
Abstract
Background
The gut microbiome changes in response to a range of environmental conditions, life events and disease states. Pregnancy is a natural life event that involves major physiological adaptation yet studies of the microbiome in pregnancy are limited and their findings inconsistent. Pregnancy with type 1 diabetes (T1D) is associated with increased maternal and fetal risks but the gut microbiome in this context has not been characterized. By whole metagenome sequencing (WMS), we defined the taxonomic composition and function of the gut bacterial microbiome across 70 pregnancies, 36 in women with T1D.
Results
Women with and without T1D exhibited compositional and functional changes in the gut microbiome across pregnancy. Profiles in women with T1D were distinct, with an increase in bacteria that produce lipopolysaccharides and a decrease in those that produce short-chain fatty acids, especially in the third trimester. In addition, women with T1D had elevated concentrations of fecal calprotectin, a marker of intestinal inflammation, and serum intestinal fatty acid-binding protein (I-FABP), a marker of intestinal epithelial damage.
Conclusions
Women with T1D exhibit a shift towards a more pro-inflammatory gut microbiome during pregnancy, associated with evidence of intestinal inflammation. These changes could contribute to the increased risk of pregnancy complications in women with T1D and are potentially modifiable by dietary means.
Video abstract
Similar content being viewed by others

Background
The gut microbiome provides essential metabolites, vitamins, co-factors and hormones, protects against pathogenic microorganisms and has a key role in the development of the immune and other systems [1, 2]. Changes in the composition of the gut microbiome are associated with ageing, environmental conditions, life events and disease states [2,3,4]. In pregnancy, women undergo significant physiological changes, but only recently has the gut microbiome been studied in this context [5, 6]. Koren et al. [5] sampled the gut microbiome in the first and third trimesters and found that the taxonomic composition in the first trimester was similar to that of non-pregnant women but in the third trimester the abundance of Actinobacteria and Proteobacteria phyla increased along with an overall decrease in bacterial richness (alpha diversity). In studies in germ-free mice, they observed that inoculation with third compared to first trimester feces led to greater weight gain, insulin resistance and gut inflammation and suggested this was an adaptive proinflammatory response to defend the fetus from pathogens and provide it with nutrients. In contrast, after analysing fecal samples weekly across pregnancy, DiGiulio et al. [6] found no significant temporal differences in diversity or composition of the gut microbiome. These contrary findings and the dearth of studies warrant further investigation of the gut microbiome in pregnancy.
Type 1 diabetes (T1D) is an autoimmune disease in which insulin-producing β cells in the islets of the pancreas are destroyed by T lymphocytes leading to insulin deficiency [7]. In pregnancy, T1D is associated with systemic and intra-uterine markers of sub-clinical inflammation and higher risks of complications for mother and fetus [8,9,10]. Alterations in the bacterial gut microbiome have been reported in T1D, mainly in children at high risk and at diagnosis (reviewed in [11], [12,13,14,15,16,17]). They include a decrease in alpha diversity (richness) [12,13,14] and in the abundance of lactate- and butyrate-producing and mucin-degrading bacteria [13,14,15,16,17], and an increase in the abundance of the Bacteroides genus [13, 14]. Functionally, these compositional changes are reflected by a decreased abundance of genes encoding related metabolic pathways and enzymes, e.g. butyryl-coenzyme A (CoA)-CoA transferase [15] and butyryl-CoA dehydrogenase for butyrate synthesis [16]. These changes are not necessarily specific for T1D but nevertheless, they may have clinical consequences, including in pregnancy. Gut butyrate is a key determinant of gut health and regulator of gene expression and homeostatic immunity [18,19,20]. It is the major energy source for the colonic mucosa, induces the synthesis of mucin and it promotes gut epithelial integrity, preventing ‘gut leakiness’. In the non-obese diabetic (NOD) mouse model of T1D, dietary butyrate supplementation promoted an increase in regulatory T cells and a decrease in the incidence of spontaneous diabetes [20]. Increased gut leakiness has been described in established T1D [21] and recently by ourselves in association with gut microbiome changes in children with islet autoimmunity who progressed to T1D [22]. Gut leakiness with translocation of toxins and dietary antigens into the blood may result in systemic inflammation, reported with T1D in pregnancy complicated by pre-eclampsia [10]. Because a consensus about the gut microbiome in pregnancy is lacking, even in the absence of T1D, we applied shotgun whole metagenomic sequencing (WMS) to analyse the gut microbiome across pregnancy in women with and without T1D participating in the Australia-wide Environmental Determinants of Islet Autoimmunity (ENDIA) study.
Results
Study population
Fecal samples were collected between February 2013 and October 2017 from women enrolled in the ENDIA study, a prospective, pregnancy-birth cohort study that follows 1500 Australian children who have a first-degree relative with T1D [23]. Thirty-five women (36 pregnancies) with T1D and 31 women (34 pregnancies) without T1D had each provided from one to three fecal samples across pregnancy (total 134 samples) for analysis by shotgun WMS (Fig. 1). Table 1 summarizes and compares characteristics of the T1D and non-T1D pregnancies.

Fecal samples obtained in pregnancy. n: number of samples; T1: trimester 1; T2: trimester 2; T3: trimester 3; T1D: women with type 1 diabetes; Non-T1D: women without T1D
Whole metagenomic sequencing
The WMS dataset, 47,766,763 ± 10,956,057 (mean ± SD) paired-end reads per sample, was obtained using an Illumina NovaSeq 6000. Raw reads (SRA accession: PRJNA604850) were pre-processed using KneadData bioBakery tool [24] to eliminate human DNA sequences and filter sequences with poor quality which on average removed 6% of the reads. After quality control and read filter steps, 44,940,628 ± 10,572,188 (mean ± SD) paired-end reads per sample were obtained (Excel file E0).
Taxonomic diversity and composition of the gut microbiome in women with and without T1D during pregnancy
Sequences were analysed with MetaPhLan2 implemented within the HUMAnN2 pipeline. Overall, 340 bacterial species were identified, with an average of 93 ± 13 (mean ± SD) species per sample. The top 25 most abundant species accounted for more than 50% of the gut microbiome composition of each subject in any given trimester (Figure S1).
Alpha diversity (observed richness or number of species) per sample was calculated and generalized estimating equations (GEE) were applied to test for differences between women without and with T1D, and between trimesters, and to determine if there was an interaction between T1D status and trimester. No differences were found in richness due to T1D status or time, or interactions (Figure S2, Excel file E1).
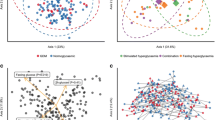
For analysis of beta diversity, Bray-Curtis coefficients were calculated between sample pairs, ordinated and plotted by principal coordinate analysis (PCoA) for each taxonomic level (Figs. 2, S3 and S4). To test for differences in beta diversity, a repeated-measure aware permutational analysis of variance (RMA-PERMANOVA) of the Bray-Curtis coefficients was performed on proportional log transformed data. This revealed a significant interaction between T1D status and time at all taxonomic levels. Therefore, differences between women with and without T1D were assessed within trimesters. No significant differences were detected in trimesters 1 and 2. However, differences were significant at the strain (P = 0.002), species (P = 0.001), genus (P = 0.070) and family (P = 0.034) levels in trimester 3 (Excel file E2).

Beta diversity analysis by T1D status. PCoA ordination plots based on Bray-Curtis distances between samples at the strain and species taxonomic levels separated by trimesters in pregnancy. T1D: women with type 1 diabetes (red); Non-T1D: women without T1D (blue)
To rule out the possibility that these results were influenced by the difference in sample size between trimesters 1 and 3, we performed a sensitivity analysis by subsampling trimester 3 to the size of trimester 1 (n = 23), using samples of trimester 3 from the same women in trimester 1, and repeated the beta diversity analysis. Similar to the complete trimester 3 dataset, differences were significant at the strain (P = 0.003), species (P = 0.003), genus (P = 0.043) and family (P = 0.047), but also phylum (P = 0.09), taxonomic levels (Excel file E2).
Differences in beta diversity reflect differences in taxonomic composition. To identify differences in specific taxa between women with and without T1D in pregnancy, differential abundance was analysed in limma. Only taxa for which the prevalence (i.e. proportion of samples with those taxa) was above 50% in at least one group and with a log2 fold-change (logFC) greater than 0.5 or less than − 0.5 were considered. Across all trimesters, the species Bacteroides caccae (FDR 0.03) and its unique strain (unclassified) in the dataset (FDR 0.03), as well as the order Enterobacteriales (FDR 0.07) were increased in women with T1D (Fig. 3; Excel file E3). On the other hand, species Bacteroidales bacterium ph8 (FDR 0.034) and its strain (GCF000311925) (FDR 0.03), the genus (FDR 0.08) and family (FDR 0.08) to which Bacteroidales bacterium ph8 belongs, and the order Bifidobacteriales (FDR 0.07), were decreased in women with T1D (Figure 3; Excel file E3).

