shown to stimulate HMGCoAR degradation through both accumulation of lanosterol and Insigs induction [84]. In contrast to these physiological features, cholesterol synthesis and regulation are altered at several levels in cancer cells to meet the unrestricted growth needs [84–87]. Indeed, tumor cells exhibit increased cholesterol levels compared to surrounding cells; moreover, cancer tissues display increased upregulation of HMGCoAR, loss of feedback inhibition, decreased expression of cholesterol exporter ATP binding cassette transporter A1 (ABCA1) and increased extracellular cholesterol uptake via LDL receptor [87]. Hence, as briefly described below (“Strategies targeting the mevalonate pathway and cholesterol synthesis in cancer” section), targeting the mevalonate pathway may be of potential relevance in cancer therapy.
Mitochondrial cholesterol trafficking in cancer
Mitochondria are cholesterol-poor organelles compared to other cell bilayers (e.g. plasma membrane). Nevertheless, the limited availability of cholesterol in the MIM plays an important physiological role, including the synthesis of bile acids in hepatocytes or steroid hormones in specialized tissues through the metabolism of mitochondrial cholesterol by CYP27A or CYP11A1, respectively. In pathological conditions, however, the accumulation of cholesterol in mitochondria alters membrane organization and the coexistence of lipid-disordered and lipid-ordered phases, which regulates membrane permeability and function of resident proteins [88]. Of relevance, increased mitochondrial cholesterol levels have been described in solid tumors. For instance, mitochondrial cholesterol levels of tumors from Buffalo rats bearing transplanted Morris hepatomas are two to fivefold higher than the content found in mitochondria prepared from host liver, and correlated with the degree of tumor growth and malignancy [89, 90]. As mitochondrial cholesterol in cancer cells contribute to the alterations in mitochondrial function and properties, understanding the mechanisms governing the trafficking of cholesterol to mitochondria may be of relevance in cancer cell biology. In this regard, given its lipophilic properties and water insolubility, non-vesicular transport by specific carriers stands as the major mechanism of cholesterol transport between organelles. In particular, mitochondrial cholesterol transport is preferentially regulated by the steroidogenic acute regulatory domain 1 (StARD1), the founding member of a family of lipid transporting proteins that contain StAR-related lipid transfer (START) domains [91]. StARD1 is a MOM protein, which was first described and best characterized in steroidogenic cells, where it plays an essential role in cholesterol transfer to MIM for metabolism by CYP11A1 to generate pregnenolone. Despite similar properties with StARD1, other StART members cannot replace StARD1, as germline StARD1 deficiency is lethal due to adrenocortical lipoid hyperplasia [92]. Moreover, targeted mutations in MLN64 (StARD3), another START member with wide tissue distribution, impair steroidogenesis while causing minor alteration in cholesterol metabolism [93]. Furthermore, analyses of the TCGA database further support a role for StARD1 and MLN64 and subsequent mitochondrial cholesterol enrichment in cancer development. Although MLN64 is an endosomal protein, it participates in the egress of cholesterol from endosomes to mitochondria [94], suggesting that MLN64 and StARD1 work in concert to ensure the trafficking of cholesterol to MIM. Increased StARD1 expression and mitochondrial cholesterol loading are causally linked as StARD1 silencing decrease mitochondrial cholesterol levels in hepatocellular carcinoma [17]. Moreover, decreased ABCA1 activity has been reported in colorectal cancer cells either through loss-of-function or gene downregulation and ABCA1 downregulation promoted cancer cell survival by increased mitochondrial cholesterol accumulation [95]. Thus, these findings indicate that the trafficking and accumulation of cholesterol in mitochondria is a characteristic feature of many types of cancer and its role in carcinogenesis may be related to the regulation of cell death and chemotherapy sensitization, which will be described below.
Role of mitochondria and cholesterol in altered cancer cell metabolism
The oncogenic transformation of cancer cells requires energy metabolism reprogramming in order to support unrestrained growth. The dependence on aerobic glycolysis despite normal oxygen tension constitutes one of the key metabolic alterations in cancer cells. This event was first described by Otto Warburg in 1930 and has ben coined since then as the Warburg Effect [96–98]. Although the glycolytic phenotype in cancer cells was proposed to be due to defective mitochondrial OXPHOS, many cancer cells exhibit competent OXPHOS activity capable to generate ATP [99]. The dependence on glycolysis is characteristic of many tumors and is widely exploited for clinical tumor imaging using positron emission tomography (PET) with a radiolabeled analog of glucose (18F-fluorodeoxyglucose) [100]. Elevated aerobic glycolysis in cancer cells serves many purposes, ensuring ATP generation without reliance on oxygen availability. Moreover, aerobic glycolysis generates bicarbonic and lactic acids, which are released to the extracellular milieu, favoring tumor invasion, angiogenesis and immunosurveillance suppression [101]. Glucose can be diverted to the pentose phosphate pathway to generate nucleotides and NADPH to fuel antioxidant defenses and biosynthetic reactions. Finally cancer cells use intermediates of glycolytic pathway for biosynthesis of de novo nucleic acids, lipids and amino acids to support their unrestrained growth and proliferation [97, 102, 103]. In line with these changes, a Warburg-like metabolism has been described in many rapidly proliferating embryonic tissues, supporting the biosynthetic programs of aerobic glycolysis in active proliferating cells [104, 105]. Given that many tumor types rely on oxidative metabolism, glucose flux is not necessarily coupled to oxidative glucose metabolism. Oxygen consumption in many cancer cells is used for mitochondrial oxidation of alternate fuels, such as glutamine [106], suggesting that the fate of glucose for mitochondrial oxidation in cancer cells is probably even lower. Cancer cells undergo a number of metabolic alterations, including the depression of oxidative mitochondrial OXPHOS and TCA cycle, which are used for anabolic reactions [107, 108]. Moreover, several transcriptional and posttranslational mechanisms have been proposed to contribute to the metabolic reprogramming and dependence on the Warburg effect in cancer cells, involving activation of oncogenes and inactivation of tumor suppressor genes. In this regard, activated oncogenes such as KRAS and MYC along with mutated tumor suppressors such as TP53 can extensively reprogram cell metabolism resulting in diversion of carbon skeletons to fuel anabolic reactions for biomass synthesis instead of being completely oxidized through mitochondrial respiration.
MYC in tumor metabolism reprogramming
MYC is an oncogene that plays a role in cell cycle progression, apoptosis and cellular transformation. In addition, MYC is important for the increased transcription of metabolic enzymes required for anabolism in cancer and fast-growing cells, regulating the conversion of glucose to pyruvate through the activation of important glycolytic genes and glucose transporters, while blocking the entry of pyruvate into the TCA cycle via pyruvate dehydrogenase kinase (PDK1). Interestingly, MYC promotes the metabolic adaptation of tumor cells [109] by activating genes important for mitochondrial biogenesis and function [110, 111]. Moreover, the AMPK-related protein kinase 5 (ARK5), which is involved in maintenance of mitochondrial integrity and bioenergetic homeostasis, was identified as a MYC target [112]. This dual role of MYC as a driver of Warburg effect and a promoter of mitochondrial biogenesis underlies the dependence of cancer cells on glutamine oxidation, an essential event for cell survival under conditions with low glucose and oxygen [113]. Moreover, MYC upregulates the glutamine transporters SLC5A1 and SLC7A1, which contribute to glutamine uptake in cancer cells. As MYC induces the flux of 3-phosphoglycerate from glycolysis to the synthesis of serine and glycine needed for nucleotide biosynthesis, MYC coordinates the synthesis of nucleotides with glutamine metabolism [114]. Indeed, the rate of glutaminolysis is greater compared to the rate of glycolysis in cells with high MYC expression and are more dependent on mitochondrial oxidative metabolism than cells with low MYC levels.
Tumors are metabolically heterogeneous, exhibiting complex metabolic profiles [115], including the dependence on aerobic glycolysis and reliance on OXPHOS [116–119]. For instance, while cancer stem cells are quiescent and exhibit high OXPHOS reliance, they may coexist with other highly cycling cancer cells that rely on glycolysis. The dependence of these cancer stem cells on mitochondrial OXPHOS prompted the use of mitochondrial OXPHOS inhibitors to selectively target these cells to prevent tumor relapse after cytotoxic treatment [120, 121]. It has been described that in pancreatic tumors MYC acts as a switch between the OXPHOS-dependent metabolism of cancer stem cells towards the highly glycolytic differentiated progeny, creating a gradient of heterogeneous oxidative/glycolytic population inside the tumor. Moreover, MYC acts as a direct transcriptional inhibitor of peroxisome proliferator-activated receptor α (PGC1-α) suppressing mitochondrial respiration while activating glycolytic programs [119]. This heterogeneity defines a scenario where therapies targeting specifically highly respiratory or highly glycolytic tumor cells may not be completely effective.
TP53 and tumor metabolism
Reduced expression of the tumor suppressor protein TP53 can also impact metabolic reprogramming in cancer cells. Defects in P53 function lead to impaired transactivation of SCO2, a mitochondrial protein required for the correct assembly of the cytochrome c oxidase in the ETC and of TIGAR, an isoform of 6-phospho-fructo-2-kinase, whose expression exerts a tumor suppressor function by inhibiting glycolytic flux [122, 123]. Moreover, TP53 activates transcription of glutaminase 2 (GLS2) to promote glutaminolysis to fuel the TCA cycle and facilitate fatty acid oxidation as an alternative source [124]. Collectively, TP53, in addition to its role in orchestrating cell cycle arrest and apoptosis, counteracts the Warburg effect by favoring OXPHOS and minimizing glycolytic metabolism, and therefore its loss-of-function is a requirement for the aerobic glycolysis in most carcinogenic processes.
Hypoxia-inducible factor (HIF1α)
Hypoxia is an inherent feature of solid tumor development that arises due to the disorganized structure and architecture of tumor vasculature resulting in irregular and inefficient oxygen delivery. Hypoxia is considered a negative prognostic factor for response to treatment and survival of cancer patients [125, 126]. Hypoxia-inducible factor (HIF) is a key transcription factor activated mainly by hypoxia due to the dependence of HIF-proly hydroxylases (PHD) on oxygen (see below). In addition to hypoxia HIF is also regulated by oxidative stress, inflammation and metabolic stress [127]. HIF1 comprises a stable β subunit (HIF-1β/Arnt) and a labile α subunit (HIF1α) encompassing three family members, HIF1α, HFI2α and HIF3α (Fig. 1). In normoxia HIF1α is rapidly degraded due to the sequential action of oxygen-dependent PHD and the Von Hippel-Landau E3-ubiquitin ligase (pVHL). PHDs primarily function as oxygen sensors so that in normoxia PHDs become activated to hydroxylate HIF1α on two highly conserved proline residues. Hydroxylated HIF1α is then recognized and ubiquitinated by the pVHL, marking HIF1α for proteasomal degradation (Fig. 1a). In low oxygen conditions, PHDs are inactivated and therefore HIF1α is stabilized, translocate to the nucleus where heterodimerize with HIF1β/Arnt to form a complex that activates hundreds of genes involved in energy metabolism, autophagy and angiogenesis [128] (Fig. 1b). Activation of HIF1α promotes the conversion of glucose to pyruvate and lactate by upregulating the transcription of glucose transporters (GLUT1), hexokinases (HK1 and HK2), lactate dehydrogenase A (LDHA) as well as the lactate-extruding monocarboxylate transporter 4 (MCT4) [129], supporting the shift to aerobic glycolysis. Activated HIF1α increase the transcription of the PDK1, which inhibits PDH, decreasing the conversion of pyruvate to acetyl-CoA, which compromises OXPHOS, therefore linking low oxygen conditions to the depression of mitochondrial function. Moreover, HIF-1 activates transcription of the cytochrome c oxidase subunit 4-2 (COX4-2) and the LON mitochondrial protease, which degrades COX4-1 subunit and allows its substitution by the less efficient COX4-2 subunit [130]. In a scenario with inhibited mitochondrial OXPHOS by genetically downregulating the master regulator of mitochondrial biogenesis PGC1α, ROS-mediated HIF1α stabilization is able to rescue cell bioenergetics by activating transcription of glycolytic genes and glycolysis, allowing cancer cells to escape from metabolic stress [131]. In addition, HIF1 α induces the expression of BCL-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L), which trigger mitochondrial autophagy, thereby decreasing the oxidative metabolism of both fatty acids and glucose [132]. Therefore, HIF1α not only counteracts the MYC-mediated suppression of mitochondrial biogenesis by reducing mitochondrial mass and function, but also cooperates with MYC to promote aerobic glycolysis by induction of HK2 and PDK1 [133]. There are three PHDs known in mammals, encoded by three genes (PHD1, PHD2 and PHD3) [134]. Although, PHDs are thought to act as true oxygen sensors due to their requirement of oxygen for hydroxylation of HIF1, they are also dependent on iron (Fe2+), ascorbate and on the TCA intermediate 2-oxoglutarate (2-OG) as cofactors. Conversely, it has been reported that several TCA intermediates, such as fumarate and succinate competitively inhibit all three PHDs, while citrate and oxaloacetate inhibit factor inhibiting HIF1 (FIH), an asparaginyl hydroxylase which is able to block the transcriptional activity of HIF1α by catalyzing the hydroxylation of an asparagine residue of HIF1α [135]. These effects have important implications as succinate dehydrogenase (SDH) inactivation and isocitrate dehydrogenase (IDH) neomorphic gain-of-function leading to accumulation of succinate and 2-hydroxyglutarate, respectively, contribute to HIF1α stabilization and cancer promotion [136].

