
Abstract
Background
The incidence of type 1 diabetes has increased worldwide, particularly in younger children and those with lower genetic susceptibility. These observations suggest factors in the modern environment promote pancreatic islet autoimmunity and destruction of insulin-producing beta cells. The Environmental Determinants of Islet Autoimmunity (ENDIA) Study is investigating candidate environmental exposures and gene-environment interactions that may contribute to the development of islet autoimmunity and type 1 diabetes.
Methods/design
ENDIA is the only prospective pregnancy/birth cohort study in the Southern Hemisphere investigating the determinants of type 1 diabetes in at-risk children. The study will recruit 1,400 unborn infants or infants less than six months of age with a first-degree relative (i.e. mother, father or sibling) with type 1 diabetes, across five Australian states. Pregnant mothers/infants will be followed prospectively from early pregnancy through childhood to investigate relationships between genotype, the development of islet autoimmunity (and subsequently type 1 diabetes), and prenatal and postnatal environmental factors. ENDIA will evaluate the microbiome, nutrition, bodyweight/composition, metabolome-lipidome, insulin resistance, innate and adaptive immune function and viral infections. A systems biology approach will be used to integrate these data. Investigation will be by 3-monthly assessments of the mother during pregnancy, then 3-monthly assessments of the child until 24 months of age and 6-monthly thereafter. The primary outcome measure is persistent islet autoimmunity, defined as the presence of autoantibodies to one or more islet autoantigens on consecutive tests.
Discussion
Defining gene-environment interactions that initiate and/or promote destruction of the insulin-producing beta cells in early life will inform approaches to primary prevention of type 1 diabetes. The strength of ENDIA is the prospective, comprehensive and frequent systems-wide profiling from early pregnancy through to early childhood, to capture dynamic environmental exposures that may shape the development of islet autoimmunity.
Trial registration
Australia New Zealand Clinical Trials Registry ACTRN12613000794707.
Similar content being viewed by others


Background
The rising incidence of T1D and gene-environment interaction
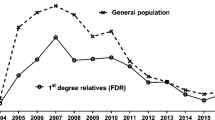
Type 1 diabetes (T1D), one of the most common childhood-onset chronic diseases, is associated with enormous human and economic costs. The incidence of T1D is increasing worldwide [1] with a younger age of onset described in European and Australian populations [2, 3]. In the 1980s the mean adjusted incidence rate of T1D in Australia was ~11 per 100,000 person-years [4, 5]. By 2006, this had increased to 21.7 per 100,000 person-years, with a 4.1% increase in the diagnosis of children less than 15 years of age from 1999 to 2006 [3].
The doubling in incidence of T1D in Australia over the past two decades is consistent with a major role for the modern environment in T1D pathogenesis. International evidence to support this includes: (a) the reduced relative frequency of high risk genotypes in newly diagnosed children; (b) a less than 40% concordance of T1D in monozygotic twins; (c) discrepancies in disease incidence among genetically similar populations living in different regions, and (d) migration studies that show T1D incidence increases as populations move from low-risk to high-risk areas [6].
The HLA region on chromosome 6p contributes approximately half of the genetic susceptibility to T1D, yet the relative frequency of high risk HLA class II genotypes in children presenting with T1D has decreased as the incidence of the disease has increased [7]. Moreover, the recognition that over 60 gene loci are associated with T1D has led to speculation that T1D is a disease with discrete genetic subtypes whereby susceptibility genes interact differently with environmental exposures [8]. Variation in gene-environment interactions may explain different effects of environmental exposures in different populations at-risk of T1D. For example, caesarean section increases the risk of progression from islet autoimmunity to T1D in individuals with T1D susceptible IFIH1 genotypes [9], whereas the early introduction of cow’s milk may increase risk of islet autoimmunity only in some at-risk genotypes [10]. As the immunological processes associated with the development of islet autoantibodies and T1D occur months to years prior to the onset of clinical disease, a long asymptomatic period provides the opportunity for prediction and, potentially, prevention [11].
Prospective studies, including our own, reveal the development of islet autoimmunity may occur at any point during the first years of life [12] through to adolescence [13], although recent data pooled from three large international cohorts have indicated the median age at seroconversion is 2.1 years [14]. Once persistent islet autoimmunity has developed the risk of progressing to T1D by 15 years of age is 12.7% in children with a single islet autoantibody, 61.6% in children with two islet autoantibodies and 79.1% in children with three islet autoantibodies compared with a 0.4% risk in children with no islet autoantibodies [14]. Moreover, progression to T1D is faster in children who: (a) seroconvert prior to three years of age, (b) have human leukocyte antigen (HLA) genotype DR3/DR4-DQ8, and (c) are female [14].
The aim of the Environmental Determinants of Islet Autoimmunity (ENDIA) study is to identify environmental factors, and gene-environment interactions, that contribute to and protect against the development of islet autoimmunity and progression to T1D in children genetically at-risk of T1D. We will follow 1,400 infants with a first-degree relative (FDR) with T1D prospectively from early pregnancy into childhood to investigate relationships between prenatal and postnatal environmental factors, and the development of islet autoimmunity and subsequent T1D. ENDIA will evaluate the microbiome, nutrition, bodyweight/composition, metabolome-lipidome, insulin resistance, innate and adaptive immune function and viral infections. A systems biology approach will integrate multi-omics analyses to explore hypotheses and mechanisms underlying the development of islet autoimmunity.
The microbiome and islet autoimmunity
Contributors to aberrant development of the microbiome
Humans live in a symbiotic relationship with trillions of microorganisms collectively known as the commensal microbiome [15]. The establishment of the microbiome begins at birth when the neonate is exposed to microorganisms derived from the mother and immediate environment [16]. The composition of the microbiome changes rapidly during early life until it is similar to that of an adult by two years of age [17].
Several environmental factors contribute to shaping the composition of microbiome including genotype, mode of delivery, antibiotic use and microbial exposure in early life, time of first fever, nutrition and weight gain in early life [18, 19]. Equally, contemporary changes in these factors have also been linked to T1D. For example, the proportion of Australian deliveries by caesarean section has increased from 21% in 1998 to 31% in 2007 [20]. Vaginally delivered infants acquire bacterial communities resembling their own mother’s vaginal microbiome whereas infants delivered by caesarean section harbour communities similar to those found on the skin [16]. This change in the initial microbiome may lead to alternative microbial succession patterns that persist over time and contribute to variations in normal physiology and/or to disease risk [16]. A meta-analysis of international observational studies showed a 20% increase in the incidence of T1D in children delivered by caesarean section [17]. As a second example, the prevalence of overweight and obesity, currently at 34-50% [21] in pregnancy and 27% [22] in childhood in Australia, has increased over the last 20 years. With excessive weight gain in pregnancy, Bifidobacterium counts are lower in the mother’s breast milk, which in turn impacts upon the microbiome of the infant [17]. Early childhood weight gain is associated with an increased risk of islet autoimmunity [12] while childhood obesity is preceded by lower counts of Bifidobacterium at six and 12 months of age [17]. Finally, antibiotic use, which has a direct effect on the gut microbiome [18], has increased in children aged over two in the last 10 years in Australia, although data during the first two years of life are lacking [20].
The gut microbiome and host immune system
The development of the mucosal immune system, and maturation of the systemic immune system, depends on bacterial colonisation of the mucosa [23]. A normal mucosal immune system is necessary to generate regulatory T cells (Treg) in response to oral antigens [24]. The essential role of the mucosal immune system in maintaining immune homeostasis is illustrated by the effect of a germ-free versus a conventional ‘dirty’ environment on the incidence of spontaneous autoimmune diabetes in the non-obese diabetic (NOD) mouse, the best animal model of T1D. The incidence of spontaneous diabetes in NOD mice differs greatly between colonies around the world and is inversely correlated with exposure to microbial infection [25]. The high incidence of diabetes in NOD mice housed under pathogen-free conditions is reduced by conventional conditions of housing and feeding [26, 27]. Under ‘dirty’ conditions, bacterial colonisation of the intestine is accompanied by maturation of mucosal immune function [28]. Emerging evidence indicates that the gut microbiome differs in composition and function between children at risk for T1D and case controls [29]. Supplementary to the microbiome, breast milk contains growth factors, cytokines and other immunomodulatory agents that promote functional maturation of the intestinal mucosa and mucosal immune system. Breast milk also contains endogenous insulin [30], probably the key autoantigen in T1D [31], and ‘oral tolerance’ to insulin conceivably might protect against the development of T1D.