Means and standard errors of the log2-transformed fitted values shown as a point in each trimester for differentially abundant taxa in women with (red) and without (blue) T1D. * in the top right corner denotes a significant difference (FDR < 0.1) between groups throughout pregnancy; * between points denotes a significant difference (FDR < 0.1) between groups in that trimester
Differences between women with and without T1D were also assessed within trimesters. In trimesters 1 and 2, taxa were not significantly different. However, several differences were found in trimester 3, in which the unique strain (unclassified) of Bacteroides caccae (FDR 0.004), the species Bacteroides caccae (FDR 0.004), the species Bacteroides vulgatus (FDR 0.04) and its unique strain (unclassified) (FDR 0.04) and Bacteroides uniformis (FDR 0.04) were increased in women with T1D, while the species Bacteroidales bacterium ph8 (FDR 0.01) and its strain (GCF000311925; FDR 0.005), and the genus (FDR 0.08) and family (FDR 0.08) of Bacteroidales bacterium ph8 and the order Bifidobacteriales (FDR 0.07), were decreased (Fig. 3; Excel file E3). A significant Spearman correlation (R2 > 0.4) was found between B. caccae and B. vulgatus (R2 = + 0.43; adj.P = 0.013).
A sensitivity analysis of differential abundance was also applied to the subset of trimester 3 samples referred to above: 13 species, 13 strains, 2 genera, 3 families, 2 orders and 3 phyla were detected as differentially abundant. From these, Bacteroides caccae and Bacteroides uniformis, an unclassified strain of Bacteroides caccae and the order Bifidobacteriales were also detected in the larger dataset of trimester 3 samples. Differential abundance results are summarised in Excel file E3.
In order to identify the bacterial species that were most abundant within the Enterobacteriales and Bifidobacteriales orders, we plotted the average relative abundance in women with and without T1D (Figure S5A). Escherichia coli was the most abundant species within Enterobacteriales and, together with an unclassified species of the genus Escherichia, accounted for almost the complete abundance of this order. In addition, a significant Spearman correlation was found between E. coli and Coprococcus sp. ART55_1 (R2 = − 0.6, adj.P = 0.09). Bifidobacterium adolescentis and Bifidobacterium longum were the most abundant species within Bifidobacteriales (Figure S5A). A lmer test applied to test differences in the abundance of these four species between women with and without T1D revealed that the abundance of E. coli in trimester 3 and of B. adolescentis in trimester 1 were significantly different between women with and without T1D (P = 0.01 for both; Figure S6).
Effect of gestation time and other factors on the gut microbiome during pregnancy
No significant differences in alpha diversity were detected in women with or without T1D according to time, analysed either by days of gestation (P value 0.5) or by trimester (P values > 0.6), i.e. as continuous or categorical variables, respectively (Excel file E1). Due to the significant interaction between T1D status and time, differences in beta diversity across time (days or trimesters) were assessed separately in women with and without T1D (Excel file E2). Differences were detected only at the strain (P value 0.03) and species (P value 0.06) levels in women without T1D with time as continuous variable (Excel file E2). However, in women with T1D, differences in beta diversity across days of gestation and between trimesters were significant at all taxonomic levels except order and phylum (Excel file E2). These observations suggested that the microbial community structure across pregnancy is less stable in women with T1D. Therefore, we sought to identify differentially abundant taxa across trimesters separately within each group.
Throughout pregnancy, in women with T1D, the abundance of an unclassified species of the family Peptostreptococcaceae (FDR 0.02), the species Odoribacter splanchnicus (FDR 0.098), the genus Prevotella (FDR 0.066) dominated by the species Prevotella copri (Figure S5B) and the phylum Verrucomicrobia (FDR 0.043) decreased, while an unclassified strain of species Streptococcus thermophilus (FDR 0.099) and the species Streptococcus thermophilus (FDR 0.04) and family Porphyromonadaceae (FDR 0.092) increased (Excel file E4; Figure S7). In women without T1D, an Anaerostipes hadrus GCF000332875 strain (FDR 0.038) and species Anaerostipes hadrus (FDR 0.059), an unclassified strain of Haemophilus parainfluenzae (FDR 0.001) and species Haemophilus parainfluenzae (FDR 0.003), genus Haemophilus (FDR 0.004), family Pasteurellaceae (FDR 0.002), strain (GCF000218445 [FDR 0.04]) and species of Lachnospiraceae bacterium 1157FAA (FDR 0.055) and an unclassified species of Veillonella genus (FDR 0.083) decreased during pregnancy (Excel file E4; Figure S7). Furthermore, in women without T1D strains Ruminococcus sp. 5139BFAA GCF000159975 and Lachnospiraceae bacterium 3157FAACT1 GCF000218405 (FDR 0.063 and 0.075, respectively) and their corresponding species (FDR 0.065 and 0.075), unclassified strains of Streptococcus thermophilus (FDR 0.06) and Bifidobacterium_animalis (FDR = 0.06) and their corresponding species (FDR 0.06 for both) increased throughout pregnancy (Excel file E4; Figure S7).
As expected, women with and without T1D differed in serum 1,5-anhydroglucitrol (1,5-AG), a marker of short-term glycemic control [25] (Table 1), but in women with T1D, serum 1,5-AG was related to beta diversity only at the phylum level (Excel file E2). Mode of delivery had an effect on the beta diversity only at the family level (Excel file E2). No significant associations were found between beta diversity and age at conception, body mass index (BMI), parity, carbohydrate or fibre intake (Excel file E2). However, a difference was observed in the microbiome composition at the strain and species levels according to the human leukocyte antigen (HLA) class II type (Excel file E2). The model used to test for differences in beta diversity between women with and without T1D was adjusted for HLA type. HLA type accounted for 3.2% of the variation [R2] in beta diversity in trimester 3 (Excel file E2). After controlling for this effect, T1D status explained 2.9% of the variation and the difference in beta diversity between women with and without T1D women was statistically significant (P = 0.004) (Excel file E2). Finally, even though for the differential abundance analysis an adjustment for HLA type was included in the model, an additional analysis was performed to detect differences in the abundance of specific taxa due to HLA type and to verify that the taxa that were detected as differentially abundant due to T1D status were not affected by HLA type. Differences due to HLA type were detected only between HLADR34 and HLADR3X and DR4X for the abundance of strain Eubacterium ramulus GCF000469345 and species Eubacterium ramulus in trimester 1 and an unclassified strain of species Eubacterium rectale and species Eubacterium rectale in trimester 3, which were decreased in women with HLA DR34 Excel file E3. None of the taxa identified as differentially abundant due to T1D status were significantly affected by HLA type.
Validation of differentially abundant species by qPCR
To validate the findings from metagenomic sequencing, we analysed the relative abundance of two of the top-ranked differentially abundant bacteria, Bacteroides caccae and Bacteroides vulgatus, in the same cohort of T1D and non-T1D mothers in trimester 3. Relative abundances obtained by metagenomic sequencing and qPCR were strongly correlated (Spearman R = + 0.91 and + 0.74 for B. caccae and B. vulgatus, respectively). By fitting linear models in lmer with conception age, BMI, parity and HLA type introduced as fixed effects, and ‘woman ID’ and processing batches as random effects, qPCR confirmed the increase in relative abundance of B. caccae (P = 0.00005) and B. vulgatus (P = 0.04) in women with T1D (Figure S8).
Functional annotation of gut microbiome taxa
Sequences processed with HUMAnN2 were annotated, complete metabolic pathways quantified, gene abundances calculated and regrouped into KO (Kegg Orthology) and MetaCyc reaction functional categories. A total of 451 complete pathways, 5628 KO and 3204 MetaCyc reaction categories were obtained. No significant interaction in richness was detected between factors T1D status and time. In the model in which time was considered as a continuous variable, richness was significantly higher in women with T1D for all three functional categories (Figure S9, Excel file E1). For beta diversity, the interaction between T1D status and time was significant. Therefore, differences between groups were assessed within each trimester, but were significant for the three functional categories only in trimester 3 (Fig. 4; Excel file E2).

Beta diversity analysis by type 1 diabetes (T1D) status. PCoA ordination plots based on Bray-Curtis distances between samples for a pathways, b KOs and c MetaCyc reactions in trimester 3. T1D: women with type 1 diabetes (red); women without T1D (blue)
Women with and without T1D displayed significant differences in the abundance of a number of features identified in pathways, KO and MetaCyc categories; these are comprehensively listed in Supplementary Excel files E5-10. Selected functions, namely LPS production, vitamin K2 synthesis, vitamin B6 synthesis, vitamin B12 synthesis, short-chain fatty acid (SCFA) synthesis and mucin degradation, and the principal bacterial species contributing to these functions, are summarised in Table 2. Examples of bacteria contributing to a functional feature are shown in Figures S10 and S11. Of interest, a pathway (PWY1269: CMP-3-deoxy-d-manno-octulosonate biosynthesis I), 17 KO gene categories and two MetaCyc reactions (DARAB5PISOM-RXN and UDPGLCNACEPIM-RXN) involved in the synthesis of bacterial lipopolysaccharides (LPSs) were enriched in women with T1D (Excel file E5-E7; Table 2; Fig. 5, Figure S10A). Seven pathways and 6 KO categories involved in vitamin K2 synthesis were also increased in women with T1D (Excel files E5-E6; Table 2; Fig. 5; Figure S10B). In addition, two KO categories increased in women with T1D in trimester 3 were involved in antibiotic tolerance (K03771) and biofilm formation and (K18831) (Excel file E6).