Regulation of HIF1α transcriptional activity. a In normoxia, PHD hydroxylates HIF1α in several proline and asparagine residues, with 2-OG, ascorbate and Fe3+, acting as cofactors for the reaction. Hydroxyted HIF1α binds with high affinity to the E3 ubiquitin-ligase pVHL and HIF1α becomes ubiquitinated, marking it for proteasomal degradation. Through this mechanism, HIF1α is kept at very low concentrations and transcriptionally inactive. b In low O2 conditions, the activity of PHD is inhibited due to lack of oxygen, resulted in no hydroxylation nor ubiquitination of HIF1α. These events lead to the HIF1α protein stabilization which can translocate to the nucleus where it forms a complex with HIF1β and recruits CBP/p300 to the promoter of HIF1α target genes, activating the transcription of a vast array of genes responsible of glycolysis, angiogenesis and cell death resistance which are involved in tumor progression
Role of ROS in cancer cell biology
The impact of ROS in cancer research is controversial due to their dual role in promoting tumor growth, angiogenesis and metastasis or supression of tumor development, depending on the context and on the type of species generated [137–140]. ROS are highly reactive molecules with the potential to target and oxidize proteins, lipids and DNA, which derive from different sources and mechanisms (Table 2) and from environmental events, such as ultraviolet or ionizing radiation [39, 139, 141].
ROS-induced damage on DNA can lead to enhanced mutation rates, driving the transformation of normal cells into a tumorigenic phenotype. In line with this association, moderate intake of antioxidants have shown to reduce the risk of cancer development and slow cancer progression [142–145], leading to the concept that antioxidants can prevent ROS-induced damage and therefore cancer incidence. Moreover, high ROS production in cancer cells can stabilize survival factors such as HIF1α, which drive tumor initiation and progression [146]. Solid tumor formation, in turn, contributes to hypoxia development due to the disorganized vasculature, and the limited oxygen supply in solid tumors stimulates mitochondrial ROS generation and HIF1α stabilization [147, 148]. HIF1α in turn activates ROS generation, establishing a feed-forward loop where HIF1α supports its stability to promote cancer cell survival and malignant progression [141]. However, transformed cells adapt to this oxidative environment by turning on strategies that control the generation of ROS to ensure their role in proliferation signaling, while containing the damaging effects of ROS overproduction. An important strategy in this regard is the modulation of mitochondrial ROS generation, which is downregulated in cancer cells by shifting to aerobic glycolysis. This scenario suggests that reducing mitochondrial oxidation not only promotes survival of cancer cells but also increases anabolic metabolism. On the other hand, the pro-apoptotic activity of mitochondrial inhibitors are reversed by antioxidants [121, 149], lending further support for the association of ROS with tumor prevention [141]. Conversely, large-scale multicenter clinical trials of antioxidant supplementation showed a significant increase in cancer incidence [150–154]. Quite intriguingly recent preclinical studies confirmed the pro-tumorigenic and pro-metastatic effects of antioxidant supplementation such as N-acetyl-l-cysteine (NAC), a GSH precursor [155, 156], thus highlighting the relevance of antioxidants in the protection of cancer cells against oxidative damage. Therefore, antioxidant supplementation can promote the growth of tumors by rescuing the viability of cells under high oxidative stress.
A key survival strategy of cancer cells is the upregulation of antioxidant systems to detoxify the production of ROS. One central factor associated to the resistance of cancer cells is the transcription factor NF-E2-related factor (NRF2). NRF2 is a master regulator of the antioxidant response and xenobiotic metabolism through the regulation of a wide range of antioxidant and detoxification genes [157]. NRF2 is sequestered in the cytoplasm by the Kelch-like ECH-associated protein 1 (KEAP1), which acts as a NRF2 repressor and plays a pivotal role in the regulation of the NRF2 pathway. KEAP1 binds and promotes NRF2 degradation through the ubiquitin–proteasome pathway. Under oxidative stress or through particular chemical inducers, cysteine residues of KEAP1 are modified and the resulting conformational change leads to the release of NRF2, which is stabilized and translocated to the nucleus, to induce the transcription of a large number of genes [158]. In this regard, NRF2 activators, such as curcumin, butylated hydroxyanisole (BHA) or the synthetic oleane triterpenoids (CDDO), have preventive properties against carcinogenesis [157]. However, given the dual role of ROS on cancer genesis and development, NRF2 activation also provides protection to cancer cells. Therefore, NRF2 is constitutively elevated in many types of cancer cells [159–162] and this increase is associated with a poor prognosis in cancer patients [163–165]. A variety of molecular mechanisms contribute to the constitutive expression and/or stabilization of NRF2 in cancer cells. Loss-of-function by somatic mutations or epigenetic silencing of KEAP1 impairs its binding to NRF2 and abrogates its repressive effect [159, 166]. The autophagy protein P62, also named sequestrosome 1, binds and sequesters KEAP1 in autophagosomes, leading to the autophagy-dependent KEAP1 degradation, resulting in increased NRF2 stability and activation of target genes [167–170]. Overexpression of P62 or increased P62 levels due to defects in autophagy leads to persistent activation of NRF2 [171, 172], contributing to carcinogenesis [173]. In addition, activation of oncogenes such as K-RAS, BRAF and MYC stimulates the transcription of NRF2 [174]. There is substantial evidence that impaired TCA cycle activates NRF2 [175], in a similar fashion as described for HIF1α. In this case, fumarate accumulation can form adducts with KEAP1 on its cysteine residues and provoke NRF2 activation. Physiological fumarate levels are low due to the activity of fumarate hydratase (FH). However, in cancer cells with loss-of-function of FH, high levels of fumarate are associated with sustained NRF2 activation [176, 177]. Nonetheless, activation of NRF2 transcriptional activity leads to the upregulation of antioxidants and detoxifying enzymes that promote not only the survival of cancer cells but also mediate chemoresistance [178, 179]. Besides these important roles of NRF2 on detoxification, it has also been shown that NRF2 can contribute to other aspects of cancer survival such as the counteraction of cell death by BCL-2 overexpression [180] and altered metabolism by redirecting glucose and glutamine to the production of ribose-5-phosphate for nucleotide synthesis and to the regeneration of NADPH through the activation of the pentose-phosphate pathway [181].
Besides the role of ROS scavenging in cancer progression, this event is also important for cancer metastasis. Hence, it can be postulated that the supplementation of antioxidants would provide an additional advantage for cancer cells to spread to distant sites by counteracting their sensitivity to anoikis and oxidative stress. For instance, metastatic cells undergo reversible metabolic changes that allow them to counteract oxidative stress [156, 181]. Indeed, it has been recently shown that increased GSH synthesis mediates the metastatic colonization of colorectal cancer cells to the liver [182]. Conversely, other reports showed that inhibition of mitochondrial oxidative stress prevents metastasis [183, 184] and this apparent paradox might be explained by the different targets of antioxidants and their effect in different types of cancer cells [184–186]. Therefore, current antioxidant strategies are not clinically effective in cancer therapies, illustrating our limited understanding on the complex role of ROS in tumor initiation, progression and metastasis, which needs to be fully characterized to identify new and more effective therapeutic venues.
Mitochondrial cholesterol in HIF1α regulation
As mentioned above, HIF-1 is the main transcription factor regulating the cellular response to hypoxia and its stabilization is known to promote cell survival and tumor progression. While HIF-1 stabilization is mainly determined through oxygen sensing by PHD and iron availability, PHD activity is also dependent on the cytosolic levels of 2-OG. Indeed, 2-OG emerges as a potential inhibitor of angiogenesis and cellular transformation by promoting the degradation of HIF1 [187, 188].
HIF-1α activation contributes to the metabolic reprogramming of cancer cells by impairing mitochondrial phosphorylation and the subsequent stimulation of aerobic glycolysis. Although the physiological levels of mitochondrial cholesterol are low, mitochondrial cholesterol accumulation impairs mitochondrial function and the activity of the mitochondrial 2-oxoglutarate carrier (2-OGC), which exchanges cytosolic GSH by matrix 2-OG. As StARD1 promotes mitochondrial cholesterol accumulation in the inner membrane, StARD1 induction thus contributes to the impairment of OGC carrier, resulting in the depletion of 2-OG in the cytosol and GSH in the mitochondrial matrix (Fig. 2). As mentioned above, mGSH is a key mitochondrial antioxidant that controls hydrogen peroxide production [39, 189, 190]. Moreover, mitochondrial ROS generation has been shown to promote HIF-1 α stabilization [147, 191]. Thus, it is conceivable that StARD1 induction and the subsequent accumulation of cholesterol in mitochondria result in the depletion of cytosolic 2-OG, impairing PHD activation and subsequent HIF-1α stabilization. Therefore, mitochondrial cholesterol loading may have an important role in cancer cell survival by a dual effect through impairment in mitochondrial function and dynamics, while promoting HIF1α stabilization via depletion of cytosolic 2-OG levels and generation of mitochondrial ROS. As a result, mitochondrial cholesterol loading in cancer cells acts as an additional mechanism governing angiogenesis and novel vessel growth via HIF1α stabilization, although this molecular link deserves to be further tested and it is currently under investigation. Finally, histone lysine demethylases have been recognized as important players in cancer cell biology by removing methyl moieties from DNA and aberrant expression of these chromatin modifying enzymes is implicated in the course of tumor initiation and progression [192]. Like PHD, histone methyl demethylases are also dependent on iron and 2-OG, and therefore mitochondrial cholesterol loading may further modulate cancer progression by the regulation of histone lysine demethylases via limitation of cytosolic 2-OG levels, which deserves further investigation.