Changes in the gut microbiome associated with islet autoimmunity and T1D
Experimental diabetes models show that specific bacteria and their products program Treg and T helper-17 (Th17) cell development [32]. However, it is yet to be confirmed if and how human gut microbes contribute to the development of islet autoimmunity. Two small European studies involving a total of 17 children with islet autoimmunity and 17 age/gender/HLA genotype matched controls reported decreased microbial diversity in stools from children with islet autoimmunity [33, 34], while metagenomic analysis revealed that children with islet autoimmunity have less butyrate-producing and mucin-degrading bacteria [35]. This has led to speculation that the normal process during the first two years of life by which a healthy microbiome becomes more diverse and stable, is not seen in children who develop islet autoimmunity and T1D. Socio-economic and cultural factors may play a role in shaping the diversity of the microbiome. For example, the incidence of T1D in Finland is 41.4 per 100,000 people per year, yet in neighbouring Russian Karelia it is only 7.4 per 100,000 people per year, despite these populations sharing a genetically similar background [36]. Karelian children show significantly higher infection rates than their Finnish peers, which alters the composition of the gut microbiome [37]. The challenge is to determine how these alterations relate to the pathogenesis of T1D.
Therapeutic implications of the microbiome for T1D
If the microbiome is shaped by the early environment and if specific changes in the microbiome are associated with susceptibility to T1D, then in theory it should be possible to re-engineer the microbiome and reduce the incidence of T1D. This might be achieved in several ways, for example by reducing the frequency of caesarean births, avoiding unnecessary administration of antibiotics, promoting breast feeding, modifying the early infant diet, avoiding excessive weight gain and administering pre- and pro-biotics. Administration of probiotics to NOD mice has been shown to reduce their incidence of spontaneous autoimmune diabetes [38].
Effects of weight gain on the development of islet autoimmunity and T1D
Pregnancy weight gain and perinatal risk factors influencing islet autoimmunity
In a retrospective study, higher maternal body mass index (BMI) before pregnancy and weight gain >15 kg during pregnancy predicted risk of islet autoimmunity in offspring with the high-risk HLA genotype, DR4-DQ8/DR3-DQ2 [39]. No prospective data during pregnancy have been published, however, perinatal risk factors have been analysed in the German BabyDiab Study. Birth weight in the middle tertile (3,250-3,700 g) was associated with increased islet autoimmunity risk, as was maternal HbA1c greater than 7% [40]. The presence of islet autoantibodies in cord blood may also be a marker of reduced risk for T1D in offspring [41, 42].
Weight gain and nutrition during childhood
The rise in childhood incidence of T1D parallels the overweight/obesity epidemic in Western childhood populations. Some evidence exists for an effect of early childhood weight gain on risk of islet autoimmunity. In the first Australian birth cohort of at-risk children (Australian BabyDiab study) weight z-score and body mass index (BMI) z-score were continuous predictors of risk of islet autoimmunity [12]. Similarly, in the at-risk Diabetes Autoimmunity Study in the Young (DAISY) birth cohort followed in Colorado, height velocity from age two was associated with risk of islet autoimmunity [43]. However, children in the German BabyDiab cohort showed no effect of weight gain on risk of islet autoimmunity [44]. Younger age of T1D onset is inconsistently associated with higher BMI at diagnosis [45, 46]. Retrospective case control studies from Scandinavia link increased linear growth and weight gain in early childhood with T1D [47].
Prospective birth cohorts show mixed data on the duration of exclusive breastfeeding and effect of solid food exposure on risk of islet autoimmunity [48–54]. An early analysis from the Trial to Reduce IDDM in the Genetically at-Risk (TRIGR) study suggests that feeding a casein hydrolysate formula to Finnish infants at-risk of T1D following cessation of breastfeeding reduced risk of islet autoimmunity [55]. Recent evidence from the DIASY study shows that both early and late first exposure to any solid food predicts development of T1D, leading to speculation that early exposure to solid foods evokes an abnormal response in the immature gut; while late exposures may relate to the cessation of breastfeeding before solid foods introduction so that the protective effects of breast milk are lost before the introduction of foreign food antigens [56].
Links between weight gain, systemic inflammation and insulin resistance
Weight gain is associated with markers of innate immune system activation and chronic low-grade inflammation, which have been linked to insulin resistance. Previous studies have shown that pro-inflammatory CD11c + adipose tissue macrophages impair insulin-stimulated glucose uptake by human adipocytes, providing a possible mechanistic link between obesity and insulin resistance [57]. In addition, adipose tissue macrophages metabolize lipid and may initiate adaptive immune responses by adipose tissue T cells, which are activated in adipose tissue in obesity. Infants born small for gestational age with catch up growth have accelerated weight gain and insulin resistance in early life. Appropriate for gestational age but premature babies also have reduced insulin sensitivity [58]. The mechanism is unknown but underscores the importance of prospective studies from pregnancy in at-risk children.
Innate and adaptive immune function and risk of islet autoimmunity and T1D
Pro-inflammatory mediators and anti-inflammatory dietary components in T1D
Circulating pro-inflammatory cytokines are increased at diagnosis in T1D [59–61] and, in a nested case–control study of 67 children enrolled in the Viruses in the Genetically at Risk (VIGR) birth cohort study (27 with islet antoantibodies; 40 age-matched antibody negative controls), pro-inflammatory cytokines and chemokines were increased in children with islet autoimmunity [62]. Whilst longitudinal studies before diagnosis are lacking, the DAISY Study found that serum C-reactive protein concentrations predicted progression to islet autoimmunity in early childhood [63]. Chronic low-grade inflammation in the mother has been linked to insulin resistance in the offspring. Thus, concentrations of serum pro-inflammatory advanced glycation end products (AGEs) at the end of healthy pregnancies were associated with lower adiponectin and higher insulin levels in the offspring [64]. We will measure both AGEs in pregnancy and AGEs and adiponectin in the offspring in ENDIA.
The omega-3 fatty acids docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) reduce inflammatory cytokines and inflammatory prostaglandins, both of which contribute to the initial islet lesion. Accordingly, higher intake of DHA and EPA might lower the risk of T1D, but definite evidence is lacking. Cod liver oil, omega-3 fatty acids and vitamin D taken during pregnancy or infancy have been associated with a lower incidence of T1D in a case control study [65] and increased dietary omega-3 fatty acid intake in infancy is weakly associated with a lower risk of islet autoimmunity [66]. Therefore, protective effects of these natural anti-inflammatory dietary components may occur during pregnancy, during infancy, or both. DHA and EPA inhibit prostaglandin E2 (PGE2) synthase with equivalent or greater potency than other fatty acid analogues.
Vitamin D is an anti-inflammatory steroid with multiple immunomodulatory effects that promotes immune tolerance. The vitamin D receptor (VDR) is expressed by all cells of the immune system, while activated macrophages and dendritic cells express 1α hydroxylase, the enzyme that converts 25-hydroxyvitamin D3 (25 OHD) to its metabolically active form, 1,25 OHD. Polymorphisms in both the VDR and the gene that encodes 1α hydroxylase, CYP27B1 are associated with T1D risk [67–69]. Two Northern European cross-sectional studies have shown an inverse relationship between vitamin D supplementation in infancy and T1D incidence [65, 70], but the DAISY Study found no relationship between 25 OHD levels and risk of islet autoimmunity or progression to T1D [71]. Population 25 OHD levels have fallen over the last 50 years with urbanisation, avoidance of or less access to sunlight, decreased intake of oily fish and a lower recommended daily intake of vitamin D. In Australia, vitamin D insufficiency is common in pregnancy [72].
Adaptive immune function
Aberrant immune responses to self-antigens are controlled by tolerance mechanisms, including suppression by Treg cells. The impaired generation or function of Treg cells, or resistance to their action, could contribute to the development of autoimmune disease [73]. Indeed, an imbalance between Treg and effector T cells has been linked to T1D [74]. Natural CD4+ Treg are generated in the thymus during development and induced CD4+ Treg (iTreg) by ‘tolerogenic’ conditions of antigen presentation postnatally in the periphery, especially in the mucosa. The microbiome is essential for development of the mucosal immune system, which in turn is required for the generation of iTreg. Treg are defined by high expression of CD25 and stable expression of the transcription factor FOXP3, which is critical to maintain expression of genes required for suppressor function and prevent expression of genes required for effector function [75]. FOXP3 is also expressed transiently by activated T cells but its stable expression in Treg is associated with demethylation at the FOXP3 locus. We have identified another marker of stable Treg, namely peptidase inhibitor 16 (CD359), which will be used to enumerate Treg in the ENDIA Study [75].
In addition to conventional Treg, we have identified a sub-population of CD4+ T cells with antigen-activated suppressor function that are required to prevent autoimmune diabetes in the NOD mouse model of T1D [76]. These cells are characterised by high expression of CD52 and exert suppression by releasing soluble CD52, which ligates inhibitory Siglec receptors. We observed that the frequency and function of CD52hi CD4+ T cells in response to the islet autoantigen, glutamic acid decarboxylase 65 (GAD65), was impaired in children at risk for T1D [76]. The characterisation of islet autoantigen-specific T cells, including CD52hi CD4+ T cells, will be a critical component of the ENDIA Study.