Means and standard errors of the log2-transformed fitted values shown as a point in each trimester for differentially abundant functional features in women with (red) and without (blue) T1D. One example for each of six broad categories is shown: lipopolysaccharide (LPS) production (CMP–3–deoxy–d–manno–octulosonate synthesis [PWY–1269]), vitamin K2 synthesis (superpathway of menaquinol–8 synthesis [PWY–5838]), vitamin B6 synthesis (pyridoxal 5′–phosphate synthase [K06215]), vitamin B12 synthesis (adenosylcobalamin salvage from cobinamide [COBALSYN–PWY]), short-chain fatty acid (SCFA) production (3–hydroxybutyryl–CoA dehydrogenase [K00074]) and mucin degradation (beta–N–acetylhexosaminidase [K01207]). * in the top right corner denotes a significant difference (FDR < 0.1) between groups throughout pregnancy; * between points denotes a significant difference (FDR < 0.1) between groups in that trimester
The enzyme pyridoxal 5′-phosphate synthase (K06215) involved in the deoxyxylulose 5-phosphate (DXP)-independent pathway for vitamin B6 synthesis, one pathway, five KO categories and six metaCyc reactions related to vitamin B12 (cobalamin) synthesis and five pathways, 11 KO categories and 13 MetaCyc reactions involved in SCFA synthesis, including pyruvate and acetyl-CoA production and butyrate synthesis from acetate or lactate, were decreased in women with T1D (Excel file E5-E7; Table 2; Fig. 5; Figure S11). The abundance of beta-N-acetylhexosaminidase (K01207) involved in the degradation of mucin was also significantly reduced in women with T1D, but again only in trimester 2 (Table 2; Excel file E6; Fig. 5; Figure S11).
Identification of bacterial clusters based on differentially abundant functional features
Differentially abundant functional features derived from HUMAnN2 were contributed not by a single species but rather a combination of species. Therefore, relative abundances of the principal contributing species in each of the six selected functions could be grouped into clusters (Table 2). For functions with three or more features, only principal contributors to at least three features were considered. For each cluster, a linear model was fitted with lmer using the same design as for the differential abundance analysis. This confirmed that women with T1D had an increased abundance of bacterial clusters contributing to production of LPS and synthesis of vitamin K2 and a decreased abundance of bacterial clusters contributing to synthesis of vitamins B6 and B12, production of SCFA and degradation of mucin (Figure S12).
Markers of gut pathology
Because the composition and function of the gut microbiome of women with T1D was suggestive of a pro-inflammatory state, we sought evidence for gut inflammation in women with T1D. Fecal calprotectin, released from neutrophils and monocytes, is a marker of intestinal inflammation that may result in increased epithelial permeability [26]. Serum intestinal fatty acid-binding protein (I-FABP) is a marker of intestinal epithelial damage [27]. Fecal calprotectin and serum I-FABP were measured in trimester 3 in 61 women (32 with T1D) and 55 women (27 with T1D), respectively. Fecal calprotectin was increased in women with T1D compared to those without T1D (112 ± 148 vs. 36 ± 28 [mean ± SD] mg/kg: P value 0.04; Mann-Whitney test). Serum I-FABP was also increased in women with T1D compared to women without T1D (587 ± 235 vs. 314 ± 185 [mean ± SD] pg/mL: P value 0.0003; Mann-Whitney test) (Figure S13). However, these markers did not significantly correlate (Spearman R > 0.4) with any of the individual taxa that were differentially abundant between T1D and non-T1D women.
Discussion
The gut microbiome in pregnancy has previously been analysed in two studies, by Koren et al. [5] and DiGiulio et al. [6], who pyrosequenced the 16S rRNA gene V1–V2 and V3–V5 regions, respectively, but with different conclusions. To our knowledge, the current study is the first to also include women with T1D, who have a higher frequency of complications and evidence of systemic and intra-uterine inflammation in pregnancy [8, 11, 12] that could conceivably be related to the gut microbiome. Koren et al. [5] compared single samples from trimesters 1 and 3 from 91 pregnancies and reported a decrease in alpha diversity and ‘remodelling of the gut microbiome’ by the third trimester, specifically a decrease in the abundance of taxa in the genus Faecalibacterium that generate the anti-inflammatory SCFA butyrate [18] and an increase in taxa in the phylum Proteobacteria recognised to be pro-inflammatory [28]. On the other hand, DiGiulio et al. [6] by weekly sampling of 49 women found no significant changes in diversity or composition across pregnancy. Similar to DiGiulio et al. [6], we observed no differences across pregnancy in alpha diversity but found differences in beta diversity at the strain and species levels in women without T1D and at all taxonomic levels in women with T1D. In addition, particularly in women with T1D, we saw changes in the relative abundance of specific taxa across pregnancy with progression to a more pro-inflammatory microbiome, similar to Koren et al. [5]. The taxonomic differences between women with and without T1D were reinforced by functional annotation, revealing differential abundance in enzymes and pathways as pregnancy progressed. These differences could not be attributed to demographic or other factors, including diet. It is important, however, to keep in mind that our findings are based on DNA analysis and they might not necessarily reflect changes at the RNA or protein level.
In examining differential abundance, we observed two main patterns: (1) taxa that were differentially abundant between women with and without T1D across all trimesters and (2) taxa that were similar in abundance in women with and without T1D in the first trimester but decreased or increased to be differentially abundant in trimester 3. In the first category, B. caccae and the order Enterobacteriales were increased in women with T1D across all trimesters. Within Enterobacteriales, Escherichia coli was the most abundant species and was enriched in women with T1D. Products of E. coli including lipopolysaccharide [29] and microcin [30] promote intestinal inflammation, intestinal permeability and low-grade systemic inflammation, and are implicated especially in the pathogenesis of inflammatory bowel disease [31, 32]. Moreover, an increase in these facultative anaerobes may displace obligate anaerobic bacteria that produce SCFAs [32], supported by the negative correlation between the abundance of E. coli and Coprococcus sp. ART55_1, further accentuating inflammation. In women with T1D, this may contribute to the decrease in abundance of the genus Prevotella comprising almost entirely Prevotella copri, a species that produces succinate and the SCFAs propionate and acetate known to be associated with improved glucose metabolism [19, 33]. Furthermore, in women with T1D, E. coli contributed to an increased abundance of enzymes involved in antibiotic tolerance (K18831) and biofilm formation (K03771). Bacterial biofilms confer increased tolerance to antibiotics and host immune responses [34] and may provide E. coli with a protective advantage over other more sensitive bacteria that compete for the same resources in the gut including SCFA-producers, which were less abundant in women with T1D.
In the second category (taxa that became differentially abundant by trimester 3), we observed that three species from the Bacteroidales order, B. caccae, B. uniformis and B. vulgatus, were increased in women with T1D. The genus Bacteroides was reported to be more abundant in children with islet autoimmunity compared to healthy controls [11, 12]. Bacteria from the Bacteroides (B. caccae, B. vulgatus, B. uniformis, B. dorei, B. fragilis, B. faecis, B. finegoldii, B. thetaiotaomicron, B. xylanisolvens, B. stercoris and B. ovatus) and Alistipes (A. finegoldii, A. onderdonkii and A. shahii) genera, all belonging to the Bacteriodales order, formed part of the LPS bacterial cluster which was enriched in women with T1D especially by trimester 3.
Twenty-nine functional features related to the production of SCFAs were decreased in women with T1D. The SCFAs bacterial cluster was composed of Faecalibacterium prausnitzii, Eubacterium rectale, Anaerostipes hadrus, Lachnospiraceae_bacterium 5 1 63FAA, Ruminococcus torques, Roseburia intestinalis and Roseburia inulinivorans all of which belong to the class Clostridia and are major butyrate producers [35]. Butyrate prevents gut inflammation and promotes gut barrier function [19]. In addition, the enzyme beta-N-acetylhexosaminidase (K01207), which degrades mucin [36], contributed by Eubacterium rectale, E. siraeum, Ruminococcus bromii, Bifidobacterium adolescentis, B. bifidum and Roseburia intestinalis, was decreased in women with T1D, but only in trimester 2. Degradation of mucins produces oligosaccharides, and acetate and propionate, which together then stimulate mucus production and enhance epithelial integrity [37], preventing ‘gut leakiness’ and translocation of toxins and dietary antigens into the systemic circulation. Because mucin degradation was lower in women with T1D, the stimulus to mucin production would be less. This would also be contributed to by the lower abundance of butyrate-producing bacteria observed in women with T1D. Thus, the gut microbiome in women with T1D exhibits pro-inflammatory features likely to be associated with low-grade systemic inflammation.
Women with T1D bacterial functions associated with vitamin K2 (menaquinone; MK-7) synthesis were increased and those associated with synthesis of the B-group vitamins B6 (pyridoxine) and B12 (cobalamine) were decreased. Mammals cannot synthesize these vitamins and must acquire them from the diet or gut microorganisms [38]. A small study based on metagenomic sequencing of the gut microbiome [39] observed a similar increase in the vitamin K2 superpathway (PWY-5838) in people with type 2 diabetes. Vitamin K2 is required for blood clotting and bone health [40] but why its synthesis by gut bacteria is increased in diabetes is unclear. All B-group vitamins contribute to regulation of immunity-inflammation and their deficiency has been associated with inflammatory disorders [41]. Vitamin B6 deficiency has been associated with inflammatory markers in population-based studies [42] and is reported to be common in T1D [43, 44]. Of interest therefore, we found that the key enzyme in B6 synthesis, pyridoxal 5′-phosphate synthase (K06215), was decreased across pregnancy in women with T1D. Vitamin B12 has several anti-oxidant properties [45] and is required for conversion of succinate to propionate by Prevotella [46]. The majority of women with and without T1D reported taking multi-B group vitamins from early pregnancy (Table 1) and in the third trimester plasma B6 and serum B12 did not differ significantly between women with and without T1D (Table 1). Nevertheless, the relative deficiency of these vitamin-synthesizing gut bacteria in women with T1D could contribute to other alterations in the gut microbiome, underscoring the importance of dietary supplementation in this group of women.