Mitochondrial cholesterol-mediated HIF1α stabilization. Mitochondrial cholesterol loading mediated by StARD1 decreases mitochondrial membrane fluidity which leads to an impaired activity of the OGC, which exchanges mitochondrial 2-OG by cytosolic GSH. Cytosolic 2-OG depletion may promote HIF1α stabilization by the impairment of PHD due to their requirement of 2-OG as a cofactor for HIF1α hydroxylation and subsequent degradation. Moreover, 2-OGC inhibition results in mGSH depletion, which in turn, limits the detoxification of ROS, in particular, H2O2. The subsequent increase in ROS and oxidative stress impact negatively on PHD, resulting in HIF1α stabilization
Role of mitochondria and cholesterol in cancer cell death and chemotherapy resistance
Mitochondria and cell death resistance
Cancer cells have evolved multiple mechanisms to disable programmed cell death to support their survival and proliferation. Given that mitochondria are key players in several pathways of programmed cell death (see above) many strategic battles regulating cell death resistance take place in mitochondria [49]. The most prominent example of this is the overexpression of pro-survival BCL-2 proteins, a common feature in diverse cancers. The gene encoding BCL-2 was first identified in a chromosomal translocation that resulted in constitutively high levels of BCL-2 in neoplastic B cells [193, 194]. Different mechanisms such as genomic copy number amplification, oncogenic transcriptional upregulation or downregulation of microRNA repressors or stabilization of BCL-2 family members contribute to the maintenance of high levels of Bcl-2 [195, 196]. On the other hand, due to genomic deletion or promoter methylation leading to transcriptional silencing, loss-of-function of several pro-apoptotic proteins such as BAK, BAX and BH3-only family members have been observed in a variety of cancer types. Although BAX and BAK can play redundant roles, recent experimental data argues that in the context of activation of BH3-only protein or anti-apoptotic BCL-2 there is a strict dependence of either BAX or BAK [197, 198]. Although cancer cells are generally resistant to apoptosis, certain stress conditions, such as hypoxia and low nutrient availability, lower the threshold for apoptosis susceptibility. Cancer cells often exhibit higher levels of pro-apoptotic BH3-only protein, which is accompanied by higher anti-apoptotic BCL-2 proteins to antagonize apoptosis. This state has been termed as cancer cells “primed for death” [199] and this dependence on anti-apoptotic BCL-2 proteins can be exploited to design more effective pro-apoptotic therapeutic strategies [200].
Loss–of-function of TP53 is found in more than 50 % of human cancers. In addition to the above-mentioned roles of its inactivation in cancer cell metabolism, TP53 is central in the orchestration of cell death pathways upon cellular stress such as DNA damage by stimulating the transcription of pro-apoptotic proteins (PUMA, BAX), autophagy and cell-cycle arrest. TP53 exerts a vast array of extranuclear functions and therefore the cytoplasmic pool of TP53 cooperates with its nuclear counterpart to activate programmed cell death in response to certain cellular stresses. TP53 is involved in various forms of cell death such as apoptosis, necrosis and necroptosis and is able to mediate both MOMP and MPT opening in response to death stimuli. After stress induced TP53 activation, a small fraction translocates to MOM, resulting in the activation of the intrinsic apoptotic pathway [201]. TP53-mediated MOMP is related to the ability to bind and inactivate anti-apoptotic BCL-2 and BCL-xL, and to transcriptionally induce the expression and activation of pro-apoptotic proteins by direct binding [202]. Moreover, TP53 regulates MPT openings of necrosis/necroptosis via cyclophillin D and dynamin-related protein 1 (DRP1) [203, 204] in response to specific cell death triggers, such as TNFα or oxidative stress. In addition, TP53 inhibits autophagy [205], resulting in impaired mitophagy, contributing to the reduced threshold for cell death. Given these protective roles against specific alterations in cell cycle and cell death resistance, many cancer-associated TP53 mutations have been identified. Although most of TP53 mutations has been described as loss-of-function, it has been proposed that some TP53 mutations may have oncogenic capabilities [206].
Although MOMP is considered a point of no return for apoptosis, cancer cells are able to inhibit caspases ensuring survival in certain conditions. This mechanism described in some post-mitotic cells, such as neurons and certain cancer cells, allows the recovery of cancer cells provided that MOMP-inducing stimuli are removed [207–209]. Caspases can be directly inhibited by XIAP or by the neutralization of its inhibitors [200]. In addition, cytochrome c released through MOMP can be targeted for proteasomal degradation thereby avoiding the assembly of the apoptosome [209]. Besides caspase inhibition, survival after MOMP requires a pool of intact mitochondria in which MOMP has not been triggered [210]. The selective maintenance of cells with intact mitochondria may contribute to carcinogenesis and cancer relapse after cytotoxic therapies due to the increased susceptibility to oncogenic transformation [211]. Moreover, limited mitochondrial permeabilization induced by sub-lethal apoptosis triggers can promote DNA damage, genomic instability and ultimately carcinogenesis [212, 213]. This mechanism would have two important implications for cancer progression. First, low-level limited apoptosis can drive mutagenesis in surviving cancer cells, serving as a driving force towards malignancy. Second, sub-lethal apoptotic anticancer therapies can increase the tumorigenic potential of surviving cancer cells by promoting new mutations that favor relapse and chemotherapy resistance.
Mitochondrial cholesterol in cell death and chemotherapy resistance
As mentioned above, cholesterol trafficking to mitochondria has been reported in tumor cells, including mitochondria from HCC due to overexpression of StARD1 [17]. Mitochondrial cholesterol loading in cancer cells may account for the recognized mitochondrial dysfunction and resistance to BAX-mediated cell death induced by chemotherapeutic agents that target mitochondria to elicit MOMP. In line with this, treatments that resulted in mitochondrial cholesterol loading in tumor cells impaired stress-induced apoptosis [17, 18], while StARD1 knockdown or treatments that resulted in downregulation of cholesterol loading sensitized HCC cells to chemotherapy [17]. Isolated mitochondria from HCC with increased cholesterol levels have been reported to be resistant to MOMP and release of cytochrome c or smac/DIABLO in response to various stimuli, such as MPT triggers and active BAX. In agreement with these findings, HeLa cells treated with the amphiphilic amine U18666, which perturbs intracellular cholesterol trafficking and stimulates mitochondrial cholesterol accumulation, impairs MOMP and the release of cytochrome c in response to BAX [18]. Furthermore, ABCA1 downregulation determines resistance to chemotherapy through increased mitochondrial cholesterol accumulation [95]. Similar behavior was observed in cholesterol-enriched mitochondria or liposomes and reversed by restoring mitochondrial membrane order or cholesterol extraction. Cholesterol inhibited the membrane-permeabilizing activity of tBID/BAX or BAX pre-oligomerized with octylglucoside in a dose-dependent manner. Similar to the effect found on BAX, cholesterol also decreased the permeabilizing activity of melittin, a widely studied antimicrobial peptide, which induces membrane permeabilization by forming lipid-containing toroidal pores rather than through the formation of protein channels [17]. These findings indicate that cholesterol-mediated decrease in membrane fluidity of the bilayer directly modulates BAX pro-apoptotic activity by reducing the capacity of BAX to insert into the lipid matrix of the membrane, underlying the anti-apoptotic role of mitochondrial cholesterol accumulation in cancer cells. Thus, mitochondrial cholesterol contributes to chemotherapy resistance in HCC by increasing membrane order and resistance of MOM to MOMP. As StARD1 regulates mitochondrial cholesterol trafficking, it is conceivable that this member of the StART family stands as a novel target to regulate cancer cell death and chemotherapy response.
Cancer biology and therapeutics
As described in the previous sections, cancer cells exhibit critical metabolic transformations induced by mutations that result in cell cycle deregulation associated with enhanced cellular stress. Adaptation to this stress phenotype is required for cancer cells to survive and involves the participation of genes that regulate metabolism, bioenergetics, cell death and ROS detoxification (Fig. 3). In this context, small molecules that selectively kill cancer cells while sparing normal surrounding cells, are the desired approach for the treatment of cancer. To this aim, cancer therapeutics should target the differential features of cancer cells. Here, we briefly summarize the therapeutic strategies that involve mitochondria and their proposed mechanism of action to selectively target transformed cells.

General summary of altered mitochondrial functions in cancer cell life and death. a In normal non-transformed cells glucose is mainly metabolized through the glycolysis pathway and the resulting pyruvate enters the mitochondrial TCA cycle, producing reduced equivalents that are fed into the ETC to generate ATP with high efficiency through the OXPHOS. Antioxidant defenses and coupled respiration through OXPHOS maintain low levels of mitochondrial ROS. b Normal cells are sensitive to apoptotic stimuli triggered by different stresses that finally converge in BAK/BAX activation and MOMP with subsequent release of cytochrome c into the cytosol, stimulating the formation of the apoptosome and apoptotic cell death. c In cancer cells gain-of-function of oncogenes and loss-of-function of tumor suppressor genes (such as MYC, HIF1α and TP53) results in altered metabolism, exemplified by the Warburg effect, characterized by high glucose consumption rates. Glucose is degraded through glycolysis to obtain biosynthetic intermediates and the resulting pyruvate is reduced to lactate to generate ATP. In this scenario, mitochondrial TCA is diverted to generate biosynthetic intermediates, which is accompanied by low OXPHOS activity and increased mitochondrial ROS production. NRF2 is upregulated in cancer cells to counteract ROS and permits cancer cells to withstand its deleterious effects. d Cancer cells, through the overexpression of anti-apoptotic proteins or inactivation of pro-apoptotic proteins, counteract the action of BAX/BAK and evade MOMP formation. Besides this effect, mitochondrial cholesterol loading shields mitochondrial membrane, impairing BAK/BAX oligomerization in MOM and subsequent MOMP formation and represents an additional mechanism of cell death resistance in tumor cells
Therapeutics aimed at cancer metabolism and bioenergetics
Cells are addictive to glucose and glutamine and their limitation can cause cell death. This dependence is driven by the activation of MYC and HIF1-α [109] and consequently, targeting pathways regulating glucose/glutamine metabolism may be of relevance for cancer treatment. The specific GLS1 inhibitor bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) inhibits proliferation of lymphoma cells but has no effect on neuroblastoma cells, which express GLS2 [214, 215], implying that the general GLS inhibitor 6-diazo-5-oxo-L-norleucine (DON) may exhibit broader antitumor effects [216, 217]. Inhibitors of glutamate dehydrogenase (GDH) are promising agents to target glutamine addiction of certain cancer cells. For instance, the green tea component epigallocatechin-3-gallate (EGCG), which inhibits GDH, has been shown to promote apoptosis in several cancers types, resulting in tumor growth inhibition, setting the basis for the exploration of its efficacy in phase II clinical trials [214, 218].
The inhibition of aerobic glycolysis in cancer cells is also of potential relevance. While inhibitors such as 2-deoxy-d-glucose and ionidamine, which targets early steps in the glycolysis pathway, exhibit severe toxic side effects [218], inhibition of distal steps in glycolysis are effective. Inhibition of lactate production by inactivation of lactate dehydrogenase (LDH) reduces tumorigenicity in several cancer models [219–221]. Additionally, the specific inhibitor of LDHA, FX11, reduces tumor progression in lymphoma in vivo [219]. In some cancers LDHB can replace LDHA, hence limiting the efficacy of the LDHA inhibitors [109, 222]. Inhibition of lactate export from cancer cells results in wide-reaching consequences, leading not only to lactate accumulation, alterations in glycolytic intermediates, reduction in glucose transport and ATP, NADP and GSH levels but also in mitochondrial damage and cell death [223], suggesting that inhibition of lactate transporter MCT1 is a suitable therapeutic approach. Moreover, the effect of small molecules that block the entry of pyruvate to the mitochondrial TCA cycle, such as dichloroacetate, a PDK1 inhibitor, may be of potential relevance [109, 218, 224].