Enhanced innate immunity promoted by the environment may drive adaptive immunity by impairing regulatory and stimulating pathogenic T cell immunity. In the ENDIA Study, we will measure cytokines and chemokines produced in whole blood in vitro in response to innate immune (TLR agonists) stimuli and adaptive immune (islet [proinsulin, GAD65] and control [childhood vaccines]) antigens [77].
Viral infection is associated with islet autoimmunity and T1D
Viruses might trigger the onset of islet autoimmunity and/or promote the progression of established islet autoimmunity. Prospective studies from birth are required to obtain evidence in support of these possibilities; direct proof of a causal role for viruses will be much harder to obtain. In a meta-analysis, we recently reported evidence for a role for enterovirus (EV) at the clinical onset of T1D [78], with an odds ratio (OR) ~10 for EV infection [79]. We have also found a significant association between EV infection and islet autoimmunity [78], with an OR of ~4 in children with EV infection. Data on the role of EV infection as an initiator [80, 81] or accelerator [82] of islet autoimmunity in longitudinal studies of genetically-predisposed children are conflicting, although some of these differences may be related to the use of different techniques for determining EV infection. The temporal relationship between EV infection and development of islet autoimmunity needs to established, as well as the interaction between EV infection and other environmental determinants. For example, it has been reported that the effect of EV infection on the development of islet autoimmunity may be increased by early exposure to cow’s milk [83]. In particular, the effect of maternal exposure to the virus prenatally and of breastfeeding duration has not been examined. We found that EV infection is more frequent in recent-onset T1D in patients with co-existing vitamin D deficiency (unpublished data).
Rotavirus (RV) infection, the most common cause of infant diarrhoea, has been associated temporally with the first appearance of islet autoantibodies [84], and molecular mimicry has been demonstrated between human T cell epitopes in RV and pancreatic islet autoantigens [85]. The live RV vaccines RotaRix (GlaxoSmithKline) and RotaTeq (Merck/CSL) are now available to all Australian infants aged 6–32 weeks, with a 2008–2009 uptake rate of >80% [86] compared with <50% in US [87]. It will therefore be important to document if RV immunization alters the development of islet autoimmunity and T1D incidence. The lack of direct evidence for viral infection in causing T1D is partly methodological but also suggests multiple other and possibly ubiquitous environmental factors are at play.
Omics studies in the development of islet autoimmunity and T1D
One of the challenges for interventional studies to prevent T1D is a lack of biomarkers that reliably correlate with disease activity. Consequently, study endpoints are limited to assessments of beta-cell function and/or diagnosis of T1D following months to years of intervention [74]. Changes in the dynamic metabolome, lipidome and/or proteome may provide suitable and sensitive biomarkers, whilst providing insight into the underlying mechanisms that initiate autoimmunity. For example, reduced levels of succinic acid at birth and reduced phospholipids and triglycerides in childhood, as measured within the blood metabolome, were reported to precede islet autoimmunity in children who progressed to T1D [88]. The same study also identified elevated serum glutamic acid prior to the appearance of GAD autoantibodies, leading to speculation that an increased glutamate load on the beta cells may increase activity of GAD65, a major beta-cell antigen, potentially triggering the autoimmune process. More recently, children who later developed islet autoimmunity and progressed to T1D were shown to have a distinct cord blood lipidomic profile with reduced major choline-containing phospholipids, implicating choline metabolism in pregnancy as a factor associated with progression to T1D, although not necessarily with development of islet autoimmunity per se [89].
Environmental factors are likely to modify the DNA template by chemical and other epigenetic changes. An epigenome-wide association study (EWAS) of DNA methylation profiles of purified CD14+ monocytes (an immune effector cell type relevant to T1D pathogenesis) from 15 pairs of monozygous twins discordant for T1D revealed 132 CpG sites that were differentially methylated in accordance with T1D status [90]. By genome-wide methylation analysis of human T cell subsets we identified previously unrecognised hypomethylated promoter regions regulated by FOXP3 in natural Treg [91]. Importantly, significantly less methylation was observed in many of these gene promoters in Treg from children with islet autoantibodies compared to healthy controls, pointing to widespread alterations in the epigenetic landscape. The ENDIA Study will allow the epigenetic landscape to be scanned across time.
ENDIA in the context of international studies investigating the environment and T1D risk
To our knowledge there are no other studies in the Southern Hemisphere or South-East Asia investigating environmental determinants of T1D from pregnancy through early life. Internationally, the ENDIA Study will differ from The Environmental Determinants of Diabetes in the Young (TEDDY) [92], the TRIGR [93] and German BabyDiab [94] studies in that:
-
ENDIA includes children on the basis of family history, irrespective of HLA genotype. This is relevant as the rising incidence of T1D is accounted for by individuals with lower risk HLA genotypes.
-
Prospective samples will be collected for analysis of the microbiome from the gut (represented by stool), oral cavity, nares, skin and vagina during pregnancy and from breast milk during lactation (including colostrum) and 3-monthly in early childhood. Pregnancy and the first two years of life are critical periods for the development and establishment of the microbiota, underscoring the importance of prospective studies from pregnancy.
-
Nutritional status and intake will be documented prospectively during pregnancy (from the first trimester) and lactation [95].
-
Weight gain and physical activity (using validated tools [96]) will be measured prospectively during pregnancy.
-
Nutritional status and intake in childhood will be documented by daily diary until weaning and then by multi-pass 24-h food recalls [97, 98] until 24 months, and from two years by the Australian Toddler Eating Survey [99].
-
Fasting blood samples will be collected in childhood to measure insulin resistance.
-
The Australian population differs in many ways from those in the USA and Europe, e.g. in diet, UV exposure, and immunisation status (Australia has a much higher RV vaccine uptake than in USA).
Hypothesis and objectives
Hypotheses
The unifying hypothesis is that gene-environment interactions during prenatal and postnatal development drive the development of islet autoimmunity and T1D in children at-risk for T1D.
The specific hypotheses are:
-
1.
The maternal microbiome during pregnancy and lactation differs in composition, diversity and functional products between mothers whose offspring do and do not develop islet autoimmunity and T1D.
-
2.
The microbiome differs in composition, diversity and functional products in children who do and do not develop islet autoimmunity and T1D during the first three years of life.
-
3.
Accelerated weight gain during pregnancy, and accelerated weight gain and insulin resistance during the first three years of life, is associated with an increased risk of islet autoimmunity.
-
4.
Viral infection during pregnancy and first three years of life modifies the risk of islet autoimmunity and T1D.
Objectives
-
1.
To follow 1,400 children who have a first-degree relative (FDR) with T1D during pregnancy and early life to determine HLA genotype together with multiple susceptibility genes, changes in the microbiome, weight gain, metabolome-lipidome, insulin sensitivity, nutritional status, inflammatory markers, the timing and frequency of viral infections, and the relationships between genetic and environmental determinants.
-
2.
To determine the relationship between changes in the microbiome and prenatal and postnatal exposures, including weight gain, metabolome-lipidome, insulin sensitivity, nutritional status, and viral infection, and the development of persistent islet autoimmunity in children with a FDR with T1D.
The long-term objective is to follow T1D at-risk children into adolescence to determine the relationship between genotype, the microbiome and the environment, and the development of islet autoimmunity and T1D.
Methods/design
Summary of design
This is a prospective cohort study of the offspring of 1,400 mothers who have T1D, or a FDR with T1D, from pregnancy through childhood. Recruitment and consent will occur during pregnancy. The first investigation occurs as soon as feasible after the mother has given consent and investigation is 3-monthly throughout the pregnancy. At birth there is investigation of the mother and child and the child is then followed 3-monthly for two years, and 6-monthly thereafter. The primary outcome measure is persistent islet autoimmunity. The secondary outcome measure is the development of T1D. An overview of the study activities across time is shown in Table 1.
Estimation of target population
The population in the five states (New South Wales (NSW), Western Australia (WA), Queensland (Qld), Victoria (Vic) and South Australia (SA)) participating in ENDIA is 20.8 million, i.e. ~92% of Australia’s total population, and of these 3,500,000 men and 3,500,000 women aged 15–40 years [100]. Conservatively, there are 90,000 people with T1D in Australia, of whom ~15,000 are aged 0–14 years and ~45,000 aged 15–40 years, affecting men and women equally. Therefore, the prevalence of T1D among Australian women aged 15–40 years is 22,500/3,500,000 or 0.6%. Every year 250,000 women give birth in Australia, of whom 0.6% (n = 1,500) have T1D. There will also be 1,500 new fathers each year with T1D. Thus, approximately 3,000 births annually are to a parent with T1D. In contrast, the prevalence of siblings with T1D is relatively low. The incidence of T1D in children aged 0–14 is 22 per 100,000 person years [3]. Of 250,000 newborns each year in Australia, approximately half (125,000) will have a sibling. If the majority of those have a sibling aged 0–14 years, only ~28 will have T1D. Therefore, there are approximately 3,000 potential participants per annum of whom we aim to recruit 400–475 per year.