Our findings reveal that the composition of the gut microbiome not only changes across pregnancy but in a distinct way in women with T1D. By the third trimester, women with T1D exhibited a more pro-inflammatory and catabolic gut microbiome profile, reflected by an increase in LPS-producing bacteria and a decrease in SCFA-producing bacteria. These changes may account for the increase in calprotectin (marker of gut inflammation) and I-FABP (marker of gut epithelial integrity) we observed in women with T1D, known to be associated with impaired epithelial barrier function and leakage of LPS and other bacterial products leading to low-grade systemic inflammation. We suggest that systemic inflammation secondary to changes in the gut microbiome in T1D may contribute to the increased risk of pregnancy complications in T1D. Furthermore, a pro-inflammatory gut microbiome in the mother may impact the infant postnatally. In an elegant study in mice, Aguero et al. [47] found that transient exposure to an auxotrophic E. coli mutant in the intestine of germ-free mothers in pregnancy accentuated innate immune development in the intestine of their germ-free offspring. This effect was mediated by the transfer, in part via maternal antibodies, of a range of E. coli products across the placenta and in the mother’s serum and milk. Thus, with a single gut bacterium, the mother primed the immune system of her offspring, before their exposure to the external environment [47]. If T1D mothers with an increased abundance of E. coli and other LPS-producing gut bacteria better prime innate immunity in their offspring, this could protect against potentially diabetogenic infections in early life [48] and account for the lower risk of T1D in infants with a maternal compared to paternal proband [49].
Conclusions
The gut microbiome changes across pregnancy but these changes are distinct in women with T1D. They include an increase in bacteria with pro-inflammatory properties, a decrease in bacteria with anti-inflammatory properties and a decrease in bacteria that synthesize essential vitamins, which together may lead to low-grade gut inflammation, epithelial barrier dysfunction, increased epithelial permeability and low-grade systemic inflammation. These are features of the gut microbiome ‘dysbiosis’ observed in a wide range of diseases [50], some of which have shown clinical benefit in response to probiotic and other dietary interventions. The relationship of these changes to the increased risk to mother and fetus in the T1D pregnancy requires further investigation. Intervention by dietary means to promote a less pro-inflammatory gut microbiome could potentially benefit both mother and fetus.
Methods
Participants and study design
This study involved 70 pregnancies in women participating in the ENDIA pregnancy-birth cohort study [23], 36 in women with established T1D on daily insulin treatment and 34 in healthy women with no history of gestational diabetes. The main criterion for participation in ENDIA was an unborn child with a first-degree relative with T1D. Table 1 provides summary statistics for participants, on 66 women with 70 pregnancies (four sibling pairs). Therefore, four women were included twice in the study population (each with two pregnancies). The unit of observation is the pregnancy, and therefore observations from the same mother but different pregnancies have been included as separate observations, as characteristics might change between pregnancies. Trimesters were categorised according to gestational age: T1 0–99 days; T2 100–196; T3 197–274.
Women provided written informed consent and were enrolled into the study between 2013 and 2016 at one of eight clinical sites. Up to three study visits occurred during pregnancy, ideally one in each trimester. The study was approved by a Human Research Ethics Committee (HREC) at each clinical site, with the Women’s and Children’s Health Network HREC in Adelaide acting as the lead HREC under the Australian National Mutual Acceptance Scheme (reference number HREC/16/WCHN/066). ENDIA is an observational study registered on the Australia New Zealand Clinical Trials Registry (ACTRN1261300794707).
Maternal and paternal demographics, medical history, past-pregnancy history, pre-pregnancy weight, assisted conception status and plurality of pregnancy were recorded at the first opportunity. Standardized questionnaires were offered at each pregnancy visit to record pregnancy complications, antibiotic and supplement usage, maternal and household smoking, household composition and pet ownership. Maternal diet during pregnancy was measured at the third trimester visit using a validated 74 item food frequency questionnaire, Dietary Questionnaire for Epidemiological Studies version 2 (DQESv2) [51]. Even though this was administered only in the third trimester, evidence for stability of dietary intake over the course of the pregnancy was obtained from a separate, purpose-built ENDIA Pregnancy Lifestyle Questionnaire administered before each of the three study visits during pregnancy. This assessed consumption of milk (dairy and non-dairy), caffeinated and decaffeinated tea and coffee, caffeine-containing soft drinks, dairy products, soy, gluten containing cereals (wheat, barley and rye) and non-gluten containing cereals (rice, corn and oats). Analysis across the study visits revealed that on 86% of occasions respondents reported either the same unit or within one-unit difference between visits 1–2, visits 1–3 and visits 2–3. Magnitude changes of four or five units were reported on < 2% of occasions. This supports the DQESv2 as being reflective of the whole pregnancy period. Women were advised to take multi B-group vitamin supplements from as early as possible in pregnancy.
Sample collections and analyses
Serum 1,5-AG, a measure of glucose control in pregnancy [25], was measured by GlycoMark (Nippon Kayaku Co. Ltd., New York, NY, USA) in a single batch. Serum vitamin D3 was measured with a Liaison Analyser by the DiaSorin method (DiaSorin, Turin, Italy). Plasma vitamin B6 was measured by the Chromsystems HPLC-based assay (Chromsystems Instruments & Chemicals, Gräfelfing, Germany). Serum vitamin B12 was measured by the Abbot Architect Chemiluminescent Microparticle Immunoassay.
Fecal samples were collected in accordance with our validated collection-processing-storage method [52]. Briefly, samples were captured in a toilet using the Easy Sampler device (Co-Vertec Ltd, Waterlooville, UK) then transferred into a sterile 70 mL collection jar. Participants were instructed to store the sample in the refrigerator prior to transport to the laboratory in an insulated bag within 24 h. Samples were divided into aliquots with a sterile spatula in a Biosafety Level 2 cabinet, then stored at − 80°C. A total of 134 fecal samples were collected from the 70 pregnancies with either two or three samples collected longitudinally in each pregnancy (Fig. 1). DNA was extracted from fecal samples at the Walter and Eliza Hall Institute of Medical Research (WEHI) with the MoBio PowerSoil kit (MoBio Laboratories, Carlsbad, CA) as per manufacturer’s instructions.
Fecal calprotectin (micrograms per kg) was measured in 31 and 26 samples collected in the third trimester from women with and without T1D, respectively, by quantitative, enzyme-linked immunoassay (CALPROTMOslo, Norway) according to the manufacturer’s instructions. Human serum intestinal fatty acid binding protein (I-FABP) (picograms/mL) was measured in 26 and 26 samples collected in the third trimester from women with and without T1D, respectively, by a commercial ELISA kit (Enzyme-Linked Immunosorbent Assay; Hycult Biotech, the Netherlands) according to the manufacturer’s instructions.
HLA DR typing was performed on DNA in saliva collected with OG-500 Oragene DNA tubes (DNA Genotek, Ontario, Canada) by TaqMan-based PCR-typing and imputation from three single-nucleotide polymorphisms (rs3104413, rs2187668 and rs9275495), as described previously [53].
Whole metagenome sequencing and generation of taxonomic and functional profiles
Whole metagenome sequencing (WMS) libraries were generated and sequenced with the 2 × 150 bp paired-end chemistry on two separate runs of an Illumina NovaSeq 6000 (Illumina, San Diego, California, USA) sequencer at the Ramaciotti Centre for Genomics (UNSW, Sydney, Australia. www.ramaciotti.unsw.edu.au). Sequencing data were quality controlled, and reads aligning to the human genome were removed using KneadData (v0.6.1) [24]. For the functional analysis, filtered reads classified using Kraken2 with the standard database [54], were further processed using HUMAnN2 (v0.11.1) [55] with the UniRef90 database to generate functional annotations (i.e. genes and metabolic pathways) and define the metabolic potential of the microbial communities. A functional profile (i.e. function-per-sample counts matrix) for metaCyc [56] complete metabolic pathways was obtained. In addition, two functional profiles were generated by grouping genes into KO [57] and MetaCyc-reactions functional categories using the humann2_regroup_table command. As part of the HUMAnN2 pipeline, MetaPhLan2 (v 2.7.5) [58] was used on the reads filtered with KneadData, to detect and quantify individual species with a library of clade-specific markers (ChocoPhlAn database) and generate whole-metagenome-based profiles at strain, species, genus, family, order and phylum taxonomic levels. Taxonomic and functional profiles were imported into the phyloseq [59] package in R [60]. An abundance filter was applied to remove all taxa and functional categories with a relative abundance across all samples of < 0.01%.
Alpha diversity (diversity within microbial communities) was obtained from the number of observed taxa (richness) using the function estimate richness from the R package phyloseq.
Beta diversity (diversity between microbial communities) was determined with phyloseq (function distance, method=‘bray’) on proportional log transformed data. This function calculates Bray-Curtis coefficients, which measure the distance between communities based on the taxa/functions that they contain and their abundances. The data were visualised using principal coordinates analysis (PCoA) plots in phyloseq.