Although targeting glycosis may be effective in a specific population of cancer cells exhibiting a highly glycolytic dependence, stem cancer cells that rely on OXPHOS might become resistant. Moreover, although mitochondrial oxidation under the Warburg effect is dramatically reduced, many cancer cells still have a central requirement on mitochondrial metabolism, strongly suggesting that OXPHOS inhibitors might represent an important target for drug-resistant cancers [121]. A key agent with potential relevance in inhibiting OXPHOS is metformin, one of the most prescribed drugs around the world for the treatment of type II diabetes [225–228]. Metformin is an indirect activator of AMP-activated Kinase (AMPK) through inhibition of mitochondrial complex I, resulting in the activation of the ATM/LKB1/AMPK axis. LKB1 is a well-characterized tumor suppressor in pancreatic, lung cancer and melanoma. AMPK activation inhibits the mTOR pathway and this effect accounts for the potential antineoplastic effects of metformin in breast and renal tumors. Moreover, metformin reduces glycolysis and increases mitochondrial respiration in tumors, and these events are associated with growth arrest [229]. In addition, metformin exhibits antiangiogenic effects, which contribute to its antineoplastic properties [230]. Other compounds with mild OXPHOS inhibition such as tamoxifen, which also inhibits complex I, resveratrol, which antagonizes complex III, and the complex V inhibitor 3,3-diindolylmethane have potential in cancer treatment. VLX600 is a novel compound targeting OXPHOS that inhibits tumor growth of colon carcinoma cells, thus exhibiting potential application in clinical trials [231]. Besides, a number of emerging mitochondrial inhibitors successfully used in experimental studies could be effective against cancer cells and might synergize with chemotherapeutics [149, 232, 233].
Therapeutics targeting cancer cell death
Given that BCL-2 is overexpressed in many tumors, most strategies to engage apoptosis pathways are based in the blockade of anti-apoptotic members of the BCL-2 family. BCL-2 inhibitors have been developed based on the structure of BH3-binding groove of BCL-xL [234], leading to the development of the prototypic BH3 mimetic that displays sub-nanomolar affinity for BCL-xL and a binding profile similar to the BH3-only protein BAD. The BH3 mimetic ABT-737 and the more soluble analogue ABT-263 bind BCL-xL, BCL-2 but not MCL1 and both show antitumor activities either as single agents or in combination [235]. However, the clinical applicability of these BH3 mimetics is limited due to severe thrombocytopenia mediated by platelet apoptosis [236, 237]. ABT-199, a novel BH3 mimetic developed from the structure of ABT-263 [238], is effective in chronic lymphocyte leukemia. A potential side effect of ABT-199 is the induction of tumor lysis syndrome [239], which can be controled by step-wise dose escalation. Although the therapeutic results with BH3 mimetics are promising, resistance is a potential drawback. For instance, BCL-2 mutations that abrogated binding of BH3 mimetics mediate resistance of ABT-199 in experimental lymphoma models [240]. As BCL-2 inhibitors do not target MCL-1, a key anti-apoptotic BCL-2 member, the efficiency of BH3 mimetics may be limited, particularly in the treatment of solid tumors [241–243]. Hence, the combination of specific MCL1 inhibitors [244, 245] with BH3 mimetics is a promising therapeutic approach to overcome chemotherapy resistance. Moreover, as MCL-1 plays a key role in mitochondrial physiology and autophagy, targeting MCL-1 may cause undesirable side effects [246, 247]. For instance, the toxicity of pan-BCL-2 inhibitors, such as Gossypol or Obatoclax, which inhibit BCL-2 and MCL1, prevented the evaluation of their efficacy in clinical settings [243]. These findings suggest that MCL1 inhibition should be fine-tuned and that the relative contribution of BCL-2-family components to the apoptosis resistance of cancer cells should be carefully evaluated through the “BH3 profiling” to determine the therapeutic window [248, 249]. In addition, incomplete cell death caused by triggers of mitochondrial apoptosis can promote genomic instability and mutagenesis derived from the incomplete MOMP and caspase-dependent DNA cleavage, contributing to tumor relapse and the acquisition of drug resistance [212, 213]. Based on the ability of TP53 to induce apoptosis, mitochondrial targeted TP53 fusion proteins have been developed to induce intrinsic apoptosis in cancer cells, which may be of relevance in adjuvant therapy for cancer treatment [201, 250]. Overall, targeting or sensitizing cancer cells to apoptosis is a promising strategy currently under development, which may lead to personalized medicine through specific tumor-profiling and fine-tuning dosage and therapy combinations.
Therapeutics targeting cancer cell ROS sensitivity
Despite generation of higher ROS levels cancer cells are more sensitive to intracellular ROS induction than untransformed cancer cells. Many cancer chemotherapeutic agents, including taxanes, vinca alkaloids, platinum coordination complexes, paclitaxel and elesclomol are currently used to induce high levels of ROS to kill cancer cells [251]. The ultimate effect of these molecules is determined by the intrinsic antioxidant capacity of cancer cells as the cytotoxic potential of these agents is lost upon antioxidant co-treatment [252–254].
A key mechanism to counteract the generation of ROS by chemotherapeutic agents is the regulation of GSH homeostasis [137, 182]. Several small molecules, which modulate ROS, such as β-phenethyl isothiocyanate (PEITC), buthionine sulphoximine (BSO), curcumin or CDDO derivatives, have potential therapeutic effects for the treatment of cancer by promoting mGSH deletion and subsequent ROS generation specifically in cancer cells [255–258]. BSO, an inhibitor of glutamate-cysteine ligase, which is the rate-limiting enzyme in GSH biosynthesis [259] is the only clinically used drug to suppress the novo GSH synthesis. The simultaneous administration of BSO and the thioredoxin inhibitor auranofin induce ROS and clonogenic killing in carcinoma cells [260]. Sulfasalazine, which inhibits cystine uptake via XcL carrier, limits cysteine availability impairing GSH biosynthesis, which leads to reduced growth and viability of cancer cells in vitro and in vivo [261, 262]. In addition, specific mGSH depletion has also been associated with apoptosis induced by chemotherapeutic drugs. For example, the triterpenoid methyl CDDO derivative (CDDO-Me), induces cytotoxicity in chemotherapy-resistant myeloid leukemia cells and this event is associated with selective depletion of mGSH, resulting in increased ROS generation [263, 264]. Moreover, PEITC depletes mGSH and consequently increases ROS and nitric oxide, contributing to inhibition of the mitochondrial complex I, suppression of mitochondrial respiration, and subsequent cytotoxicity of leukemia cells [265]. Using a cell-based small-molecule screening and quantitative proteomics, piperlongumine has emerged as a cytotoxic agent that triggers apoptosis and necrosis in leukemia cells [266]. Interestingly, piperlongumine induces ROS generation and cell death in transformed cells but not primary normal cells [267]. Piperlongumine also decreases GSH and increases GSSG levels in cancer cells without effects in nontransformed cells, and these effects parallel the ability of piperlongumine to alter mitochondrial morphology and function. Consequently, co-treatment with piperlongumine and NAC prevented piperlongumine-mediated GSH depletion and cell death in cancer cells. These findings support the concept that cancer cells have high levels of ROS, and hence, have a strong reliance on the ROS stress-response pathway driven by NRF2.
At present, radiotherapy is widely used in various types of cancer treatments, and the therapeutic effect is mainly determined by ROS generation. The induction of water radiolysis occurs in seconds after ionizing radiation, lasts several hours after exposure and enhances ROS generation and oxidative stress [268, 269]. Some studies suggested that antioxidant supplementation could sensitize cancer cells to chemo- or radio-therapy and reduce their side effects by protecting the normal cells [270]. However, other studies indicated that antioxidants may also protect cancer cells against these therapies [252, 271, 272]. Therefore, the safety and efficacy of the combined treatment of antioxidants with radiotherapy or ROS-inducing chemotherapy remain controversial.
Strategies targeting the mevalonate pathway and cholesterol synthesis in cancer
As described above, cancer cells exhibit alterations in the regulation of cholesterol homeostasis and de novo synthesis in the mevalonate pathway. Despite that the main therapeutic benefit of statins is the prevention of cardiovascular diseases and heart attacks, the use of statins has been associated with lower incidence of colorectal carcinoma, melanoma, prostate cancer and HCC, although the benefit of statins in other types of cancer has been disappointing [273]. While statins inhibit cholesterol synthesis, they also affect other intermediates of the mevalonate pathway, including isoprenoids, and therefore the beneficial effects of statins in cancer may be independent of cholesterol synthesis. For instance, statins inhibit the activation of the proteasome pathway, contributing to the maintenance of proteins that block cell cycle. Through cholesterol downregulation, statins regulate the function of Hedgehog, a signaling pathway involved in carcinogenesis [273]. Besides these wide-reaching effects of statins, their benefit in cancer treatment is limited due to the complex regulation of HMGCoAR and the metabolites generated in the mevalonate pathway. Reduction of isoprenoid and cholesterol levels in cancer by chronic treatment with statins leads to upregulation of HMG-CoAR levels and eventually development of resistance [274]. In vitro mechanistic studies of statins used significantly higher concentrations than those that were therapeutically achievable in phase I trials. Dose-limiting toxicities, including gastrointestinal side effects, myelotoxicity, myalgias, elevation of creatine phosphokinase and hepatotoxicity, precluded further dose increase in clinical trials [275]. Inhibition of SS has attracted much interest as a pharmacological target as it implies the inhibition of cholesterol synthesis without depressing isoprenoid levels. For instance, lapaquistat (TAK-475, Takeda), a SS inhibitor, progressed to phase III clinical trials, although its outcome in cancer remains to be established due to hepatotoxic effects at high dosing [276]. As mentioned before, prenylation is a key postranslational mechanism of targeted proteins, and many prenylated proteins are involved in various aspects of carcinogenesis, including cellular proliferation, apoptosis, angiogenesis and metastasis. Farnesylation is catalyzed by farnesyltransferase (FTase) and geranylgeranylation by geranylgeranyltransferase, GGTase. Given the role of protein prenylation in carcinogenesis, FTase inhibitors (FTIs) and GGTase inhibitors (GGTIs) have been developed for cancer treatment. GGTI–FTI combinations synergistically inhibit proliferation of multiple myeloma cell lines and primary cells, and induce apoptosis. Interestingly, dual prenylation inhibitors (DPIs) that block both FTase and GGTase enzymatic activities have been shown to induce apoptosis in PSN-1 pancreatic tumor cells by blocking K-Ras prenylation compared to either FTI or GGTI agents alone [277]. H and N-Ras prenynation is effectively inhibited by FTIs and only partially by GGTIs, whereas K-Ras prenylation requires both FTIs and GGTIs inhibition [278]. Thus, combined inhibition of geranylgeranylation and farnesylation can overcome the resistance conferred by cross-prenylation, thus potentiating the activity of either FTIs or GGTIs alone. Finally, targeting the specific targeting of cholesterol to mitochondria may be an additional approach of potential benefit in cancer treatment by modulating cell death and chemotherapy resistance. This specific field is currently under investigation to identify potential specific inhibitors of StARD1 and MLN64 to sensitize cancer cells to cell death triggers and chemoterapeutic agents.
Conclusions and future approaches
Cancer cells undergo an array of genetic and epigenetic modifications that lead to a phenotype characterized by high proliferation, death resistance, rapid growth and invasiveness. Mitochondria play an essential role in metabolism, bioenergetics and cell death regulation and consequently oncogenic modifications characteristic of many cancer types mediate the array of metabolic alterations of cancer cells by impairing key mitochondrial functions. This continuum evolving process in the adquisition of a highly proliferative phenotype requires the selection of cells with decreased mitochondrial oxidation of fuels, relying on the oxidation of glucose for ATP generation, resulting secondarily in the engagement of the pentose phosphate pathway as a source of reducing equivalents needed for anabolism and antioxidant defense. These metabolic alterations are accompanied by the involvement of mitochondria in biosynthetic pathways to support continuous growth, while reducing the deleterious effects of high-rate production of ROS, a characteristic feature of cancer cells. Furthermore, mitochondria undergo changes in membrane dynamics, exemplified by the decrease in membrane fluidity to protect cancer cells against the induction of programmed cell death triggered by the immune system or by metabolic or xenobiotic stresses. A key player in this event is the accumulation of cholesterol in mitochondria of cancer cells, which increases the threshold for MOMP by restructuring mitochondrial membrane bilayers. Besides this function, mitochondrial cholesterol accumulation may indirectly contribute to the metabolic changes of cancer cells by impairing mitochondria function and activation of survival programs turned on by HIF1α activation. Given these functions of mitochondrial cholesterol, preventing or reversing this process may be of relevance in cancer cell biology to shift the balance towards increased apoptosis susceptibility and sensitization to chemotherapy. In addition, oncogenes, transcription factors (e.g. MYC, HIF1α, NRF2) and inactivation of tumor suppressors, such as TP53, allow invasiveness and chemoresistance, in part, by regulating mitochondrial function and metabolism as well as by controling the outcome of ROS generation. Metabolic stress, immune surveillance and chemotherapy act as a selective pressure that allows only the survival of cells with specific features, driving cancer cells towards a highly glycolytic, apoptosis-incompetent and invasive phenotype. Given the complexity in the metabolic alterations of cancer cells mediated largely through alterations in mitochondrial function, further research is required to identify more efficient strategies for cancer treatment involving the use of small molecules targeting mitochondrial metabolism.