Determination of sample size
We estimate recruitment of 1,400 participants from the five states over three years, followed for a median of two years with a 9% rate of persistent islet autoimmunity. Maximum total dropout /lost to follow-up rate is estimated at 15% with the majority of dropouts occurring in the first six months of follow-up. These estimates are based on data from the Australian BabyDiab [12] and TRIGR [93] at-risk birth cohorts in which dropout rates were 15% and 7%, respectively.
Using nQuery Advisor, with 600 participants above the median and 600 below (1200 total after 15% drop out from 1,400), for a given exposure variable, power is 90% to detect a difference between survival (i.e. no islet autoimmunity) of 93% in one group and survival of 88% in the other – with 108 events (islet autoimmunity) or 9% of the total sample of 1,200 required [101]. For examining interactions between uncorrelated exposure variables with approximately 375 participants per combination of above/below the median, the power is 77% to detect a difference in survival (no islet autoimmunity) between 93% and 87% (70 events in 600 participants required).
Recruitment, retention and withdrawal of participants
Inclusion criteria
The inclusion criteria for ENDIA are an unborn child with an FDR with T1D, targeting the mother for recruitment in first, second or third trimester of pregnancy, or an infant less than six months of age with a FDR with T1D.
Exclusion criteria
The only exclusion criterion is the incapacity for the pregnant woman (or in the case of postnatal recruitment, the child’s primary caregiver) to understand the requirements of her and/or her child’s participation. This may be due to illiteracy, an intellectual disability or a mental illness.
Withdrawal criteria
Participants will be withdrawn from the study if they develop T1D in the case of the child, or request to discontinue participation in the case of the mother on behalf of herself and the child. The reason for withdrawal from the study will be captured on the case report form. Every effort will be made to continue to follow all participants enrolled in the study except for those who withdraw consent. Participants who are withdrawn from the study will be contacted by telephone annually to determine their T1D status.
Recruitment
Participants will be identified during pregnancy or within six months of birth in NSW, WA, Qld, Vic and SA. FDRs will be mothers, fathers or siblings with T1D. Since majority will be a mother or a father with T1D, pregnant women with T1D or whose partner has T1D are the main focus of recruitment. Eligible participants will be identified from the following sources: (1) prospective mothers attending public and private antenatal clinics prior to delivery, (2) prospective parents attending public and private diabetes clinics, (3) pregnant mothers of existing patients attending paediatric diabetes clinics, (4) diabetes organisations, and (5) local and national mainstream advertising.
Private endocrinologists and obstetricians known to manage T1D in young adults will be targeted with visits from research staff and personalised letters, brochures and posters. Paediatric T1D clinics will recruit families in which a sibling has T1D. Our research team includes endocrinologists and a perinatologist, all with substantial experience in recruitment of participants during pregnancy.
State and national diabetes networks and advocacy groups will be used to promote the study, as well as a dedicated website (http://www.endia.org.au) and social media such as Facebook (https://www.facebook.com/endiastudy) and Twitter. All advertising material, media releases and other articles intended for the general public will be approved by the relevant Human Research Ethics Committees in each state prior to distribution.
Participant retention
Our past experience indicates that families affected by T1D are highly motivated to participate in research. The previous Australian BabyDiab birth cohort in SA and Victoria recruited 548 children followed over a median of 5.7 years, while the participant retention rate in the NSW arm of the TRIGR study was 87% after five years (80% planned) [93]. This high rate is attributed to frequent family phone contacts, newsletters, distribution of calendars with reminders and incentives such as birthday cards, the set-up of parent’s groups for T1D families and active assistance with surveillance of the health of the participating child. Similar strategies will be implemented in ENDIA.
Participant engagement in the study will be central to high retention rates. More than half of people within the target population (men and women aged 18–34) have a Facebook page [102]. The site will be monitored daily and updated regularly to maintain relevancy. We will also use Facebook to link with existing T1D networks and advocacy groups, including JDRF International, JDRF Australia and Diabetes Australia, thereby generating further awareness of the study.
Study activities
Pregnancy visits 1 and 2: first and second trimesters
The following will be undertaken before 13 weeks gestation and again before 26 weeks gestation:
-
Documentation of maternal and paternal demographics including medical and obstetric history
-
Maternal anthropometry: height, weight, BMI
-
Collection of blood sample for investigations defined in Table 1
-
Collection of nasal, buccal, tongue and throat swabs, urine and stool specimens for microbiome studies
-
Administration of the Pregnancy Lifestyle Questionnaire and Pregnancy Physical Activity Questionnaire [96]
-
Documentation of proband history of T1D and other significant medical history
-
Blood and saliva will also be collected from the proband on one occasion for HLA genotyping and SNP analysis, respectively
Pregnancy visit 3: third trimester
Between 24 and 38 weeks gestation investigations will be as for first/second trimester PLUS
-
Documentation of results of the oral GTT, as routinely performed in pregnancy
-
Explanation by research nurse (trained by study dietician) as to how to complete the DQES version 2 (DQESv2) questionnaire for nutrition analysis [95]
-
Vaginal and skin swabs for analysis of the microbiome
-
Documentation of pregnancy complications: hypertension, preeclampsia, HELLP, hyperemesis gravidarum
Birth and week 1 of life
The following will be undertaken across two study visits within the first week of life. The first, termed the B1 visit, will be within the first two days post-partum; the second, termed the B2 visit, will be 3–5 days post-partum.
-
Collection of cord blood samples for investigations defined in Table 1
-
Completion of the documentation of the pregnancy complications, and /or of any congenital abnormalities in the child
-
Documentation of birth date, gestation, gender, birth weight, placental weight, birth length, APGAR score, method of delivery and any complications of the birth, perinatal and neonatal periods
-
Record of maternal weight gain during pregnancy
-
Viral studies, including serology and qPCR, will be performed from urine taken at B2 visit
-
Collection of colostrum samples at B1 and B2 visits for microbiome studies
-
Collection of skin swabs from infant and mother B1 and B2 visits for microbiome studies
-
Collection of meconium (first nappy) at B1 visit and stool samples, nasal, buccal, tongue and throat swabs from infant at B2 visit, all for microbiome studies
Follow-up visits at 3, 6, 9, 12, 15, 18, 21, 24, 30 and 36 months of age
The following will be performed once:
-
Saliva sampling from the infant for SNP genotype
-
Documentation of maternal nutrition during lactation using the DQESv2 questionnaire at three months
The following will be undertaken at each visit:
-
Collection of blood sample for investigations outlined in Table 1. If a child develops persistent islet autoimmunity, as defined above, HbA1c will be also measured 6-monthly
-
Anthropometrics: length (height at 30 and 36 months), weight, BMI, waist circumference (at umbilicus)
-
Collection of nasal, buccal, tongue, throat and skin swabs and stool samples for microbiome studies
-
Nutrition and Lifestyle assessments (Refer to Additional file 1)
Data collection, sampling procedures and laboratory investigations
A detailed description of the data collection, sampling procedures and laboratory investigations can be found in Additional file 1. Data collection tools developed specifically for the ENDIA Study evaluating infant feeing, maternal lifestyle in pregnancy, and maternal/infant lifestyle postpartum are provided in Additional files 2, 3 and 4, respectively.
Analysis of the primary outcome measure
The primary outcome measure is islet autoimmunity defined as elevation of ≥ one islet autoantibody on consecutive tests. Time-to-event analyses will explore the effect of variables on the risk of islet autoimmunity. Unadjusted and adjusted Hazard Ratios (HR) will be calculated using parametric and non-parametric (proportional hazard) survival models. The models will account for right censored data due to variable length of follow up and loss of follow up of participants. Variables measured over time (continuous and categorical) will be entered into survival models as time-dependent covariates as these measurements will be repeatedly obtained from individual participants. Adjusted HRs will be used to analyse the development of islet autoimmunity while controlling for associated variables as we have described [12]. Viral infections will also be modelled using survival analysis techniques. A fixed (time-independent) covariate will be used to test for associations with antenatal/postnatal viral infections/events, and time-dependent variables will be constructed to indicate exposure to viral infections throughout the follow-up period for each child.
Logistic regression analysis will be used to compare the relationship between presence or absence of islet autoimmunity and other variables. For analysis of nested case controls and longitudinal changes in the microbiome, islet autoimmune and non-islet autoimmune participants will be matched for age, gender, HLA type, ethnicity, proband relationship, parity, mode of delivery and gestation/birth weight.