Statistical analyses
For continuous responses, where appropriate, the summary tables present mean and standard deviation derived from fitting a linear mixed model. The model fit for each continuous response adjusts for the fact that the observations from women with more than one pregnancy are not fully independent but may be correlated. For some response variables, the assumption of normally distributed residuals was not met. In these analyses, the response variable was transformed using a square root or log transformation, as appropriate. For transformed responses, the back transformed means and approximated standard deviations are presented. A Wald’s test is used to determine whether the groups are significantly different.
For categorical responses, summary tables show numbers and percentages. The percentage was calculated using the total number of pregnancies or samples as the denominator. To determine whether the distribution of observations between groups for categorical data were similar or not, a generalized linear mixed model was fitted, with a random effect for woman (i.e. subject). Such models adjust for correlated women observations. To determine whether groups were significantly different, the change in deviation of the final mode (i.e. a likelihood ratio test), which includes and excludes the treatment term, was examined. A pre-set P value of 0.05 was used as a cut-off for determining statistical significance for all models. Data analysis was performed in R (R Core Team, 2018 [61];), with the R packages lme4 v1.1.21 [62], car v3.0.3 [63], predictmeans v1.0.4 [64] and nnet v7.3.12 [65].
For testing differences in alpha diversity between groups of interest, GEEs [66] were applied using the R function geeglm from package geepack v1.2-1 ([67]; parameter family set to default ‘Gaussian’) to account for possible correlation of multiple measurements within a woman over time. The default empirical (robust or ‘sandwich’) estimator was used to ensure that estimates are robust to misspecification of the correlation structure. The model used for the regression included T1D status and time (i.e. two models were tested considering time as a continuous [gestational days] or categorical [trimesters] variable) as well as their interaction term (T1D × days or trimester; to test if differences in alpha diversity between women with and without T1D change across days or trimesters) and was adjusted for sample processing batches (which includes sequencing run), conception age, BMI, parity and HLA type. Mean-centred values were used for gestational days, conception age and BMI to ensure that the model coefficients are meaningful.
Differences in beta diversity were evaluated by PERMANOVA using Bray-Curtis dissimilarities with the Adonis function from the vegan [68] R package. For tests that included multiple samples across trimesters from the same participant (i.e. longitudinal analysis), a modified version of Adonis, which performs a RMA-PERMANOVA test [32], was employed. This statistical model included T1D status and time with their interaction adjusted as in the alpha diversity model (i.e. adjusting for sequencing run, conception age, BMI, parity and HLA type). In addition, interactions between time and other factors were also tested as described in the results section. When an interaction was significant (i.e. FDR < 0.1), statistical analysis was performed within trimester (i.e. when testing for differences between women with and without T1D) or by separating data from women with and without T1D (i.e. when testing for differences in time).
Differential abundance of taxa from MetaPhlan2 and gene categories and metabolic pathways from HUMAnN2 was analysed with the R package limma [69]. First, taxonomic relative abundances from MetaPhlan were multiplied by the library size of each sample, whereas for the functional analysis of data generated with HUMAnN2 counts (CPM) were used. Taxonomic and functional data were filtered using the filterByExpr function with default parameters with an additional general abundance filter that removed all those taxa or functions with a relative abundance across all samples of < 0.01% and a prevalence filter that removed those taxa present in less than 33 samples (i.e. ~ 25% of the samples). Library sizes were normalized using the trimmed mean of log expression ratios with singleton pairing (TMMwsp) method [70] in edgeR which is expected to perform better for data with a high proportion of zeros. Counts were transformed to log2-counts per million (logCPM), voom precision weights were calculated and limma linear models were fitted while allowing for loss of residual degrees of freedom due to exact zeros using the voomLmFit function [71] [72]. Here, ‘women IDs’ were considered as blocks to calculate the consensus correlation and account for multiple measurements while estimating contrasts statistics using the contrasts.fit function and empirical Bayes moderated t statistics. Since we have samples for all possible combinations of T1D status and trimester, this is a factorial design. Therefore, in order to build our model, factors T1D status and trimester were combined into a single factor with six levels and the comparisons of interests were defined as contrasts. In addition, the model design included an adjustment for sample processing batches, conception age, BMI, parity and HLA type. For this, groups means were computed by mean-correcting covariates and factors before performing the test: for numerical covariates, the mean was subtracted and for factors the contr.sum function was used (e.g. contrasts(ExtractionBatch) <- contr.sum(levels(ExtractionBatch). The mean abundance of each taxon in each group is computed by limma when fitting the model and are contained in the coefficients component (fit$coef). The standard errors are obtained as following: fit$stdev.unscaled[,] * sqrt(fitc$s2.post). Note that after normalization, the data are on the log2 scale. For testing for differences in specific taxa due to HLA type, the limma model included, similarly to the previous model, ‘women IDs’ as blocks and adjustments for sample processing batches, conception age, BMI and parity. Four contrasts were fitted: (1) High (DR34) vs. low (DRXX, DR3X and DR4X) risk HLA types, (2) DR34 vs. DR3X and DR4X grouped into a factor, (3) DRXX vs. DR3X and DR4X grouped into a factor and (4) DR34 vs. DRXX. P values were adjusted with the Benjamini and Hochberg method to control the FDR. FDR < 0.1 were considered significant. Taxa or functions significantly different with an abundance logFC greater than 0.5 or less than − 0.5 and present in at least 50% of the samples in either of the groups being compared were regarded as biologically significant. For identifying the ‘principal bacterial contributors’ to each differentially abundant function, first, the HUMAnN2-generated files with functions (Kegg orthology, MetaCyc reaction and complete pathways) stratified by contributing species were obtained. Next, the functions of interest with contributing species were disaggregated into individual files using grep. Finally, limma was applied as explained above to each subset of function with contributing species and only species with a larger log2-FC in the group of interest for the specific function were considered principal contributors belonging to the same bacterial cluster. The significance of the difference between measurements of serum markers was tested with a Wilcoxon rank sum test equivalent to the Mann-Whitney test using the wilcox.test function from the stats R package with parameter paired set to FALSE.
Real-time quantitative PCR analysis
The qPCR reaction comprised 10 μL Sybr Green GoTaq qPCR Master Mix (2×) (Promega), 0.3 mM of each primer, 8.4 μL of water and 1 ng of DNA in 20 μL. Assays were performed in triplicate using the QuantStudio 12K Flex Real-Time PCR System (Thermofisher) with the following protocol: one cycle at 95 °C for 10 min, followed by 40 cycles of a two-stage temperature profile at 95 °C for 15 s and 60 °C for 1 min. Primers were Bacteroides vulgatus (BV-1) 5′-GCATCATGAGTCCGCATGTTC-3′, BV-2 5′-TCCATACCCGACTTTATTCCTT-3′; Bacteroides caccae (BaCA-1) 5′-GGGCATCAGTTTGTTTGCTT-3′, BaCA-2 5′-GAACGCATCCCCATCTCATA-3′; universal 16S V4 primers Univ-1 5′-GTGYCAGCMGCCGCGGTAA-3′, Univ-2 5′-GGACTACNVGGGTWTCTAAT-3′. Standard curves were generated by 2-fold dilutions ranging from 10 to 0.02 ng of a pooled human fecal DNA.
Data from each triplicate fell within a 0.5 threshold cycle (Ct); outliers (> 1 standard deviation) were removed before calculating the average Ct of each sample. Amplification efficiency (E) was determined from the slope of the standard curves for each primer pair using the formula E = (10−1/slope)-1. Efficiencies ranged from 97 to 102%. The abundances (N) for Bacteroides vulgatus and Bacteroides caccae were determined relative to the total bacterial load measured with the universal 16S primers, where N (B. vulgatus) = (Efficiency_B. vulgatus + 1)Ct_B.vulgatus, N (B. caccae) = (Efficiency_B. caccae + 1)Ct_B.caccae, N universal = (Efficiency_16S universal + 1)Ct_Universal, B. vulgatus relative abundance = N (B. vulgatus)/ N Universal, B. caccae_relative abundance = N (B. caccae)/ N Universal.
Relative abundances were log10-transformed and used as input for the regression models. The association between the relative abundance of Bacteroides caccae and Bacteroides vulgatus and T1D status was determined using a linear mixed effects model (lmer) with conception age, BMI, parity and HLA type as fixed effects, and ‘woman ID’ and processing batches as random effects.
Availability of data and materials
The demultiplexed raw datasets supporting the conclusions of this study can be accessed in the NCBI SRA https://www.ncbi.nlm.nih.gov/sra (project number PRJNA604850). All the python commands used to run HUMAnN2 and the R code used to perform statistical analyses are available at GitHub (https://github.com/PapenfussLab/RothSchulze_pregnancy-gut-microbiome-T1D) as R markdown coding and knitr html files along with the necessary R objects which contain taxonomic and functional profiles with metadata.