Abbreviations
- ROS:
-
reactive oxygen species
- OXPHOS:
-
oxidative phosphorylation
- HMGCoAR:
-
3-hydroxy-3-methyl-glutaryl-CoA reductase
- HCC:
-
hepatocellular carcinoma
- TCGA:
-
cancer genome atlas
- mtDNA:
-
mitochondrial DNA
- MIM:
-
mitochondrial inner membrane
- MOM:
-
mitochondrial outer membrane
- ALR:
-
augmenter of liver regeneration
- ETC:
-
electron transport chain
- Δψm :
-
mitochondrial transmembrane potential
- TCA:
-
tricarboxylic acid cycle
- ACLY:
-
ATP citrate lyase
- mGSH:
-
mitochondrial glutathione
- H2O2 :
-
hydrogen peroxide
- SOD:
-
superoxide dismutases
- 2-OG:
-
2-oxoglutrarate
- GPX:
-
glutathione peroxidase
- mGSSG:
-
mitochondrial oxidized glutathione
- Prx/Trx:
-
peroxiredoxin/thioredoxin
- MOMP:
-
mitochondrial outer membrane permeabilization
- IMS:
-
mitochondrial intermembrane space
- BOK:
-
BCL-2 ovarian killer
- MPT:
-
mitochondrial permeability transition
- VDAC:
-
voltage-dependent anion channel
- ANT:
-
adenine nucleotide translocase
- TSPO:
-
translocator protein
- RIPK1:
-
receptor-interacting serine/threonine-protein kinase 1
- MLKL:
-
mixed lineage kinase domain-like protein
- IPP:
-
isopentenyl pyrophosphate
- DMAPP:
-
dimethyl allyl pyrophosphate
- GPP:
-
geranyl pyrophosphate
- FFP:
-
farnesyl pyrophosphate
- GGPP:
-
geranyl geranyl pyrophosphate
- SS:
-
squalene synthase
- ER:
-
endoplasmic reticulum
- ABCA1:
-
ATP binding cassette transporter A1
- StARD1:
-
steroidogenic acute regulatory domain 1
- PET:
-
positron emission tomography
- PDK1:
-
pyruvate dehydrogenase kinase
- ARK5:
-
AMPK-related protein kinase 5
- PGC1α:
-
peroxisome proliferator-activated receptor gamma coactivator 1 alpha
- GLS2:
-
glutaminase 2
- HIF1α:
-
hypoxia-inducible factor 1 alpha
- PHD:
-
HIF-proly-hydroxylase
- pVHL:
-
Von Hippel-Landau ubiquitin ligase
- LDHA:
-
lactate dehydrogenase A
- MCT4:
-
monocarboxylate transporter 4
- COX4-2:
-
cytochrome c oxidase subunit 4–2
- BNIP3:
-
BCL-2/adenovirus E1B 19-kDa interacting protein 3
- 2-OG:
-
2-oxoglutarate
- FIH:
-
factor inhibiting HIF1
- SDH:
-
succinate dehydrogenase
- IDH:
-
isocitrate dehydrogenase
- NAC:
-
N-acetyl-l-cysteine
- NRF2:
-
NF-E2-related factor
- KEAP1:
-
Kelch-like ECH-associated protein 1
- BHA:
-
butylated hydroxyanisole
- CDDO:
-
2-cyano-3,12-dioxooleana-1,9-diene-28-oic acid
- FH:
-
fumarate hydratase
- 2-OGC:
-
2-oxoglutarate carrier
- DRP1:
-
dynamin-related protein 1
- BPTES:
-
bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- DON:
-
6-diazo-5-oxo-L-norleucine
- GDH:
-
glutamate dehydrogenase
- EGCG:
-
epigallocatechin-3-gallate
- LDH:
-
lactate dehydrogenase
- AMPK:
-
AMP-activated Kinase
- PEITC:
-
β-phenethyl isothiocyanate
- BSO:
-
buthionine sulfoximine
- FTase:
-
farnesyltransferase
- FTI:
-
farnesyltransferase inhibitors
- GGTI:
-
geranyl geranyl transferase inhibitor
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC et al (2009) Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 1793(9):1524–1532. doi:10.1016/j.bbamcr.2009.01.006
Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E et al (2010) Mitochondrial gateways to cancer. Mol Aspects Med 31(1):1–20. doi:10.1016/j.mam.2009.08.002
Raimundo N (2014) Mitochondrial pathology: stress signals from the energy factory. Trend Mol Med. doi:10.1016/j.molmed.2014.01.005
Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA et al (2008) LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134(1):97–111. doi:10.1016/j.cell.2008.04.052
Lo Sasso G, Celli N, Caboni M, Murzilli S, Salvatore L, Morgano A et al (2010) Down-regulation of the LXR transcriptome provides the requisite cholesterol levels to proliferating hepatocytes. Hepatology 51(4):1334–1344. doi:10.1002/hep.23436
Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA et al (2010) Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA 107(34):15051–15056. doi:10.1073/pnas.0910258107
Dang CV (2012) Links between metabolism and cancer. Gene Dev 26(9):877–890. doi:10.1101/gad.189365.112
Borena W, Strohmaier S, Lukanova A, Bjorge T, Lindkvist B, Hallmans G et al (2012) Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int J Cancer 131(1):193–200. doi:10.1002/ijc.26338
Singh S, Singh PP (2014) Statins for prevention of hepatocellular cancer: one step closer? Hepatology 59(2):724–726. doi:10.1002/hep.26614
Stryjkowska-Gora A, Karczmarek-Borowska B, Gora T, Krawczak K (2015) Statins and cancers. Contemp Oncol. 19(3):167–175. doi:10.5114/wo.2014.44294
Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J (2014) Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol Syst Biol. 10:721. doi:10.1002/msb.145122
Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q et al (2011) MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 71(6):2286–2297. doi:10.1158/0008-5472.CAN-10-3367
Mansourian PG, Yoneda M, Krishna Rao M, Martinez FJ, Thomas E, Schiff ER (2014) Effects of statins on the risk of hepatocellular carcinoma. Gastroenterol & Hepatol. 10(7):417–426
Cancer Genome Atlas Research, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA et al (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45(10):1113–1120. doi:10.1038/ng.2764
Kuzu OF, Noory MA, Robertson GP (2016) The role of cholesterol in cancer. Cancer Res 76(8):2063–2070. doi:10.1158/0008-5472.CAN-15-2613
Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basanez G et al (2008) Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res 68(13):5246–5256. doi:10.1158/0008-5472.CAN-07-6161
Lucken-Ardjomande S, Montessuit S, Martinou JC (2008) Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ 15(3):484–493. doi:10.1038/sj.cdd.4402280
Mishra P, Chan DC (2014) Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15(10):634–646. doi:10.1038/nrm3877
Sukhorukov VM, Meyer-Hermann M (2015) Structural Heterogeneity of mitochondria induced by the microtubule cytoskeleton. Sci Rep. 5:13924. doi:10.1038/srep13924
Patergnani S, Missiroli S, Marchi S, Giorgi C (2015) Mitochondria-associated endoplasmic reticulum membranes microenvironment: targeting autophagic and apoptotic pathways in cancer therapy. Front Oncol. 5:173. doi:10.3389/fonc.2015.00173
Ghosh S, Singh KK, Sengupta S, Scaria V (2015) Mitoepigenetics: the different shades of grey. Mitochondrion 25:60–66. doi:10.1016/j.mito.2015.09.003
Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A et al (2004) Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J 23(19):3735–3746. doi:10.1038/sj.emboj.7600389
Fischer M, Horn S, Belkacemi A, Kojer K, Petrungaro C, Habich M et al (2013) Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol Biol Cell 24(14):2160–2170. doi:10.1091/mbc.E12-12-0862
Gandhi CR, Chaillet JR, Nalesnik MA, Kumar S, Dangi A, Demetris AJ et al (2014) Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology 148(2):379–391. doi:10.1053/j.gastro.2014.10.008
Maehara Y, Fernandez-Checa JC (2015) Augmenter of liver regeneration links mitochondrial function to steatohepatitis and hepatocellular carcinoma. Gastroenterology 148(2):285–288. doi:10.1053/j.gastro.2014.12.013
Calvo SE, Clauser KR, Mootha VK (2015) MitoCarta 2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. doi:10.1093/nar/gkv1003
Venditti P, Di Stefano L, Di Meo S (2013) Mitochondrial metabolism of reactive oxygen species. Mitochondrion 13(2):71–82. doi:10.1016/j.mito.2013.01.008
Sun F, Zhou Q, Pang X, Xu Y, Rao Z (2013) Revealing various coupling of electron transfer and proton pumping in mitochondrial respiratory chain. Curr Opin Struct Biol 23(4):526–538. doi:10.1016/j.sbi.2013.06.013
Kulawiak B, Hopker J, Gebert M, Guiard B, Wiedemann N, Gebert N (2013) The mitochondrial protein import machinery has multiple connections to the respiratory chain. Biochim Biophys Acta 1827(5):612–626. doi:10.1016/j.bbabio.2012.12.004
Cheng Z, Ristow M (2013) Mitochondria and metabolic homeostasis. Antioxid Redox Signal 19(3):240–242. doi:10.1089/ars.2013.5255
Hammerman PS, Fox CJ, Thompson CB (2004) Beginnings of a signal-transduction pathway for bioenergetic control of cell survival. Trends Biochem Sci 29(11):586–592. doi:10.1016/j.tibs.2004.09.008
Renault TT, Chipuk JE (2013) Inter-organellar communication with mitochondria regulates both the intrinsic and extrinsic pathways of apoptosis. Commun Integr Biol. 6(2):e22872. doi:10.4161/cib.22872
Arnould T, Michel S, Renard P (2015) Mitochondria retrograde signaling and the UPR mt: where are we in mammals? Int J Mol Sci 16(8):18224–18251. doi:10.3390/ijms160818224
Guha M, Avadhani NG (2013) Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 13(6):577–591. doi:10.1016/j.mito.2013.08.007
Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45(7–8):466–472. doi:10.1016/j.exger.2010.01.003
Quinlan CL, Perevoschikova IV, Goncalves RL, Hey-Mogensen M, Brand MD (2013) The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzymol 526:189–217. doi:10.1016/B978-0-12-405883-5.00012-0
Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1:304–312. doi:10.1016/j.redox.2013.04.005
Ribas V, Garcia-Ruiz C, Fernandez-Checa JC (2014) Glutathione and mitochondria. Front Pharmacol doi:10.3389/fphar.2014.00151
Cadenas E, Davies KJ (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biol Med 29(3–4):222–230
Mari M, Colell A, Morales A, von Montfort C, Garcia-Ruiz C, Fernandez-Checa JC (2010) Redox control of liver function in health and disease. Antioxid Redox Signal 12(11):1295–1331. doi:10.1089/ars.2009.2634
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48(2):158–167. doi:10.1016/j.molcel.2012.09.025
Mari M, Morales A, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC (2013) Mitochondrial glutathione: features, regulation and role in disease. Biochim Biophys Acta 1830(5):3317–3328. doi:10.1016/j.bbagen.2012.10.018
McCommis KS, McGee AM, Laughlin MH, Bowles DK, Baines CP (2011) Hypercholesterolemia increases mitochondrial oxidative stress and enhances the MPT response in the porcine myocardium: beneficial effects of chronic exercise. Am J Physiol Regul Integr Comp Physiol 301(5):R1250–R1258. doi:10.1152/ajpregu.00841.2010
Sies H (2014) Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem 289(13):8735–8741. doi:10.1074/jbc.R113.544635
Kil IS, Ryu KW, Lee SK, Kim JY, Chu SY, Kim JH et al (2015) Circadian oscillation of sulfiredoxin in the mitochondria. Mol Cell 59(4):651–663. doi:10.1016/j.molcel.2015.06.031
Taylor RC, Cullen SP, Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9(3):231–241. doi:10.1038/nrm2312
LiM X, Dewson G (2015) Mitochondria and apoptosis: emerging concepts. F1000Prime Rep 7:42. doi:10.12703/P7-42
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87(1):99–163. doi:10.1152/physrev.00013.2006
Martinou JC, Green DR (2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2(1):63–67. doi:10.1038/35048069
Westphal D, Kluck RM, Dewson G (2014) Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ 21(2):196–205. doi:10.1038/cdd.2013.139
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S et al (2016) BOK Is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell. doi:10.1016/j.cell.2016.02.026
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9(1):47–59. doi:10.1038/nrm2308
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91(4):479–489
Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412(6842):95–99. doi:10.1038/35083620
Munoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR (2006) Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci USA 103(31):11573–11578. doi:10.1073/pnas.0603007103
Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102(1):33–42
Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23(16):2861–2874. doi:10.1038/sj.onc.1207523
Modjtahedi N, Giordanetto F, Madeo F, Kroemer G (2006) Apoptosis-inducing factor: vital and lethal. Trend Cell Biol 16(5):264–272. doi:10.1016/j.tcb.2006.03.008
Lee HJ, Pyo JO, Oh Y, Kim HJ, Hong SH, Jeon YJ et al (2007) AK2 activates a novel apoptotic pathway through formation of a complex with FADD and caspase-10. Nat Cell Biol 9(11):1303–1310. doi:10.1038/ncb1650
Gonzalvez F, Gottlieb E (2007) Cardiolipin: setting the beat of apoptosis. Apoptosis Int J Program Cell Death 12(5):877–885. doi:10.1007/s10495-007-0718-8