A systems biology approach will be used to reveal inter-relationships between prenatal and postnatal environmental exposures that may condition the microbiome, metabolome and epigenome – these include maternal T1D, nutrition, weight gain, physical activity and viral infection during pregnancy; and mode of delivery, accelerated weight gain, nutrition, immune function, early fever, antibiotic use and viral infection during early life.
Safety parameters
Ethics committee review
In accordance with the Australian National Statement [103], in line with the ICH Guideline for Good Clinical Practice, the investigators have obtained written approval of the study protocol and Participant Information and Consent Form from the Human Research Ethics Committee (HREC) prior to commencement of the study. The study has been registered with the Australian New Zealand Clinical Trials Registry (ANZCTR) with registration number ACTRN12613000794707.
Subject information and consent
As this study involves mothers and children aged less than 12 years, the mother will be invited to provide written consent for herself and the child. The informed consent to participate in the study must be obtained by the investigator, or a person designated by the investigator, in accordance with the ICH Guidelines for Good Clinical Practice. Written informed consent will be obtained for the child using the HREC approved Participant Information and Consent Form. The parent/guardian will be advised in a timely manner of any new information that may be relevant to their willingness to participate.
Monitoring
The Investigator(s)/institution(s) will permit study-related monitoring, audits, HREC review, and direct access to case record forms, source documents and study files at all study sites for monitoring and audit purposes, at reasonable times, during the course of the study and after completion. The Project Manager will undertake an annual visit to at all five study sites to review case record forms, source documents, sample storage and study files. The investigators will participate in regular teleconferences and an annual face-to-face meeting.
Adverse event reporting
The investigators are responsible for conduct of the study in accordance with GCP regulations, which includes the recording and reporting of adverse events observed during and after the study. For all adverse events, the severity, onset, duration, determination of seriousness, action taken, any treatment given, outcome, and the investigator’s assessment of the relationship to study procedure will be recorded by the investigator. The most likely physical adverse event will be related to venipuncture. Reaction to venipuncture will be elicited by direct questioning of the mother or the primary caregiver. There is also potential for adverse psychological events associated with anxiety and grief the parents are likely to feel if their child develops islet autoimmunity. Each state has investigator(s) experienced in counselling families about the significance of islet autoimmunity and knowledgeable about the care of children with T1D. The investigators have combined experience in following over 1,000 children for the development of islet autoimmunity over the last 20 years.
Data handling and record keeping
Data protection and confidentiality
All provided biological samples will be labelled with a re-identifiable study number. The electronic study information will be stored in a dedicated, limited access clinical registry developed by the Melbourne eResearch Group of the University of Melbourne. The registry features electronic Case Report Forms and integrated electronic questionnaires for real-time data processing. Key capabilities support for fine grained access control systems with strong data encryption, secure data enclaves with advanced data management as well as secure back-up of the data entered. As such, the registry is compliant with international standards for electronic systems in clinical studies.
Discussion
The central aim of the ENDIA Study is to identify the gene-environment interactions occurring during prenatal and/or postnatal development that drive the development of islet autoimmunity and T1D in children genetically at-risk of T1D. Recruitment will begin prospectively during pregnancy with 3-monthly follow-up until two years of age then 6-monthly thereafter in an ongoing cohort study. The cohort will be well characterised in terms of demographics, maternal and proband medical history, obstetric history, parturition and anthropometry. An in-depth analysis of maternal and infant nutrition will inform a comprehensive sampling strategy involving the longitudinal collection of blood and DNA for immune and multi-omics analyses, and stool, urine, breast milk, oral cavity, nares and skin samples/swabs for microbiome analysis. The rapid change of the microbiome after birth in relation to prenatal and postnatal exposures, geographical variations and the onset of islet autoimmunity before two years of age underscores the importance of studying the cohort from pregnancy into early childhood with frequent sampling during the first two years of life, and analysis in relation to the environmental, cultural and genetic influences that are likely to be relevant to an Australian population.
Defining modifiable host and environment factors that initiate and/or promote destruction of insulin-producing cells in early life will have enormous implications for individuals with and at risk of T1D and their families, and for the health care system given the annual cost of approximately $5,000 per person with T1D in Australia [104]. In the short term, outcome data generated from this study aims to provide education programs for T1D risk reduction during pre-conception, pregnancy and early infancy in families with children at risk. In the medium term, the translation of data for gene-environment interactions may allow testing of families for T1D susceptibility genes and education about their relative risk of environmental factors according to genotype. In the longer term, outcomes of the ENDIA Study may lead to a means of primary prevention of T1D before the autoimmune process begins.
Authors’ information
ENDIA Study Group:
Peter A Baghurst1, Simon C Barry2, Fergus J Cameron3, Jodie M Dodd4, Chris Duran5, Josephine M Forbes6, Maria Makrides2,7, Grant Morahan8, Karen E Nelson9, Alison J Nankervis10, Richard O Sinnott5, John M Wentworth11,12
1School of Population Health, University of Adelaide, Adelaide, SA 5005
2Discipline of Paediatrics, School of Paediatrics and Reproductive Health, University of Adelaide, Adelaide, SA 5005
3Department of Endocrinology and Diabetes and Centre for Hormone Research, Royal Children’s Hospital and Murdoch Children’s Research Institute, Parkville, Victoria 3052
4School of Obstetrics and Gynaecology, University of Adelaide, Adelaide, SA 5005
5Department of Computing and Information Systems, University of Melbourne, Parkville, Victoria 3010
6Mater Medical Research Institute, South Brisbane, Queensland 4101
7Women’s and Children’s Health Research Institute, King William Road, North Adelaide, SA, 5006
8Centre for Diabetes Research, Western Australian Medical Research Institute, Perth, WA 6000
9J. Craig Venter Institute, Rockville, MD 20850 USA
10Royal Women’s Hospital, Parkville, Victoria 3052
11Royal Melbourne Hospital, Parkville, Victoria 3050
12Walter and Eliza Hall Institute, Parkville, Victoria 3050
Abbreviations
- AGEs:
-
Advanced glycation end products
- BMI:
-
Body mass index
- DHA:
-
Docosahexaenoic acid
- ENDIA:
-
Environmental determinants of islet autoimmunity
- EPA:
-
Eicosapentaenoic acid
- EV:
-
Enterovirus
- FDR:
-
First-degree relative
- GADAb:
-
Glutamic acid decarboxylase 65 autoantibodies
- HLA:
-
Human leukocyte antigen
- IA2Ab:
-
Tyrosine phosphatase-like insulinoma antigen autoantibodies
- IAA:
-
Insulin autoantibodies
- NOD:
-
non-obese diabetic
- PGE2:
-
Prostaglandin E2
- RV:
-
Rotavirus
- T1D:
-
Type 1 diabetes
- Treg:
-
Regulatory T cell
- tTGAb:
-
Tissue transglutaminase autoantibodies
- ZnT8Ab:
-
Beta cell-specific zinc transporter 8 autoantibodies.
References
Diamond Project Group: Incidence and trends of childhood type 1 diabetes worldwide 1990–1999. Diabet Med. 2006, 23 (8): 857-866.
Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G: Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet. 2009, 373 (9680): 2027-2033. 10.1016/S0140-6736(09)60568-7.
Catanzariti L, Faulks K, Moon L, Waters AM, Flack J, Craig ME: Australia’s national trends in the incidence of type 1 diabetes in 0–14-year-olds, 2000–2006. Diabet Med. 2009, 26 (6): 596-601. 10.1111/j.1464-5491.2009.02737.x.
Haynes A, Bower C, Bulsara MK, Jones TW, Davis EA: Continued increase in the incidence of childhood type 1 diabetes in a population-based Australian sample (1985–2002). Diabetologia. 2004, 47 (5): 866-870. 10.1007/s00125-004-1385-8.
Taplin CE, Craig ME, Lloyd M, Talyor C, Crock P, Silink M, Howard NJ: The rising incidence of childhood type 1 diabetes in New South Wales, 1990–2002. Med J Aust. 2005, 183 (5): 243-246.
Knip M: Pathogenesis of type 1 diabetes: implications for incidence trends. Horm Res Paediatr. 2011, 76 (Suppl 1): 57-64.
Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC, Colman PG, Harrison LC: The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care. 2008, 31 (8): 1546-1549. 10.2337/dc08-0239.
Morahan G: Insights into type 1 diabetes provided by genetic analyses. Curr Opin Endocrinol Diabetes Obes. 2012, 19 (4): 263-270.
Bonifacio E, Warncke K, Winkler C, Wallner M, Ziegler AG: Cesarean section and interferon-induced helicase gene polymorphisms combine to increase childhood type 1 diabetes risk. Diabetes. 2011, 60 (12): 3300-3306. 10.2337/db11-0729.