Abbreviations
- AG:
-
Anhydroglucitrol
- ASV:
-
Amplicon sequence variants
- BMI:
-
Body mass index
- BCAA:
-
Branched chain amino acid
- CoA:
-
Coenzyme A
- CPM:
-
Counts per million
- ENDIA:
-
Environmental determinants of islet autoimmunity
- FDR:
-
False discovery rate
- GEE:
-
Generalized estimating equations
- HLA:
-
Human leukocyte antigen
- HPLC:
-
High-performance liquid chromatography
- HREC:
-
Human research ethics committee
- KO:
-
Kegg orthology
- LCBD:
-
Local contribution to beta diversity
- LogFC:
-
Log2 fold-change
- LPS:
-
Lipopolysaccharide
- NOD:
-
Non-obese diabetic
- OTU:
-
Operational taxonomic unit
- PCoA:
-
Principal components analysis
- PWY:
-
Pathway (Metacyc)
- RMA-PERMANOVA:
-
Repeated measure-aware permutation analysis of variance
- SCFA:
-
Short-chain fatty acid
- SD:
-
Standard deviation
- T:
-
Trimester
- TCA:
-
Tricarboxylic acid
- T1D:
-
Type 1 diabetes
- TMM:
-
Trimmed mean of log expression ratios
- WEHI:
-
Walter and Eliza Hall Institute of Medical Research
- WMS:
-
Whole metagenomic sequencing
References
Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. 2016;375(24):2369–79. https://doi.org/10.1056/NEJMra1600266.
Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative View. Cell. 2012;148(6):1258–70. https://doi.org/10.1016/j.cell.2012.01.035.
Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140(6):1713–9. https://doi.org/10.1053/j.gastro.2011.02.011.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–7. https://doi.org/10.1038/nature11053.
Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Kling Bäckhed H, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150(3):470–80. https://doi.org/10.1016/j.cell.2012.07.008.
DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci. 2015;112(35):11060–5. https://doi.org/10.1073/pnas.1502875112.
Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314(21):1360–8. https://doi.org/10.1056/NEJM198605223142106.
Jawerbaum A, Gonzalez E. Diabetic pregnancies: the challenge of developing in a pro-inflammatory environment. Curr Med Chem. 2006;13(18):2127–38. https://doi.org/10.2174/092986706777935302.
Nelson SM, Sattar N, Freeman DJ, Walker JD, Lindsay RS. Inflammation and endothelial activation is evident at birth in offspring of mothers with type 1 diabetes. Diabetes. 2007;56(11):2697–704. https://doi.org/10.2337/db07-0662.
Du M, Basu A, Fu D, Wu M, Centola M, Jenkins AJ, et al. Serum inflammatory markers and preeclampsia in type 1 diabetes: a prospective study. Diabetes Care. 2013;36(7):2054–61. https://doi.org/10.2337/dc12-1934.
Knip M, Siljander H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol. 2016;12(3):154–67. https://doi.org/10.1038/nrendo.2015.218.
Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5(1):82–91. https://doi.org/10.1038/ismej.2010.92.
de Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes. 2013;62(4):1238–44. https://doi.org/10.2337/db12-0526.
Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17(2):260–73. https://doi.org/10.1016/j.chom.2015.01.001.
de Groot PF, Belzer C, Aydin Ö, Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS One. 2017;12(12):e0188475. https://doi.org/10.1371/journal.pone.0188475.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6(10):e25792. https://doi.org/10.1371/journal.pone.0025792.
Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature. 2018;562(7728):589–94. https://doi.org/10.1038/s41586-018-0620-2.
Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8. https://doi.org/10.1111/j.1574-6968.2009.01514.x.
Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165(6):1332–45. https://doi.org/10.1016/j.cell.2016.05.041.
Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. 2017;18(5):552–62. https://doi.org/10.1038/ni.3713.
Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia. 2006;49(12):2824–7. https://doi.org/10.1007/s00125-006-0465-3.
Harbison JE, Roth-Schulze AJ, Giles LC, Tran CD, Ngui KM, Penno MA, et al. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes: A prospective cohort study. Pediatr Diabetes. 2019;20(5):574–83. https://doi.org/10.1111/pedi.12865.
Penno MAS, Couper JJ, Craig ME, Colman PG, Rawlinson WD, Cotterill AM, et al. Environmental determinants of islet autoimmunity (ENDIA): a pregnancy to early life cohort study in children at-risk of type 1 diabetes. BMC Pediatr. 2013;13(1):124. https://doi.org/10.1186/1471-2431-13-124.
McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, et al. bioBakery: a meta’omic analysis environment. Bioinforma Oxf Engl. 2018;34(7):1235–7. https://doi.org/10.1093/bioinformatics/btx754.
Nowak N, Skupien J, Cyganek K, Matejko B, Malecki MT. 1,5-Anhydroglucitol as a marker of maternal glycaemic control and predictor of neonatal birthweight in pregnancies complicated by type 1 diabetes mellitus. Diabetologia. 2013;56(4):709–13. https://doi.org/10.1007/s00125-013-2830-3.
Berstad A, Arslan G, Folvik G. Relationship between intestinal permeability and calprotectin concentration in gut lavage fluid. Scand J Gastroenterol. 2000;35(1):64–9. https://doi.org/10.1080/003655200750024551.
Thuijls G, Derikx JP, van Wijck K, Zimmermann LJ, Degraeuwe PL, Mulder TL, et al. Non-invasive markers for early diagnosis and determination of the severity of necrotizing enterocolitis. Ann Surg LWW. 2010;251(6):1174–80. https://doi.org/10.1097/SLA.0b013e3181d778c4.
Shreiner AB, Kao JY, Young VB. The gut microbiome in health and in disease. Curr Opin Gastroenterol. 2015;31(1):69–75. https://doi.org/10.1097/MOG.0000000000000139.
Jang SE, Lim SM, Jeong JJ, Jang HM, Lee HJ, Han MJ, et al. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018;11(2):369–79. https://doi.org/10.1038/mi.2017.49.
Iyer SS, Gensollen T, Gandhi A, Oh SF, Neves JF, Collin F, et al. Dietary and microbial oxazoles induce intestinal inflammation by modulating aryl hydrocarbon receptor responses. Cell. 2018;173(5):1123–34. https://doi.org/10.1016/j.cell.2018.04.037.
Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339(6120):708–11. https://doi.org/10.1126/science.1232467.
IBDMDB Investigators, Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–62.
Flint HJ, Duncan SH, Scott KP, Louis P. Links between diet, gut microbiota composition and gut metabolism. Proc Nutr Soc. 2015;74(1):13–22. https://doi.org/10.1017/S0029665114001463.
Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. 2010;35(4):322–32. https://doi.org/10.1016/j.ijantimicag.2009.12.011.
Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. https://doi.org/10.1111/1462-2920.13589.
Wang M, Zhang X-Y, Guo R-R, Cai Z-P, Hu X-C, Chen H, et al. Cloning, purification and biochemical characterization of two β-N-acetylhexosaminidases from the mucin-degrading gut bacterium Akkermansia muciniphila. Carbohydr Res. 2018;457:1–7. https://doi.org/10.1016/j.carres.2017.12.007.
Belzer C, de Vos WM. Microbes inside—from diversity to function: the case of Akkermansia. ISME J. 2012;6(8):1449–58. https://doi.org/10.1038/ismej.2012.6.
LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160–8. https://doi.org/10.1016/j.copbio.2012.08.005.
Dash NR, Al Bataineh MT. Metagenomic analysis of the gut microbiome reveals enrichment of menaquinones (vitamin K2) pathway in diabetes mellitus. Diabetes Metab J. 2021;45(1):77–85. https://doi.org/10.4093/dmj.2019.0202.
Shearer MJ, Fu X, Booth SL. Vitamin K nutrition, metabolism, and requirements: current concepts and future research. Adv Nutr. 2012;3(2):182–95. https://doi.org/10.3945/an.111.001800.
Yoshii K, Hosomi K, Sawane K, Kunisawa J. Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front Nutr. 2019;6:48. https://doi.org/10.3389/fnut.2019.00048.
Sakakeeny L, Roubenoff R, Obin M, Fontes JD, Benjamin EJ, Bujanover Y, et al. Plasma pyridoxal-5-phosphate is inversely associated with systemic markers of inflammation in a population of U.S. adults. J Nutr. 2012;142(7):1280–5. https://doi.org/10.3945/jn.111.153056.
Wilson RG, Davis RE. Serum pyridoxal concentrations in children with diabetes mellitus. Pathology. 1977;9(2):95–8. https://doi.org/10.3109/00313027709085244.
Massé PG, Boudreau J, Tranchant CC, Ouellette R, Ericson KL. Type 1 diabetes impairs vitamin B6 metabolism at an early stage of women’s adulthood. Appl Physiol Nutr Metab. 2012;37(1):167–75. https://doi.org/10.1139/h11-146.
van de Lagemaat EE, de Groot LCPGM, van den Heuvel EGHM. Vitamin B12 in relation to oxidative stress: A systematic review. Nutrients. 2019;11(2):482. https://doi.org/10.3390/nu11020482.
Strobel HJ. Vitamin B12-dependent propionate production by the ruminal bacterium Prevotella ruminicola 23. Appl Environ Microbiol. 1992;58(7):2331–3. https://doi.org/10.1128/aem.58.7.2331-2333.1992.
Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science. 2016;351:1296–302.
Perrett KP, Jachno K, Nolan TM, Harrison LC. Association of rotavirus vaccination with the incidence of type 1 diabetes in children. JAMA Pediatr. 2019;173(3):280–2. https://doi.org/10.1001/jamapediatrics.2018.4578.