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA et al (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1(4):223–232. doi:10.1038/nchembio727
Mari M, Colell A, Morales A, Caballero F, Moles A, Fernandez A et al (2008) Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology 134(5):1507–1520. doi:10.1053/j.gastro.2008.01.073
Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC (2009) Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 11(11):2685–2700. doi:10.1089/ARS.2009.2695
Montero J, Mari M, Colell A, Morales A, Basanez G, Garcia-Ruiz C et al (2010) Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim Biophys Acta 1797(67):1217–1224. doi:10.1016/j.bbabio.2010.02.010
Landeta O, Landajuela A, Gil D, Taneva S, Di Primo C, Sot B et al (2011) Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J Biol Chem 286(10):8213–8230. doi:10.1074/jbc.M110.165852
Frisch SM, Ruoslahti E (1997) Integrins and anoikis. Curr Opin Cell Biol 9(5):701–706
Gilmore AP (2005) Anoikis. Cell Death Differ 12(Suppl 2):1473–1477. doi:10.1038/sj.cdd.4401723
Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY et al (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461(7260):109–113. doi:10.1038/nature08268
Kamarajugadda S, Stemboroski L, Cai Q, Simpson NE, Nayak S, Tan M et al (2012) Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol 32(10):1893–1907. doi:10.1128/MCB.06248-11
Kamarajugadda S, Cai Q, Chen H, Nayak S, Zhu J, He M et al (2013) Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis 4:e504. doi:10.1038/cddis.2013.20
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA et al (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434(7033):658–662. doi:10.1038/nature03434
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H et al (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434(7033):652–658. doi:10.1038/nature03317
Gutierrez-Aguilar M, Baines CP (2015) Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim Biophys Acta 1850(10):2041–2047. doi:10.1016/j.bbagen.2014.11.009
McCommis KS, Baines CP (2012) The role of VDAC in cell death: friend or foe? Biochim Biophys Acta 1818(6):1444–1450. doi:10.1016/j.bbamem.2011.10.025
Sileikyte J, Blachly-Dyson E, Sewell R, Carpi A, Menabo R, Di Lisa F et al (2014) Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (TSPO). J Biol Chem. doi:10.1074/jbc.M114.549634
Christofferson DE, Yuan J (2010) Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22(2):263–268. doi:10.1016/j.ceb.2009.12.003
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11(10):700–714. doi:10.1038/nrm2970
Ardestani S, Deskins DL, Young PP (2013) Membrane TNF-alpha-activated programmed necrosis is mediated by ceramide-induced reactive oxygen species. J Mol Signal 8(1):12. doi:10.1186/1750-2187-8-12
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343(6257):425–430. doi:10.1038/343425a0
Edwards PA, Ericsson J (1999) Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Ann Rev Biochem 68:157–185. doi:10.1146/annurev.biochem.68.1.157
Brusselmans K, Timmermans L, Van de Sande T, Van Veldhoven PP, Guan G, Shechter I et al (2007) Squalene synthase, a determinant of Raft-associated cholesterol and modulator of cancer cell proliferation. J Biol Chem 282(26):18777–18785. doi:10.1074/jbc.M611763200
Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11(1):25–33
Nguyen AD, McDonald JG, Bruick RK, DeBose-Boyd RA (2007) Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J Biol Chem 282(37):27436–27446. doi:10.1074/jbc.M704976200
Garcia-Ruiz C, Mari M, Colell A, Morales A, Caballero F, Montero J et al (2009) Mitochondrial cholesterol in health and disease. Histol Histopathol 24(1):117–132
Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D et al (2011) An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer discovery. 1(5):442–456. doi:10.1158/2159-8290.CD-11-0102
Casey SC, Amedei A, Aquilano K, Azmi AS, Benencia F, Bhakta D et al (2015) Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin Cancer Biol 35(Suppl):S199–S223. doi:10.1016/j.semcancer.2015.02.007
Maxfield FR, Tabas I (2005) Role of cholesterol and lipid organization in disease. Nature 438(7068):612–621. doi:10.1038/nature04399
Crain RC, Clark RW, Harvey BE (1983) Role of lipid transfer proteins in the abnormal lipid content of Morris hepatoma mitochondria and microsomes. Cancer Res 43(7):3197–3202
Feo F, Canuto RA, Garcea R, Gabriel L (1975) Effect of cholesterol content on some physical and functional properties of mitochondria isolated from adult rat liver, fetal liver, cholesterol-enriched liver and hepatomas AH-130, 3924A and 5123. Biochim Biophys Acta 413(1):116–134
Miller WL (2013) Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol 379(1–2):62–73. doi:10.1016/j.mce.2013.04.014
Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ, Parker KL (1997) Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Natl Acad Sci USA 94(21):11540–11545
Kishida T, Kostetskii I, Zhang Z, Martinez F, Liu P, Walkley SU et al (2004) Targeted mutation of the MLN64 START domain causes only modest alterations in cellular sterol metabolism. J Biol Chem 279(18):19276–19285. doi:10.1074/jbc.M400717200
Charman M, Kennedy BE, Osborne N, Karten B (2010) MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J Lipid Res 51(5):1023–1034. doi:10.1194/jlr.M002345
Smith B, Land H (2012) Anticancer activity of the cholesterol exporter ABCA1 gene. Cell reports. 2(3):580–590. doi:10.1016/j.celrep.2012.08.011
Warburg O (1956) On respiratory impairment in cancer cells. Science 124(3215):269–270
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13(6):472–482. doi:10.1016/j.ccr.2008.05.005
Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J Gen Physiol 8(6):519–530
Alam MM, Lal S, FitzGerald KE, Zhang L (2016) A holistic view of cancer bioenergetics: mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin Trans Med 5(1):1–14. doi:10.1186/s40169-016-0082-9
Mankoff DA, Eary JF, Link JM, Muzi M, Rajendran JG, Spence AM et al (2007) Tumor-specific positron emission tomography imaging in patients: [18F] fluorodeoxyglucose and beyond. Clin Cancer Res 13(12):3460–3469. doi:10.1158/1078-0432.CCR-07-0074
Levine AJ, Puzio-Kuter AM (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330(6009):1340–1344. doi:10.1126/science.1193494
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21(3):297–308. doi:10.1016/j.ccr.2012.02.014
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Ann Rev Cell Dev Biol 27:441–464. doi:10.1146/annurev-cellbio-092910-154237
VanderHeiden MG, Cantley LC, Thompson CB (2009) Understanding the warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033. doi:10.1126/science.1160809
Vincent M (2012) Cancer: a de-repression of a default survival program common to all cells?: a life-history perspective on the nature of cancer. BioEssays News Rev Mol Cell Develop Biol 34(1):72–82. doi:10.1002/bies.201100049
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105(48):18782–18787. doi:10.1073/pnas.0810199105
Gogvadze V, Zhivotovsky B, Orrenius S (2010) The warburg effect and mitochondrial stability in cancer cells. Mol Aspects Med 31(1):60–74. doi:10.1016/j.mam.2009.12.004
Ksiezakowska-Lakoma K, Zyla M, Wilczynski JR (2014) Mitochondrial dysfunction in cancer. Menopause Rev 13(2):136–144. doi:10.5114/pm.2014.42717
Wahlstrom T, Henriksson MA (2015) Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta 1849(5):563–569. doi:10.1016/j.bbagrm.2014.07.004
Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA et al (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25(14):6225–6234. doi:10.1128/MCB.25.14.6225-6234.2005
Kim J, Lee JH, Iyer VR (2008) Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One 3(3):e1798. doi:10.1371/journal.pone.0001798
Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR et al (2012) Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 483(7391):608–612. doi:10.1038/nature10927
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J et al (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15(1):110–121. doi:10.1016/j.cmet.2011.12.009
Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N et al (2008) Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3(7):e2722. doi:10.1371/journal.pone.0002722
Viale A, Corti D, Draetta GF (2015) Tumors and mitochondrial respiration: a neglected connection. Cancer Res. doi:10.1158/0008-5472.CAN-15-0491
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M et al (2013) BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12(3):329–341. doi:10.1016/j.stem.2012.12.013
Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D et al (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23(6):811–825. doi:10.1016/j.ccr.2013.05.003
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M et al (2014) Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514(7524):628–632. doi:10.1038/nature13611
Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M et al (2015) MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 22(4):590–605. doi:10.1016/j.cmet.2015.08.015
Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z et al (2011) Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20(5):674–688. doi:10.1016/j.ccr.2011.10.015
Wolf DA (2014) Is reliance on mitochondrial respiration a “chink in the armor” of therapy-resistant cancer? Cancer Cell 26(6):788–795. doi:10.1016/j.ccell.2014.10.001
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O et al (2006) p53 regulates mitochondrial respiration. Science 312(5780):1650–1653. doi:10.1126/science.1126863
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R et al (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126(1):107–120. doi:10.1016/j.cell.2006.05.036
Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH (2013) Metabolic regulation by p53 family members. Cell Metab 18(5):617–633. doi:10.1016/j.cmet.2013.06.019
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3(10):721–732. doi:10.1038/nrc1187
Melillo G (2006) Inhibiting hypoxia-inducible factor 1 for cancer therapy. Mol Cancer Res 4(9):601–605. doi:10.1158/1541-7786.MCR-06-0235
Brocato J, Chervona Y, Costa M (2014) Molecular responses to hypoxia-inducible factor 1alpha and beyond. Mol Pharmacol 85(5):651–657. doi:10.1124/mol.113.089623
Kaelin WG Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30(4):393–402. doi:10.1016/j.molcel.2008.04.009
Semenza GL (2011) Regulation of metabolism by hypoxia-inducible factor 1. Quant Biol. 76:347–353. doi:10.1101/sqb.2011.76.010678
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129(1):111–122. doi:10.1016/j.cell.2007.01.047
Lim JH, Luo C, Vazquez F, Puigserver P (2014) Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res 74(13):3535–3545. doi:10.1158/0008-5472.CAN-13-2893-T
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J et al (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581. doi:10.1128/MCB.00166-09
Dang CV, Kim JW, Gao P, Yustein J (2008) The interplay between MYC and HIF in cancer. Nat Rev Cancer 8(1):51–56. doi:10.1038/nrc2274
Myllyharju J, Koivunen P (2013) Hypoxia-inducible factor prolyl 4-hydroxylases: common and specific roles. Biol Chem 394(4):435–448. doi:10.1515/hsz-2012-0328
Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J (2007) Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem 282(7):4524–4532. doi:10.1074/jbc.M610415200
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17(3):225–234. doi:10.1016/j.ccr.2010.01.020
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12(12):931–947. doi:10.1038/nrd4002
Sabharwal SS, Schumacker PT (2014) Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14(11):709–721. doi:10.1038/nrc3803
Castaldo SA, Freitas JR, Conchinha NV, Madureira PA (2016) The tumorigenic roles of the cellular REDOX regulatory systems. Oxid Med Cell Longev 2016:8413032. doi:10.1155/2016/8413032
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res. 44(5):479–496. doi:10.3109/10715761003667554
Tong L, Chuang CC, Wu S, Zuo L (2015) Reactive oxygen species in redox cancer therapy. Cancer Lett 367(1):18–25. doi:10.1016/j.canlet.2015.07.008
Cabello CM, Bair WB 3rd, Wondrak GT (2007) Experimental therapeutics: targeting the redox Achilles heel of cancer. Curr Opin Investig Drugs 8(12):1022–1037