Lempainen J, Vaarala O, Makela M, Veijola R, Simell O, Knip M, Hermann R, Ilonen J: Interplay between PTPN22 C1858T polymorphism and cow’s milk formula exposure in type 1 diabetes. J Autoimmun. 2009, 33 (2): 155-164. 10.1016/j.jaut.2009.04.003.
Purohit S, She JX: Biomarkers for type 1 diabetes. Int J Clin Exp Med. 2008, 1 (2): 98-116.
Couper JJ, Beresford S, Hirte C, Baghurst PA, Pollard A, Tait BD, Harrison LC, Colman PG: Weight gain in early life predicts risk of islet autoimmunity in children with a first-degree relative with type 1 diabetes. Diabetes Care. 2009, 32 (1): 94-99. 10.2337/dc08-0821.
Colman PG, McNair PD, Gellert S, Kewming K, Schmidli RS, Steele CE, Harrison LC: Development of autoantibodies to islet antigens during childhood: implications for preclinical type 1 diabetes screening. Pediatr Diabetes. 2002, 3 (3): 144-148. 10.1034/j.1399-5448.2002.30304.x.
Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, et al: Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA. 2013, 309 (23): 2473-2479. 10.1001/jama.2013.6285.
Littman DR, Pamer EG: Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011, 10 (4): 311-323. 10.1016/j.chom.2011.10.004.
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R: Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010, 107 (26): 11971-11975. 10.1073/pnas.1002601107.
Isolauri E: Development of healthy gut microbiota early in life. J Paediatr Child Health. 2012, 48 (Suppl 3): 1-6.
Johnson CL, Versalovic J: The human microbiome and its potential importance to pediatrics. Pediatrics. 2012, 129 (5): 950-960. 10.1542/peds.2011-2736.
Rigon G, Vallone C, Lucantoni V, Signore F: Maternal factors pre- and during delivery contribute to gut microbiota shaping in newborns. Front Cell Infect Microbiol. 2012, 2: 93-
Australian Institute for Health and Welfare 2011: Australia's welfare 2011. Australia’s welfare series. Volume 10, Cat. no. AUS 142. 2011, Canberra: Australian Institute for Health and Welfare
Kannieappan LM, Deussen AR, Grivell RM, Yelland L, Dodd JM: Developing a tool for obtaining maternal skinfold thickness measurements and assessing inter-observer variability among pregnant women who are overweight and obese. BMC Pregnancy Childbirth. 2013, 13 (1): 42-10.1186/1471-2393-13-42.
Australian Government Department of Health and Aging: 2007 Australian National Children’s Nutrition and Physical Activity Survey- Main Findings. http://www.commcarelink.health.gov.au/internet/main/publishing.nsf/Content/66596E8FC68FD1A3CA2574D50027DB86/$File/childrens-nut-phys-survey.pdf
Macpherson AJ, Harris NL: Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol. 2004, 4 (6): 478-485. 10.1038/nri1373.
Locke NR, Stankovic S, Funda DP, Harrison LC: TCR gamma delta intraepithelial lymphocytes are required for self-tolerance. J Immunol. 2006, 176 (11): 6553-6559.
Pozzilli P, Signore A, Williams AJ, Beales PE: NOD mouse colonies around the world–recent facts and figures. Immunol Today. 1993, 14 (5): 193-196. 10.1016/0167-5699(93)90160-M.
Suzuki T: Diabetogenic effects of lymphocyte transfusion on the NOD or NOD nude mouse. Immune deficient animals in biomedical research. Edited by: Rygaard J, Brunner N, Graem N, Spang-Thomson M. 1987, Basel: Karger, 112-116.
Funda DP, Fundova P, Harrison LC: Environmental-mucosal interactions in the natural history of type 1 diabetes: the germ-free (GF) NOD mouse model. Proceedings of the 7th Immunology of Diabetes Society Meeting. vol. 41. Edited by: Sanjeevi CB, Gale EAM. 2005, New York: Ann N Y Acad Sci
Kawaguchi-Miyashita M, Shimizu K, Nanno M, Shimada S, Watanabe T, Koga Y, Matsuoka Y, Ishikawa H, Hashimoto K, Ohwaki M: Development and cytolytic function of intestinal intraepithelial T lymphocytes in antigen-minimized mice. Immunology. 1996, 89 (2): 268-273. 10.1046/j.1365-2567.1996.d01-740.x.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, et al: Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011, 6: e25792-10.1371/journal.pone.0025792.
Shehadeh N, Gelertner L, Blazer S, Perlman R, Etzioni A: The importance of insulin content in infant diet: suggestion for a new infant formula period. Acta Pediatr. 2001, 90: 93-95. 10.1111/j.1651-2227.2001.tb00262.x.
Narendran P, Mannering SI, Harrison LC: Proinsulin-a pathogenic autoantigen in type 1 diabetes. Autoimmun Rev. 2003, 2 (4): 204-210. 10.1016/S1568-9972(03)00009-0.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al: Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009, 139 (3): 485-498. 10.1016/j.cell.2009.09.033.
Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, Drew JC, Ilonen J, Knip M, Hyoty H, et al: Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5 (1): 82-91. 10.1038/ismej.2010.92.
de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, Orivuori L, Hakala S, Welling GW, Harmsen HJ, Vaarala O: Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes. 2013, 62: 1238-1244. 10.2337/db12-0526.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, et al: Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011, 6 (10): e25792-10.1371/journal.pone.0025792.
Kondrashova A, Reunanen A, Romanov A, Karvonen A, Viskari H, Vesikari T, Ilonen J, Knip M, Hyoty H: A six-fold gradient in the incidence of type 1 diabetes at the eastern border of Finland. Ann Med. 2005, 37 (1): 67-72. 10.1080/07853890410018952.
Seiskari T, Kondrashova A, Viskari H, Kaila M, Haapala AM, Aittoniemi J, Virta M, Hurme M, Uibo R, Knip M, et al: Allergic sensitization and microbial load–a comparison between Finland and Russian Karelia. Clin Exp Immunol. 2007, 148 (1): 47-52. 10.1111/j.1365-2249.2007.03333.x.
Calcinaro F, Dionisi S, Marinaro M, Candeloro P, Bonato V, Marzotti S, Corneli RB, Ferretti E, Gulino A, Grasso F, et al: Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia. 2005, 48 (8): 1565-1575. 10.1007/s00125-005-1831-2.
Rasmussen T, Stene LC, Samuelsen SO, Cinek O, Wetlesen T, Torjesen PA, Ronningen KS: Maternal BMI before pregnancy, maternal weight gain during pregnancy, and risk of persistent positivity for multiple diabetes-associated autoantibodies in children with the high-risk HLA genotype: the MIDIA study. Diabetes Care. 2009, 32 (10): 1904-1906. 10.2337/dc09-0663.
Bonifacio E, Pfluger M, Marienfeld S, Winkler C, Hummel M, Ziegler AG: Maternal type 1 diabetes reduces the risk of islet autoantibodies: relationships with birthweight and maternal HbA(1c). Diabetologia. 2008, 51 (7): 1245-1252. 10.1007/s00125-008-1022-z.
Koczwara K, Bonifacio E, Ziegler AG: Transmission of maternal islet antibodies and risk of autoimmune diabetes in offspring of mothers with type 1 diabetes. Diabetes. 2004, 53 (1): 1-4.
Elfving M, Lindberg B, Lynch K, Mansson M, Sundkvist G, Lernmark A, Ivarsson SA: Number of islet autoantibodies present in newly diagnosed type 1 diabetes children born to non-diabetic mothers is affected by islet autoantibodies present at birth. Pediatr Diabetes. 2008, 9 (2): 127-134. 10.1111/j.1399-5448.2007.00349.x.
Lamb MM, Yin X, Zerbe GO, Klingensmith GJ, Dabelea D, Fingerlin TE, Rewers M, Norris JM: Height growth velocity, islet autoimmunity and type 1 diabetes development: the diabetes autoimmunity study in the young. Diabetologia. 2009, 52 (10): 2064-2071. 10.1007/s00125-009-1428-2.
Winkler C, Marienfeld S, Zwilling M, Bonifacio E, Ziegler AG: Is islet autoimmunity related to insulin sensitivity or body weight in children of parents with type 1 diabetes?. Diabetologia. 2009, 52 (10): 2072-2078. 10.1007/s00125-009-1461-1.
Knerr I, Wolf J, Reinehr T, Stachow R, Grabert M, Schober E, Rascher W, Holl RW: The ‘accelerator hypothesis’: relationship between weight, height, body mass index and age at diagnosis in a large cohort of 9,248 German and Austrian children with type 1 diabetes mellitus. Diabetologia. 2005, 48 (12): 2501-2504. 10.1007/s00125-005-0033-2.