Warram JH, Krolewski AS, Gottlieb MS, Kahn CR. Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N Engl J Med. 1984;311(3):149–52. https://doi.org/10.1056/NEJM198407193110304.
Fukui H. Increased intestinal permeability and decreased barrier function: does it really influence the risk of inflammation? Inflamm Intest Dis. 2016;1(3):135–45. https://doi.org/10.1159/000447252.
Hodge A, Patterson AJ, Brown WJ, Ireland P, Giles G. The Anti Cancer Council of Victoria FFQ: relative validity of nutrient intakes compared with weighed food records in young to middle-aged women in a study of iron supplementation. Aust N Z J Public Health. 2000;24(6):576–83. https://doi.org/10.1111/j.1467-842X.2000.tb00520.x.
ENDIA Study Group*, Penington JS, Penno MAS, Ngui KM, Ajami NJ, Roth-Schulze AJ, et al. Influence of fecal collection conditions and 16S rRNA gene sequencing at two centers on human gut microbiota analysis. Sci Rep. 2018;8(1):1–10.
Nguyen C, Varney MD, Harrison LC, Morahan G. Definition of high-risk type 1 diabetes HLA-DR and HLA-DQ types using only three single nucleotide polymorphisms. Diabetes. 2013;62(6):2135–40. https://doi.org/10.2337/db12-1398.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46. https://doi.org/10.1186/gb-2014-15-3-r46.
Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15(11):962–8. https://doi.org/10.1038/s41592-018-0176-y.
Caspi R, Billington R, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46(D1):D633–9. https://doi.org/10.1093/nar/gkx935.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–62. https://doi.org/10.1093/nar/gkv1070.
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–3. https://doi.org/10.1038/nmeth.3589.
McMurdie PJ, Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Team RC. A language and environment for statistical computing. Vienna: R Foundation for statistical computing; 2015. URL http://CRAN.R-project.org. 2018
Team, R. Core. R: A language and environment for statistical computing R foundation for statistical computing. Vienna: R Foundation for Statistical Computing; 2017. (2020)
Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48.
Fox J, Weisberg S. An R Companion to Applied Regression, Third edition. Thousand Oaks: Sage; 2019.
Luo D, Ganesh S, Koolaard J. Predictmeans: Calculate predicted means for linear models R Package Version, 1.0. 1. 2018: https://CRAN.R-project.org/package=predictmeans.
Venables WN, Ripley BD. Modern applied statistics with S. Statistics and computing. New York: Springer; 2002. https://doi.org/10.1007/978-0-387-21706-2.
Hardin JW. Generalized estimating equations (GEE). Encyclopedia of statistics in behavioral science; 2005.
Halekoh U, Højsgaard S, Yan J. The R package geepack for generalized estimating equations. J Stat Softw. 2006;15:1–11.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package. R package version 2.5-2. 2018.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. https://doi.org/10.1093/nar/gkv007.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. https://doi.org/10.1186/gb-2010-11-3-r25.
Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29.
Lun ATL, Smyth GK. No counts, no variance: allowing for loss of degrees of freedom when assessing biological variability from RNA-seq data. Stat Appl Genet Mol Biol. 2017;16(2):83–93. https://doi.org/10.1515/sagmb-2017-0010.
Acknowledgements
The authors thank Stephen A. Wilcox (Genomics Hub, WEHI) for advice with DNA sequencing, Alexandra Garnham (Bioinformatics Division, WEHI), Curtis Huttenhower, Jason Loyd-Price, Himel Mallick and Eric Franzosa (Broad Institute of MIT and Harvard School of Public Health, Boston) for statistical advice and Enrique Zozaya-Valdes for bioinformatics analysis advice.
ENDIA Study Group (May 2021) is composed of the following authors:
Investigators: Simon C Barry, Maria E Craig, Peter G Colman, Jennifer J Couper, Elizabeth A Davis, Mark Harris, Leonard C Harrison, Aveni Haynes, Grant Morahan, Helena Oakey, Megan A S Penno, William D Rawlinson, Richard O Sinnott, Georgia Soldatos, Rebecca L Thomson, Peter J Vuillermin, John M Wentworth. Associate investigators: Emma Hamilton-Williams, Ki Wook Kim. Project and Data Managers: Amanda J Anderson, Pat Ashwood, James Brown, Kelly J McGorm, William Hu. Coordinators: Sarah Beresford, Samantha Bertram, Debra Bezuidenhout, Susan Brandrick, Carlie Butterworth, Jacki Catteau, Nakita Clements, Kyana Gartrell, Alison Gwiazdzinski, Candice Hall, Gail Harper, Amanda Hulley, Mikayla Hoffman, Renee Kludas, Christine Monagle, Belinda Moore, Benjamin Ramoso, Alison Roberts, Georgina Thompson, Alexandra Tully, Isabelle Vicary, Rosemary Wood. Dietitians: Rachel Battersby, Stephanie Savio. Post-doctoral Fellows: Esther Bandala Sanchez, Naiara Bediaga, Chris Hope, Tim Sadlon, Alexandra Roth Schulze. Research Officers: Sabrina Binkowski, Bek Brittain, Minh Bui, Dylan Foskett, Dexing Huang, Dao Huynh, Stuti Kapadia, Asma Minhaj, Gaetano Naselli, Katrina Ngui, Trung Nguyen, Yeon Park, Kelly Watson, Cynthia Yau. Administrator: Leanne Cavenett
The ENDIA Study Group would like to thank the following institutions and individuals for their contribution to ENDIA recruitment and follow-up:
Lead Clinical Recruitment/Follow-up Sites: The Women’s and Children’s Hospital, SA (Jennifer Couper). Royal Melbourne Hospital, Vic (Peter Colman, John Wentworth, Leonard Harrison). Barwon Health, Vic (Peter Vuillermin). Monash Health, Vic (Georgia Soldatos). Children’s Hospital at Westmead, NSW (Maria Craig). Royal Hospital for Women, NSW (Maria Craig). St George Hospital, NSW (Maria Craig). Princess Margaret Hospital/Perth Children’s Hospital, WA (Elizabeth Davis, Aveni Haynes). Mater Mother’s Hospital/Queensland Children’s Hospital, QLD (Mark Harris, Andrew Cotterill)
Lead Academic Sites: The University of Adelaide/Robinson Research Institute, SA (Jennifer Couper, Megan Penno, Rebecca Thomson, Kelly McGorm, Helena Oakey, Simon Barry). WEHI, Vic (Leonard Harrison, John Wentworth). University of New South Wales, NSW (Maria Craig, William Rawlinson). University of Sydney, NSW (Maria Craig). University of Western Australia/Telethon Kids Institute/Harry Perkins Institute, WA (Elizabeth Davis, Aveni Haynes, Grant Morahan), University of Melbourne, Vic (Richard Sinnott). University of Queensland, QLD (Mark Harris)
Satellite Recruitment/Birthing Sites (SA): Country Health SA (Jennifer Couper), Flinders Medical Centre (Brian Coppin), Lyell McEwin Hospital (Jennifer Couper), Ashford Hospital, Flinders Private Hospital, North Eastern Community Hospital.
Satellite Recruitment/Birthing Sites (VIC): Royal Women’s Hospital (Alison Nankervis), Ballarat Base Hospital (David Song), Bendigo Health (Mark Savage), Epworth Geelong Hospital (Peter Vuillermin), Mercy Hospital for Women (Christine Houlihan, Peter Colman), St. John of God Geelong (Peter Vuillermin), Sunshine Hospital/ Joan Kirner Women's and Children's Hospital (Balasubramanian Krishnamurthy), Werribee Mercy Hospital (Sheetal Tipnis).
Satellite Recruitment/Birthing Sites (NSW): Hunter Diabetes Centre (Claire Morbey), John Hunter Hospital (Maria Craig), John Hunter Children’s Hospital (Maria Craig), Sydney Children’s Hospital (Maria Craig), The Sutherland Hospital (Maria Craig), Westmead Hospital (Maria Craig), North Shore Private Hospital (Maria Craig).
Satellite Recruitment/Birthing Sites (WA, all under Elizabeth Davis): Armidale Hospital, Bentley Hospital, Joondalup Health Campus, King Edward Memorial Hospital, Rockingham General Hospital, St John of God Mt. Lawley, St John of God Murdoch, St John of God Subiaco.
Satellite Recruitment/Birthing Sites (QLD): Royal Brisbane and Women’s Hospital (Michael d’Emden), Wesley Hospital (Stephen Cook, Andrew Cotterill)
Satellite Recruitment/Birthing Sites (NT): Royal Darwin Hospital (Louise Maple-Brown).
Referring Physicians in Private: Natalie Harrison/Geelong Diabetes & Endocrinology (Vic), Dorothy Graham (WA), Linda McKendrick (SA), Amanda Terry (SA).