Buchner FL, Bueno-de-Mesquita HB, Linseisen J, Boshuizen HC, Kiemeney LA, Ros MM et al (2010) Fruits and vegetables consumption and the risk of histological subtypes of lung cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Cause Cont 21(3):357–371. doi:10.1007/s10552-009-9468-y
Ostrakhovitch EA (2011) Redox environment and its meaning for breast cancer cells fate. Curr Cancer Drug Targets 11(4):479–495
Glasauer A, Chandel NS (2014) Targeting antioxidants for cancer therapy. Biochem Pharmacol 92(1):90–101. doi:10.1016/j.bcp.2014.07.017
Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V et al (2007) HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12(3):230–238. doi:10.1016/j.ccr.2007.08.004
Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD et al (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1(6):401–408. doi:10.1016/j.cmet.2005.05.001
Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT et al (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1(6):393–399. doi:10.1016/j.cmet.2005.05.003
Rico-Bautista E, Zhu W, Kitada S, Ganapathy S, Lau E, Krajewski S et al (2013) Small molecule-induced mitochondrial disruption directs prostate cancer inhibition via UPR signaling. Oncotarget 4(8):1212–1229. doi:10.18632/oncotarget.1130
Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ et al (2011) Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306(14):1549–1556. doi:10.1001/jama.2011.1437
Chandel NS, Tuveson DA (2014) The promise and perils of antioxidants for cancer patients. N Engl J Med 371(2):177–178. doi:10.1056/NEJMcibr1405701
van Zandwijk N, Dalesio O, Pastorino U, de Vries N, van Tinteren H (2000) EUROSCAN, a randomized trial of vitamin A and N-acetylcysteine in patients with head and neck cancer or lung cancer. For the European Organization for Research and Treatment of Cancer Head and Neck and Lung Cancer Cooperative Groups. J Natl Cancer Inst 92(12):977–986
Watson J (2013) Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol 3(1):120144. doi:10.1098/rsob.120144
Dunn BK, Richmond ES, Minasian LM, Ryan AM, Ford LG (2010) A nutrient approach to prostate cancer prevention: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Nutri Cancer 62(7):896–918. doi:10.1080/01635581.2010.509833
Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO (2014) Antioxidants accelerate lung cancer progression in mice. Sci Trans Med 6(221):221. doi:10.1126/scitranslmed.3007653
LeGal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C et al (2015) Antioxidants can increase melanoma metastasis in mice. Sci Trans Med 7(308):308. doi:10.1126/scitranslmed.aad3740
Huang Y, Li W, Su ZY, Kong AN (2015) The complexity of the Nrf2 pathway: beyond the antioxidant response. J Nutr Biochem 26(12):1401–1413. doi:10.1016/j.jnutbio.2015.08.001
Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT (2010) Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 13(11):1713–1748. doi:10.1089/ars.2010.3221
Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M et al (2008) Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res 68(5):1303–1309. doi:10.1158/0008-5472.CAN-07-5003
Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY et al (2010) Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol 220(4):446–451. doi:10.1002/path.2653
Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M et al (2010) Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther 9(2):336–346. doi:10.1158/1535-7163.MCT-09-0589
Lister A, Nedjadi T, Kitteringham NR, Campbell F, Costello E, Lloyd B et al (2011) Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol Cancer 10:37. doi:10.1186/1476-4598-10-37
Sasaki H, Suzuki A, Shitara M, Hikosaka Y, Okuda K, Moriyama S et al (2013) Genotype analysis of the NRF2 gene mutation in lung cancer. Int J Mol Med 31(5):1135–1138. doi:10.3892/ijmm.2013.1324
Hu XF, Yao J, Gao SG, Wang XS, Peng XQ, Yang YT et al (2013) Nrf2 overexpression predicts prognosis and 5-FU resistance in gastric cancer. Asian Pac J Cancer Prev 14(9):5231–5235
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R et al (2016) Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63(1):173–184. doi:10.1002/hep.28251
Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K et al (2008) Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA 105(36):13568–13573. doi:10.1073/pnas.0806268105
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–223. doi:10.1038/ncb2021
Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R et al (2010) Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem 285(22):16782–16788. doi:10.1074/jbc.M109.096545
Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A et al (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 285(29):22576–22591. doi:10.1074/jbc.M110.118976
Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T et al (2012) Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA 109(34):13561–13566. doi:10.1073/pnas.1121572109
Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T et al (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30(13):3275–3285. doi:10.1128/MCB.00248-10
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131(6):1149–1163. doi:10.1016/j.cell.2007.10.035
Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O et al (2011) Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 193(2):275–284. doi:10.1083/jcb.201102031
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K et al (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475(7354):106–109. doi:10.1038/nature10189
Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchell SJ et al (2012) Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab 15(3):361–371. doi:10.1016/j.cmet.2012.01.017
Kinch L, Grishin NV, Brugarolas J (2011) Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 20(4):418–420. doi:10.1016/j.ccr.2011.10.005
Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H et al (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20(4):524–537. doi:10.1016/j.ccr.2011.09.006
Villeneuve NF, Lau A, Zhang DD (2010) Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal 13(11):1699–1712. doi:10.1089/ars.2010.3211
Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y et al (2008) Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29(6):1235–1243. doi:10.1093/carcin/bgn095
Shelton P, Jaiswal AK (2013) The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene? FASEB J 27(2):414–423. doi:10.1096/fj.12-217257
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z et al (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527(7577):186–191. doi:10.1038/nature15726
Nguyen A, Loo JM, Mital R, Weinberg EM, Man FY, Zeng Z et al (2016) PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J Clin Invest 126(2):681–694. doi:10.1172/JCI83587
Goh J, Enns L, Fatemie S, Hopkins H, Morton J, Pettan-Brewer C et al (2011) Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 11:191. doi:10.1186/1471-2407-11-191
Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T et al (2014) A mitochondrial switch promotes tumor metastasis. Cell Rep 8(3):754–766. doi:10.1016/j.celrep.2014.06.043
Nazarewicz RR, Dikalova A, Bikineyeva A, Ivanov S, Kirilyuk IA, Grigor’ev IA et al (2013) Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid Redox Signal 19(4):344–349. doi:10.1089/ars.2013.5185
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 107(19):8788–8793. doi:10.1073/pnas.1003428107
Matsumoto K, Imagawa S, Obara N, Suzuki N, Takahashi S, Nagasawa T et al (2006) 2-Oxoglutarate downregulates expression of vascular endothelial growth factor and erythropoietin through decreasing hypoxia-inducible factor-1alpha and inhibits angiogenesis. J Cell Physiol 209(2):333–340. doi:10.1002/jcp.20733
Kaelin WG Jr (2011) Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb Symp Quant Biol 76:335–345. doi:10.1101/sqb.2011.76.010975
Bosch M, Mari M, Gross SP, Fernandez-Checa JC, Pol A (2011) Mitochondrial cholesterol: a connection between caveolin, metabolism, and disease. Traffic 12(11):1483–1489. doi:10.1111/j.1600-0854.2011.01259.x
Colell A, Fernandez A, Fernandez-Checa JC (2009) Mitochondria, cholesterol and amyloid beta peptide: a dangerous trio in Alzheimer disease. J Bioenerg Biomembr 41(5):417–423. doi:10.1007/s10863-009-9242-6
Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M et al (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1(6):409–414. doi:10.1016/j.cmet.2005.05.002
Rotili D, Mai A (2011) Targeting histone demethylases: a new avenue for the fight against Cancer. Genes Cancer 2(6):663–679. doi:10.1177/1947601911417976
Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM (1984) Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science 224(4656):1403–1406
Pegoraro L, Palumbo A, Erikson J, Falda M, Giovanazzo B, Emanuel BS et al (1984) A 14;18 and an 8;14 chromosome translocation in a cell line derived from an acute B-cell leukemia. Proc Natl Acad Sci USA 81(22):7166–7170
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463(7283):899–905. doi:10.1038/nature08822
Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ et al (2011) Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471(7336):110–114. doi:10.1038/nature09779
Wang C, Youle RJ (2012) Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1's inhibitory effect on Bak. Oncogene 31(26):3177–3189. doi:10.1038/onc.2011.497
Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L et al (2013) BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51(6):751–765. doi:10.1016/j.molcel.2013.08.048
Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA et al (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9(5):351–365. doi:10.1016/j.ccr.2006.03.027
Lopez J, Tait SW (2015) Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer 112(6):957–962. doi:10.1038/bjc.2015.85
Wickramasekera NT, Das GM (2014) Tumor suppressor p53 and estrogen receptors in nuclear–mitochondrial communication. Mitochondrion 16:26–37. doi:10.1016/j.mito.2013.10.002
Dashzeveg N, Yoshida K (2015) Cell death decision by p53 via control of the mitochondrial membrane. Cancer Lett 367(2):108–112. doi:10.1016/j.canlet.2015.07.019
Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149(7):1536–1548. doi:10.1016/j.cell.2012.05.014
Guo X, Sesaki H, Qi X (2014) Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem J 461(1):137–146. doi:10.1042/BJ20131438
Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A et al (2008) Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 7(19):3056–3061
Soussi T, Wiman KG (2015) TP53: an oncogene in disguise. Cell Death Differ 22(8):1239–1249. doi:10.1038/cdd.2015.53
Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L et al (2007) GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129(5):983–997. doi:10.1016/j.cell.2007.03.045
Deshmukh M, Kuida K, Johnson EM Jr (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 150(1):131–143
Gama V, Swahari V, Schafer J, Kole AJ, Evans A, Huang Y et al (2014) The E3 ligase PARC mediates the degradation of cytosolic cytochrome c to promote survival in neurons and cancer cells. Sci Signal. 7(334):67. doi:10.1126/scisignal.2005309
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11(9):621–632. doi:10.1038/nrm2952
Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM et al (2012) Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell 23(12):2240–2252. doi:10.1091/mbc.E11-11-0926
Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME et al (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57(5):860–872. doi:10.1016/j.molcel.2015.01.018
Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP et al (2015) Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell 58(2):284–296. doi:10.1016/j.molcel.2015.03.003
Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X et al (2012) ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22(5):631–644. doi:10.1016/j.ccr.2012.09.021
Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK et al (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 406(3):407–414. doi:10.1042/BJ20070039
Ahluwalia GS, Grem JL, Hao Z, Cooney DA (1990) Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Therap 46(2):243–271