Clarke SL, Craig ME, Garnett SP, Chan AK, Cowell CT, Cusumano JM, Kordonouri O, Sambasivan A, Donaghue KC: Increased adiposity at diagnosis in younger children with type 1 diabetes does not persist. Diabetes Care. 2006, 29 (7): 1651-1653. 10.2337/dc06-0277.
Hypponen E, Virtanen SM, Kenward MG, Knip M, Akerblom HK: Obesity, increased linear growth, and risk of type 1 diabetes in children. Diabetes Care. 2000, 23 (12): 1755-1760. 10.2337/diacare.23.12.1755.
Colman PG, Steele C, Couper JJ, Beresford SJ, Powell T, Kewming K, Pollard A, Gellert S, Tait B, Honeyman M, et al: Islet autoimmunity in infants with a type I diabetic relative is common but is frequently restricted to one autoantibody. Diabetologia. 2000, 43 (2): 203-209. 10.1007/s001250050030.
Couper JJ, Steele C, Beresford S, Powell T, McCaul K, Pollard A, Gellert S, Tait B, Harrison LC, Colman PG: Lack of association between duration of breast-feeding or introduction of cow’s milk and development of islet autoimmunity. Diabetes. 1999, 48 (11): 2145-2149. 10.2337/diabetes.48.11.2145.
Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E: Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA. 2003, 290 (13): 1721-1728. 10.1001/jama.290.13.1721.
Virtanen SM, Knip M: Nutritional risk predictors of beta cell autoimmunity and type 1 diabetes at a young age. Am J Clin Nutr. 2003, 78 (6): 1053-1067.
Norris JM, Barriga K, Klingensmith G, Hoffman M, Eisenbarth GS, Erlich HA, Rewers M: Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA. 2003, 290 (13): 1713-1720. 10.1001/jama.290.13.1713.
Stene LC, Witso E, Torjesen PA, Rasmussen T, Magnus P, Cinek O, Wetlesen T, Ronningen KS: Islet autoantibody development during follow-up of high-risk children from the general Norwegian population from three months of age: design and early results from the MIDIA study. J Autoimmun. 2007, 29 (1): 44-51. 10.1016/j.jaut.2007.04.003.
Virtanen SM, Kenward MG, Erkkola M, Kautiainen S, Kronberg-Kippila C, Hakulinen T, Ahonen S, Uusitalo L, Niinisto S, Veijola R, et al: Age at introduction of new foods and advanced beta cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes. Diabetologia. 2006, 49 (7): 1512-1521. 10.1007/s00125-006-0236-1.
Knip M, Virtanen SM, Seppa K, Ilonen J, Savilahti E, Vaarala O, Reunanen A, Teramo K, Hamalainen AM, Paronen J, et al: Dietary intervention in infancy and later signs of beta-cell autoimmunity. New England Journal of Medicine. 2010, 363 (20): 1900-1908. 10.1056/NEJMoa1004809.
Frederiksen B, Kroehl M, Lamb MM, Seifert J, Barriga K, Eisenbarth GS, Rewers M, Norris JM: Infant exposures and development of type 1 diabetes mellitus: the diabetes autoimmunity study in the young (DAISY). JAMA Pediatr. 2013, doi:10.1001/jamapediatrics.2013.317.
Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE, Harrison LC: Pro-inflammatory CD11c + CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010, 59 (7): 1648-1656. 10.2337/db09-0287.
Hofman PL, Regan F, Jackson WE, Jefferies C, Knight DB, Robinson EM, Cutfield WS: Premature birth and later insulin resistance. New England Journal of Medicine. 2004, 351 (21): 2179-2186. 10.1056/NEJMoa042275.
Petrovsky N, Harrison LC: Diurnal rhythmicity of human cytokine production: a dynamic disequilibrium in T helper cell type 1/T helper cell type 2 balance?. J Immunol. 1997, 158 (11): 5163-5168.
Petrovsky N, Kyvik KO, Bonnevie-Nielsen V, Beck-Nielsen H, Green A, Harrison LC: Evidence from twins for acquired cellular immune hyperactivity in type 1 diabetes. Immunology. 2002, 106 (4): 584-589. 10.1046/j.1365-2567.2002.01449.x.
Antonelli A, Fallahi P, Ferrari SM, Pupilli C, d’Annunzio G, Lorini R, Vanelli M, Ferrannini E: Serum Th1 (CXCL10) and Th2 (CCL2) chemokine levels in children with newly diagnosed Type 1 diabetes: a longitudinal study. Diabet Med. 2008, 25 (11): 1349-1353.
Yeung WC, Al-Shabeeb A, Pang CN, Wilkins MR, Catteau J, Howard NJ, Rawlinson WD, Craig ME: Children with islet autoimmunity and enterovirus infection demonstrate a distinct cytokine profile. Diabetes. 2012, 61 (6): 1500-1508. 10.2337/db11-0264.
Chase HP, Cooper S, Osberg I, Stene LC, Barriga K, Norris J, Eisenbarth GS, Rewers M: Elevated C-reactive protein levels in the development of type 1 diabetes. Diabetes. 2004, 53 (10): 2569-2573. 10.2337/diabetes.53.10.2569.
Mericq V, Piccardo C, Cai W, Chen X, Zhu L, Striker GE, Vlassara H, Uribarri J: Maternally transmitted and food-derived glycotoxins: a factor preconditioning the young to diabetes?. Diabetes Care. 2010, 33 (10): 2232-2237. 10.2337/dc10-1058.
Stene LC, Joner G: Use of cod liver oil during the first year of life is associated with lower risk of childhood-onset type 1 diabetes: a large, population-based, case–control study. Am J Clin Nutr. 2003, 78 (6): 1128-1134.
Norris JM, Yin X, Lamb MM, Barriga K, Seifert J, Hoffman M, Orton HD, Baron AE, Clare-Salzler M, Chase HP, et al: Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. JAMA. 2007, 298 (12): 1420-1428. 10.1001/jama.298.12.1420.
Bailey R, Cooper JD, Zeitels L, Smyth DJ, Yang JH, Walker NM, Hypponen E, Dunger DB, Ramos-Lopez E, Badenhoop K, et al: Association of the vitamin D metabolism gene CYP27B1 with type 1 diabetes. Diabetes. 2007, 56 (10): 2616-2621. 10.2337/db07-0652.
Ramos-Lopez E, Bruck P, Jansen T, Herwig J, Badenhoop K: CYP2R1 (vitamin D 25-hydroxylase) gene is associated with susceptibility to type 1 diabetes and vitamin D levels in Germans. Diabetes/Metabolism Research Reviews. 2007, 23 (8): 631-636. 10.1002/dmrr.719.
Zhang J, Li W, Liu J, Wu W, Ouyang H, Zhang Q, Wang Y, Liu L, Yang R, Liu X, et al: Polymorphisms in the vitamin D receptor gene and type 1 diabetes mellitus risk: an update by meta-analysis. Mol Cell Endocrinol. 2012, 355 (1): 135-142. 10.1016/j.mce.2012.02.003.
Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM: Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001, 358 (9292): 1500-1503. 10.1016/S0140-6736(01)06580-1.
Simpson M, Brady H, Yin X, Seifert J, Barriga K, Hoffman M, Bugawan T, Baron AE, Sokol RJ, Eisenbarth G, et al: No association of vitamin D intake or 25-hydroxyvitamin D levels in childhood with risk of islet autoimmunity and type 1 diabetes: the diabetes autoimmunity study in the young (DAISY). Diabetologia. 2011, 54 (11): 2779-2788. 10.1007/s00125-011-2278-2.
Bowyer L, Catling-Paull C, Diamond T, Homer C, Davis G, Craig ME: Vitamin D, PTH and calcium levels in pregnant women and their neonates. Clin Endocrinol (Oxf). 2009, 70 (3): 372-377. 10.1111/j.1365-2265.2008.03316.x.
Zhang Y, Bandala-Sanchez E, Harrison LC: Revisiting regulatory T cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2012, 19 (4): 271-278.
Bluestone JA, Herold K, Eisenbarth G: Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010, 464 (7293): 1293-1300. 10.1038/nature08933.
Sadlon TJ, Wilkinson BG, Pederson S, Brown CY, Bresatz S, Gargett T, Melville EL, Peng K, D’Andrea RJ, Glonek GG, et al: Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol. 2010, 185 (2): 1071-1081. 10.4049/jimmunol.1000082.
Bandala-Sanchez E, Zhang Y, Reinwald S, Dromey JA, Lee BH, Qian J, Bohmer RM, Harrison LC: T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat Immunol. 2013, 14: 741-748. 10.1038/ni.2610.
Petrovsky N, Harrison LC: Cytokine-based human whole blood assay for the detection of antigen-reactive T cells. J Immunol Methods. 1995, 186 (1): 37-46. 10.1016/0022-1759(95)00127-V.