Other Birthing Hospitals: Albury-Wodonga Hospital, Angliss Hospital, Auburn Public Hospital, Bankstown Hospital, Bathurst Base Hospital, Beaudesert Hospital, Beijing United Family Hospital (China), Belmont Hospital, Berri Hospital, Blacktown Hospital, Box Hill Hospital, Broome Hospital, Buderim Private Hospital, Bunbury Hospital, Burnside War Memorial Hospital, Caboolture Hospital, Cabrini Hospital Malvern, Cairns Hospital, Cairns Private Hospital, Calvary Hospital, Calvary Hospital Bruce, Calvary Hospital Lenah Valley, Calvary Hospital Wagga, Calvary John James Hospital, Campbelltown Hospital, Canberra Hospital, Casey Hospital, Centenary Hospital for Women and Children, Coffs Harbour Base Hospital, Dandenong Hospital, Darwin Birth Centre, Darwin Private Hospital, Dubbo Base Hospital, Echuca Regional Health, Emerald Hospital, Epworth Freemasons Hospital, Fiona Stanley Hospital, Frances Perry House, Frankston Hospital, Gawler Hospital, Gippsland Health, Glengarry Private Hospital, Gold Coast Private Hospital, Gold Coast University Hospital, Gosford Hospital, Gosford Private Hospital, Goulburn Valley Health, Grafton Base Hospital, Greenslopes Private Hospital, Griffith Base Hospital, Gunnedah Hospital, Hawkes Bay Hospital (New Zealand), Hervey Bay Hospital, Hobart Private Hospital, Hornsby Hospital, Hurstville Private Hospital, Ipswich Hospital, Jessie McPherson Private Hospital, John Flynn Private Hospital, Kalgoorlie Hospital, Kapunda Hospital, Kareena Private Hospital, Katoomba Hospital, Launceston General Hospital, Lismore Base Hospital, Liverpool Hospital, Logan Hospital, Mackay Base Hospital, Maitland Hospital, Manly Hospital, Mater Hospital Sydney, Mater Mothers’ Hospital, Mater Mothers’ Private Hospital, Mater Private Hospital Mackay, Mater Private Hospital Redland, Mater Women's and Children's Hospital Townsville, Mitcham Private Hospital, Moruya District Hospital, Mount Barker Hospital, Narrabri Hospital, Nepean Hospital, Nepean Private Hospital, Newcastle Private Hospital, North Gosford Private Hospital, North West Hospital, North West Private Hospital, Northeast Health Wangaratta, Northern Beaches Hospital, Northern Hospital, Northpark Private Hospital, Norwest Private Hospital, Orange Base Hospital, Osborne Park Hospital, Peel Health Campus, Peninsula Private Hospital, Pindara Private Hospital, Port Augusta Hospital, Port Macquarie Hospital, Prince of Wales Hospital, Prince of Wales Private Hospital, Queanbeyan District Hospital, Queen Victoria Hospital, Redcliffe Hospital, Redland Hospital, Riverland General Hospital, Rockhampton Base Hospital, Royal Hobart Hospital, Royal Hospital for Women, Royal North Shore Hospital, Royal Prince Alfred Hospital, Sandringham Hospital, St George Private Hospital, St John of God Ballarat Hospital, St John of God Bendigo Hospital, St John of God Berwick Hospital, St John of God Bunbury Hospital, St John of God Midland Hospital, St Vincent's Hospital, St Vincent's Private Hospital, St Vincent’s Private Hospital Toowoomba, Stanthorpe Hospital, Sunnybank Private Hospital, Sunshine Coast University Hospital, Swan District Hospital, Sydney Adventist Hospital, Tamworth Hospital, Tanunda Hospital, The Canberra Hospital, The Tweed Hospital, Toowoomba Base Hospital, Toowoomba Private Hospital, Townsville University Hospital, Wallaroo Hospital, Wangaratta Hospital, Waverley Private Hospital, Westmead Private Hospital, Wodonga Hospital, Wollongong Hospital, Wollongong Private Hospital.
We also gratefully acknowledge the participants and their families who contribute to the ENDIA Study.
Funding
This research was supported by JDRF Australia, the recipient of the Commonwealth of Australia grant for Accelerated Research under the Medical Research Future Fund and with funding from the Leona M. and Harry B. Helmsley Charitable Trust (grant key 3-SRA-2020-966-M-N). In addition, support was provided by The National Health and Medical Research Council of Australia (NHMRC) Centre of Research Excellence for the Protection of Pancreatic Beta Cells (1078106), and JDRF International (1-SRA-2018-543-S-B). Additional support was provided by a NHMRC Program Grant (LCH 1037321), Victorian State Government Operational Infrastructure Support, Diabetes South Australia and the NHMRC Research Institute Infrastructure Support Scheme. MEC was supported by a NHMRC Practitioner Fellowship (1136735), ATP by an NHMRC Senior Research Fellowship (1116955) and LCH by a NHMRC Senior Principal Research Fellowship (1080887).
Author information
Authors and Affiliations
Consortia
Contributions
AJR-S performed bioinformatic analyses under the supervision of ATP and LCH, and wrote the manuscript with LCH. MASP contributed to study conception and design, compiled metadata and edited the manuscript. KMN performed DNA extraction and sequencing. HO prepared maternal summary tables and advised on statistical analysis. RLT analysed dietary and physical activity data. PJV contributed to study design, recruitment and sample collection. MEC contributed to study design, participant recruitment and sample collection. WDR contributed microbiology expertise and edited the manuscript. EAD contributed to participant recruitment and sample collection. MH contributed to participant recruitment and sample collection. GS contributed to participant recruitment and sample collection. PCG contributed to participant recruitment and sample collection. JMW contributed to participant recruitment and sample collection. AH contributed to participant recruitment and sample collection. GM analysed HLA types. NGB contributed to bioinformatic analysis. GKS contributed to bioinformatic analysis. ATP contributed to bioinformatic analysis. JJC contributed to study design, participant recruitment and sample collection and edited the manuscript. LCH designed and supervised the study, analysed data and wrote the manuscript with AJR-S. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ENDIA study is approved by Women’s and Children’s Health Network Human Research Ethics Committee (HREC) under the National Mutual Acceptance Scheme for conduct in South Australia, Victoria, New South Wales, and Queensland (HREC/16/WCHN/66). Conduct in Western Australia is approved by the Child and Adolescent Health Service HREC (RGS0000002402). The investigators and study staff conduct the study in accordance with International Conference on Harmonisation Good Clinical Practice guidelines, the Declaration of Helsinki, the Australian National Statement on Ethical Conduct in Human Research and the Australian Code of the Responsible Conduct of Research. All participants provided written informed consent and were free to withdraw from the study at any time.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. Taxonomic composition of the 25 most abundant species as measured by WMS in fecal samples collected in trimesters 1, 2 and 3 of 70 pregnancies from 66 women (35 with T1D). X-axis depicts the non-informative study ID in the format womanID_pregnancy number_trimester. LCBD: local contribution to beta diversity (a measure of the uniqueness of communities). T1D: women with type 1 diabetes, non-T1D: women without T1D. Figure S2. Alpha diversity (Richness), by T1D status of women across trimesters. Figure S3. Beta diversity analysis by T1D status of women. PCoA ordination plots based on Bray-Curtis distances between samples at the Genus and Family taxonomic levels separated by trimesters in pregnancy. Figure S4. Beta diversity analysis by T1D status of women. PCoA ordination plots based on Bray-Curtis distances between samples at the Order and Phylum taxonomic levels separated by trimesters in pregnancy. Figure S5. Relative abundance and species composition of A) two orders differentially abundant between women with and without T1D, and B) one genus differentially abundant between trimesters in women with T1D. Figure S6. Log2 transformation of the relative abundance (1000 + 0.01) of bacterial species within the Enterobacteriales and Bifidobacteriales orders in women with and without T1D (mean ± SEM). P: P-value. Figure S7. Taxa differentially abundant between trimesters in women with and without T1D (mean ± SEM). Transformed data are on a log2 scale with the mean from fitted abundances shown as a point in each trimester for each group of women. The between trimesters denotes a significant difference between those trimesters while the after the trend line denotes significant difference between trimesters 1 and 3. The color denotes if differences between trimesters are within women with or without T1D. Figure S8. Boxplots representing the abundance distribution obtained by real-time quantitative PCR (qPCR) in women and with without T1D. P: P-value. Figure S9. Boxplots representing the functional alpha diversity distribution in women with and without T1D). P: P-value. Figure S10. Taxa contributing to pathways that are differentially more abundant in women with compared to women without T1D and non-T1D. A) PWY1269: CMP-3-deoxy-D-manno-octulosonate pathway I involved in LPS biosynthesis, and B) PWY−5838: Superpathway of menaquinol−8 synthesis involved in vitamin K2 synthesis. Y-axis: log2 of CPM (counts per million). Figure S11. Taxa contributing to functional features that are differentially more abundant in women with compared to women without T1D. A) Pyridoxal 5'-phosphate synthase (K06215) involved in vitamin B6 synthesis, B) COBALSYN−PWY: Adenosylcobalamin salvage from cobinamide involved in vitamin B12 synthesis, C) 3−hydroxybutyryl−CoA dehydrogenase (K00074) involved in short chain fatty acid (SCFA) production, and D) beta−N−acetylhexosaminidase (K01207), only differentially abundant in trimester 2, involved in mucin degradation. Y-axis: log2 of CPM (counts per million). Figure S12. Log2 transformed relative abundance (+ 0.01) of bacterial clusters based on broader functions in women with and without T1D across trimesters. P: P-value. Figure S13. Boxplots representing the distribution of measures for calprotectin and I-FABP in women with and without T1D. P: P-value.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Roth-Schulze, A.J., Penno, M.A.S., Ngui, K.M. et al. Type 1 diabetes in pregnancy is associated with distinct changes in the composition and function of the gut microbiome. Microbiome 9, 167 (2021). https://doi.org/10.1186/s40168-021-01104-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01104-y