Ovejera AA, Houchens DP, Catane R, Sheridan MA, Muggia FM (1979) Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Res 39(8):3220–3224
Li B, Simon MC (2013) Molecular Pathways: targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin Cancer Res 19(21):5835–5841. doi:10.1158/1078-0432.CCR-12-3629
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM et al (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107(5):2037–2042. doi:10.1073/pnas.0914433107
Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM et al (2009) LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther 8(3):626–635. doi:10.1158/1535-7163.MCT-08-1049
Wang ZY, Loo TY, Shen JG, Wang N, Wang DM, Yang DP et al (2012) LDH-A silencing suppresses breast cancer tumorigenicity through induction of oxidative stress mediated mitochondrial pathway apoptosis. Breast Cancer Res Treat 131(3):791–800. doi:10.1007/s10549-011-1466-6
Nilsson LM, Forshell TZ, Rimpi S, Kreutzer C, Pretsch W, Bornkamm GW et al (2012) Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet 8(3):e1002573. doi:10.1371/journal.pgen.1002573
Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W et al (2014) Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res 74(3):908–920. doi:10.1158/0008-5472.CAN-13-2034
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R et al (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1):37–51. doi:10.1016/j.ccr.2006.10.020
Dasgupta B, Chhipa RR (2015) Evolving lessons on the complex role of AMPK in normal physiology and cancer. Trends Pharmacol Sci. doi:10.1016/j.tips.2015.11.007
Cazzaniga M, Bonanni B (2015) Breast Cancer Metabolism and Mitochondrial Activity: the Possibility of Chemoprevention with Metformin. BioMed research Int 2015:972193. doi:10.1155/2015/972193
Jara JA, Lopez-Munoz R (2015) Metformin and cancer: between the bioenergetic disturbances and the antifolate activity. Pharmacol Res 101:102–108. doi:10.1016/j.phrs.2015.06.014
Morales DR, Morris AD (2015) Metformin in cancer treatment and prevention. Annu Rev Med 66:17–29. doi:10.1146/annurev-med-062613-093128
Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe R, Howell A et al (2011) Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther 12(12):1101–1113. doi:10.4161/cbt.12.12.18703
Del Barco S, Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J et al (2011) Metformin: multi-faceted protection against cancer. Oncotarget 2(12):896–917. doi:10.18632/oncotarget.387
Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH et al (2014) Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun 5:3295. doi:10.1038/ncomms4295
Rohlena J, Dong LF, Ralph SJ, Neuzil J (2011) Anticancer drugs targeting the mitochondrial electron transport chain. Antioxid Redox Signal 15(12):2951–2974. doi:10.1089/ars.2011.3990
Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C et al (2013) Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 4(11):1986–1998. doi:10.18632/oncotarget.1420
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435(7042):677–681. doi:10.1038/nature03579
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68(9):3421–3428. doi:10.1158/0008-5472.CAN-07-5836
Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ et al (2011) Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 118(6):1663–1674. doi:10.1182/blood-2011-04-347849
Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR et al (2011) Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med 208(10):2017–2031. doi:10.1084/jem.20110750
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19(2):202–208. doi:10.1038/nm.3048
Ng SY, Davids MS (2014) Selective Bcl-2 inhibition to treat chronic lymphocytic leukemia and non-Hodgkin lymphoma. Clin Adv Hematol Oncol 12(4):224–229
Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA (2014) Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 123(26):4111–4119. doi:10.1182/blood-2014-03-560284
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10(5):389–399. doi:10.1016/j.ccr.2006.08.027
Modugno M, Banfi P, Gasparri F, Borzilleri R, Carter P, Cornelius L et al (2015) Mcl-1 antagonism is a potential therapeutic strategy in a subset of solid cancers. Exp Cell Res 332(2):267–277. doi:10.1016/j.yexcr.2014.11.022
Vela L, Marzo I (2015) Bcl-2 family of proteins as drug targets for cancer chemotherapy: the long way of BH3 mimetics from bench to bedside. Curr Opin Pharmacol 23:74–81. doi:10.1016/j.coph.2015.05.014
Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B et al (2012) A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol 19(9):1175–1186. doi:10.1016/j.chembiol.2012.07.018
Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J et al (2015) Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis 6:e1590. doi:10.1038/cddis.2014.561
Thomas RL, Gustafsson AB (2013) MCL1 is critical for mitochondrial function and autophagy in the heart. Autophagy 9(11):1902–1903. doi:10.4161/auto.26168
Zhou L, Qiu T, Xu J, Wang T, Wang J, Zhou X et al (2013) miR-135a/b modulate cisplatin resistance of human lung cancer cell line by targeting MCL1. Pathol Oncol Res 19(4):677–683. doi:10.1007/s12253-013-9630-4
Suryani S, Carol H, Chonghaile TN, Frismantas V, Sarmah C, High L et al (2014) Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin Cancer Res 20(17):4520–4531. doi:10.1158/1078-0432.CCR-14-0259
Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB et al (2014) Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov 4(9):1074–1087. doi:10.1158/2159-8290.CD-14-0353
Marchenko ND, Zaika A, Moll UM (2000) Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 275(21):16202–16212
Liu J, Wang Z (2015) Increased oxidative stress as a selective anticancer therapy. Oxid Med Cell Longev 2015:294303. doi:10.1155/2015/294303
Ramanathan B, Jan KY, Chen CH, Hour TC, Yu HJ, Pu YS (2005) Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res 65(18):8455–8460. doi:10.1158/0008-5472.CAN-05-1162
Hasinoff BB, Wu X, Yadav AA, Patel D, Zhang H, Wang DS et al (2015) Cellular mechanisms of the cytotoxicity of the anticancer drug elesclomol and its complex with Cu(II). Biochem Pharmacol 93(3):266–276. doi:10.1016/j.bcp.2014.12.008
Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang CY, Zhang M et al (2008) Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther 7(8):2319–2327. doi:10.1158/1535-7163.MCT-08-0298
Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H et al (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10(3):241–252. doi:10.1016/j.ccr.2006.08.009
Schumacker PT (2006) Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 10(3):175–176. doi:10.1016/j.ccr.2006.08.015
Ravindran J, Prasad S, Aggarwal BB (2009) Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J 11(3):495–510. doi:10.1208/s12248-009-9128-x
Yue P, Zhou Z, Khuri FR, Sun SY (2006) Depletion of intracellular glutathione contributes to JNK-mediated death receptor 5 upregulation and apoptosis induction by the novel synthetic triterpenoid methyl-2-cyano-3, 12-dioxooleana-1, 9-dien-28-oate (CDDO-Me). Cancer Biol Ther 5(5):492–497
Griffith OW (1982) Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem 257(22):13704–13712
Sobhakumari A, Love-Homan L, Fletcher EV, Martin SM, Parsons AD, Spitz DR et al (2012) Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One 7(10):e48175. doi:10.1371/journal.pone.0048175
Lo M, Ling V, Low C, Wang YZ, Gout PW (2010) Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol 17(3):9–16
Gout PW, Buckley AR, Simms CR, Bruchovsky N (2001) Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)− cystine transporter: a new action for an old drug. Leukemia 15(10):1633–1640
Samudio I, Konopleva M, Hail N Jr, Shi YX, McQueen T, Hsu T et al (2005) 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J Biol Chem 280(43):36273–36282. doi:10.1074/jbc.M507518200
Samudio I, Kurinna S, Ruvolo P, Korchin B, Kantarjian H, Beran M et al (2008) Inhibition of mitochondrial metabolism by methyl-2-cyano-3,12-dioxooleana-1,9-diene-28-oate induces apoptotic or autophagic cell death in chronic myeloid leukemia cells. Mol Cancer Ther 7(5):1130–1139. doi:10.1158/1535-7163.MCT-07-0553
Chen G, Chen Z, Hu Y, Huang P (2011) Inhibition of mitochondrial respiration and rapid depletion of mitochondrial glutathione by beta-phenethyl isothiocyanate: mechanisms for anti-leukemia activity. Antioxid Redox Signal 15(12):2911–2921. doi:10.1089/ars.2011.4170
Bezerra DP, Militao GC, de Castro FO, Pessoa C, de Moraes MO, Silveira ER et al (2007) Piplartine induces inhibition of leukemia cell proliferation triggering both apoptosis and necrosis pathways. Toxicol In Vitro 21(1):1–8. doi:10.1016/j.tiv.2006.07.007
Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X et al (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475(7355):231–234. doi:10.1038/nature10167
Tateishi Y, Sasabe E, Ueta E, Yamamoto T (2008) Ionizing irradiation induces apoptotic damage of salivary gland acinar cells via NADPH oxidase 1-dependent superoxide generation. Biochem Biophys Res Commun 366(2):301–307. doi:10.1016/j.bbrc.2007.11.039
Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H et al (2012) Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med 53(2):260–270. doi:10.1016/j.freeradbiomed.2012.04.033
Ghosh P, Singha Roy S, Basu A, Bhattacharjee A, Bhattacharya S (2015) Sensitization of cisplatin therapy by a naphthalimide based organoselenium compound through modulation of antioxidant enzymes and p53 mediated apoptosis. Free Radic Res 49(4):453–471. doi:10.3109/10715762.2015.1012079
Lawenda BD, Kelly KM, Ladas EJ, Sagar SM, Vickers A, Blumberg JB (2008) Should supplemental antioxidant administration be avoided during chemotherapy and radiation therapy? J Natl Cancer Inst 100(11):773–783. doi:10.1093/jnci/djn148
Filippova M, Filippov V, Williams VM, Zhang K, Kokoza A, Bashkirova S et al (2014) Cellular levels of oxidative stress affect the response of cervical cancer cells to chemotherapeutic agents. BioMed research international. 2014:574659. doi:10.1155/2014/574659
Garcia-Ruiz C, Morales A, Fernandez-Checa JC (2012) Statins and protein prenylation in cancer cell biology and therapy. Anticancer Agents Med Chem 12(4):303–315
Elson CE, Peffley DM, Hentosh P, Mo H (1999) Isoprenoid-mediated inhibition of mevalonate synthesis: potential application to cancer. Proc Soc Exp Biol Med 221(4):294–311
Hindler K, Cleeland CS, Rivera E, Collard CD (2006) The role of statins in cancer therapy. Oncologist 11(3):306–315. doi:10.1634/theoncologist.11-3-306
Elsayed RK, Evans JD (2008) Emerging lipid-lowering drugs: squalene synthase inhibitors. Expert Opin Emerg Drugs 13(2):309–322. doi:10.1517/14728214.13.2.309
Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA et al (2001) Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res 61(24):8758–8768
Sun J, Qian Y, Hamilton AD, Sebti SM (1998) Both farnesyltransferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K-Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene 16(11):1467–1473. doi:10.1038/sj.onc.1201656
Authors' contributions
VR, CGR and JCFC participated in the design of the study, revised literature, and discussed data jointly. VR, CGR and JCFC drafted Figures and the writing of the text. All authors read and approved the final manuscript.
Acknowledgements
Vicent Ribas is recipient of an IDIBAPS Postdoctoral Fellowship-BIOTRACK, supported by the European Community’s Seventh Framework Programme (EC FP7/2007-2013) under the Grant agreement number 229673 and the Spanish Ministry of Economy and Competitiveness (MINECO) through the Grant COFUND2013-40261. The work was supported by CIBEREHD, Fundació la Marató de TV3 and Grants PI11/0325 (META) from the Instituto Salud Carlos III and Grants, SAF2011-23031, SAF2012-34831, SAF2014-57674R and SAF2015-69944-R from Plan Nacional de I+D, Spain; Fundación Mutua Madrileña and the center Grant P50-AA-11999 (Research Center for Liver and Pancreatic Diseases, NIAAA/NIH).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ribas, V., García-Ruiz, C. & Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin Trans Med 5, 22 (2016). https://doi.org/10.1186/s40169-016-0106-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-016-0106-5