Yeung WC, Rawlinson WD, Craig ME: Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. Br Med J. 2011, 342: d35-10.1136/bmj.d35.
Craig ME, Howard NJ, Silink M, Rawlinson WD: Reduced frequency of HLA DRB1*03-DQB1*02 in children with type 1 diabetes associated with enterovirus RNA. J Inf Dis. 2003, 187 (10): 1562-1570. 10.1086/374742.
Oikarinen S, Martiskainen M, Tauriainen S, Huhtala H, Ilonen J, Veijola R, Simell O, Knip M, Hyoty H: Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes. 2011, 60 (1): 276-279. 10.2337/db10-0186.
Tapia G, Cinek O, Rasmussen T, Witso E, Grinde B, Stene LC, Ronningen KS: Human enterovirus RNA in monthly fecal samples and islet autoimmunity in Norwegian children with high genetic risk for type 1 diabetes: the MIDIA study. Diabetes Care. 2011, 34 (1): 151-155. 10.2337/dc10-1413.
Stene LC, Oikarinen S, Hyoty H, Barriga KJ, Norris JM, Klingensmith G, Hutton JC, Erlich HA, Eisenbarth GS, Rewers M: Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the diabetes and autoimmunity study in the young (DAISY). Diabetes. 2010, 59 (12): 3174-3180. 10.2337/db10-0866.
Lempainen J, Tauriainen S, Vaarala O, Makela M, Honkanen H, Marttila J, Veijola R, Simell O, Hyoty H, Knip M, et al: Interaction of enterovirus infection and cow’s milk-based formula nutrition in type 1 diabetes-associated autoimmunity. Diabetes Metab Res Rev. 2012, 28 (2): 177-185. 10.1002/dmrr.1294.
Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, Couper JJ, Tait BD, Colman PG, Harrison LC: Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes. 2000, 49 (8): 1319-1324. 10.2337/diabetes.49.8.1319.
Honeyman MC, Stone NL, Falk BA, Nepom G, Harrison LC: Evidence for molecular mimicry between human T cell epitopes in rotavirus and pancreatic islet autoantigens. J Immunol. 2010, 184 (4): 2204-2210. 10.4049/jimmunol.0900709.
Hull B, Dey A, Mahajan D, Menzies R, McIntyre PB: Immunisation coverage annual report, 2009. Commun Dis Intell. 2011, 35 (2): 132-148.
Tate JE, Curns AT, Cortese MM, Weintraub ES, Hambidge S, Zangwill KM, Patel MM, Baggs JM, Parashar UD: Burden of acute gastroenteritis hospitalizations and emergency department visits in US children that is potentially preventable by rotavirus vaccination: a probe study using the now-withdrawn rotashield vaccine. Pediatrics. 2009, 123 (3): 744-749. 10.1542/peds.2008-1200.
Simell O, Niinikoski H, Ronnemaa T, Raitakari OT, Lagstrom H, Laurinen M, Aromaa M, Hakala P, Jula A, Jokinen E, et al: Cohort profile: the STRIP study (Special Turku Coronary Risk Factor Intervention Project), an infancy-onset dietary and life-style intervention trial. Int J Epidemiol. 2009, 38 (3): 650-655. 10.1093/ije/dyn072.
Oresic M, Gopalacharyulu P, Mykkanen J, Lietzen N, Makinen M, Nygren H, Simell S, Simell V, Hyoty H, Veijola R, et al: Cord serum lipidome in prediction of islet autoimmunity and type 1 diabetes. Diabetes. 2013, -doi:10.2337/db13-0159
Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, Daunay A, Busato F, Mein CA, Manfras B, et al: Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7 (9): e1002300-10.1371/journal.pgen.1002300.
Zhang Y, Maksimovic J, Naselli G, Qian J, Blewitt ME, Oshlack A, Harrison LC: Genome-wide DNA methylation analysis identifies hypomethylated genes with the forkhead-binding motif in human regulatory T cells. Blood. in press
Teddy Study Group: The environmental determinants of diabetes in the young (TEDDY) study: study design. Pediatr Diabetes. 2007, 8 (5): 286-298.
Akerblom HK: The trial to reduce IDDM in the genetically at risk (TRIGR) study: recruitment, intervention and follow-up. Diabetologia. 2011, 54 (3): 627-633. 10.1007/s00125-010-1964-9.
Roll U, Christie MR, Fuchtenbusch M, Payton MA, Hawkes CJ, Ziegler AG: Perinatal autoimmunity in offspring of diabetic parents. The German Multicenter BABY-DIAB study: detection of humoral immune responses to islet antigens in early childhood. Diabetes. 1996, 45 (7): 967-973. 10.2337/diabetes.45.7.967.
Hodge A, Patterson AJ, Brown WJ, Ireland P, Giles G: The anti cancer council of Victoria FFQ: relative validity of nutrient intakes compared with weighed food records in young to middle-aged women in a study of iron supplementation. Aust N Z J Public Health. 2000, 24 (6): 576-583. 10.1111/j.1467-842X.2000.tb00520.x.
Chasan-Taber L, Schmidt MD, Roberts DE, Hosmer D, Markenson G, Freedson PS: Development and validation of a pregnancy physical activity questionnaire. Med Sci Sports Exerc. 2004, 36 (10): 1750-1760. 10.1249/01.MSS.0000142303.49306.0D.
Johnson RK, Driscoll P, Goran MI: Comparison of multiple-pass 24-hour recall estimates of energy intake with total energy expenditure determined by the doubly labeled water method in young children. J Am Diet Assoc. 1996, 96 (11): 1140-1144. 10.1016/S0002-8223(96)00293-3.
Golley RK, Hendrie GA: The impact of replacing regular- with reduced-fat dairy foods on children’s wider food intake: secondary analysis of a cluster RCT. Eur J Clin Nutr. 2012, 66 (10): 1130-1134. 10.1038/ejcn.2012.113.
Duncanson K, Burrows T, Collins C: Study protocol of a parent-focused child feeding and dietary intake intervention: the feeding healthy food to kids randomised controlled trial. BMC Publ Health. 2012, 12: 564-10.1186/1471-2458-12-564.
Australian Bureau of Statistics: Australian Demographic Statistics Tables. Australian Demographics Statistics, Dec 2012. Volume 31010DO002_201212. Edited by: Australian Bureau of Statistics. 2013, Canberra
Collett D: Modelling Survival Data in Medical Research. 2003, Florida: Chapman & Hall/CRC Press LLC, 2
SocialBaker: Australia Facebook Statistics. [http://www.socialbakers.com/facebook-statistics/australia]
National Health and Medical Research Council 2007: National Statement on Ethical Conduct in Human Research 2007 - Updated 2009. NHMRC Publications. vol. E72. Edited by: Australian Government. 2007, Canberra
Colagiuri SBA, Gomez M, Fitzgerald B, Buckley A, Colagiuri R: DiabCo$t Australia Type 1: Assessing the burden of Type 1 Diabetes in Australia. 2009, Canberra: Diabetes Australia
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2431/13/124/prepub
Acknowledgements
This study is funded by the Australian National Health and Medical Research Council (NHMRC Project Grant 1025083). The authors wish to acknowledge the contributions of A/Prof Karen Walker (Monash University), Ms Bronwen D’Arcy (Women’s and Children’s Hospital), Dr Gilly Hendrie (CSIRO) and Prof Clare Collins (University of Newcastle) in the development of tools for evaluating diet and nutrition. We also acknowledge the participation of Ms Felicity Healy (Melbourne Health), Mr Wayne Soon (Princess Margaret Hospital), Ms Sherrell Cardinal (Mater Health Service), and Ms Jacki Catteau (Children’s Hospital at Westmead) for the preparation of documents for ethics/research governance submissions and the local study coordinators.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The ENDIA Study writing group is comprised of MP, JC, MC, PC, WR, AC, TJ and LH. All assume responsibility for the production of this manuscript. The study was conceived by JC and LH. The study was designed by JC, LH, MC, PC and WR. The study Project Manager is MP. State Principal Investigators are JC (SA), MC (NSW), TJ (WA), PC (Vic) and AC (Qld). PB is an additional Principal Investigator. The Associated Investigators are FC, JD, MM, GM, AN and JW. The Significant Collaborators are CD, JF, KN and RS. All authors have read and approved the final manuscript.
Electronic supplementary material
12887_2013_858_MOESM1_ESM.pdf
Additional file 1: A detailed description of the data collection, sampling procedures and laboratory investigations employed in the ENDIA study.(PDF 300 KB)
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Penno, M.A., Couper, J.J., Craig, M.E. et al. Environmental determinants of islet autoimmunity (ENDIA): a pregnancy to early life cohort study in children at-risk of type 1 diabetes. BMC Pediatr 13, 124 (2013). https://doi.org/10.1186/1471-2431-13-124
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2431-13-124