
Abstract
Background
Large clonal populations of cells bearing PIG-A mutations are the sine qua non of PNH, but the PIG-A mutation itself is insufficient for clonal expansion. The association between PNH and aplastic anemia supports the immune escape model, but not all PNH patients demonstrate a history of aplasia; therefore, second genetic hits driving clonal expansion have been postulated. Based on the previous identification of JAK2 mutations in patients with a myeloproliferative/PNH overlap syndrome, we considered TET2 as a candidate gene in which mutations might be contributing to clonal expansion.
Methods
Here we sequenced the TET2 and JAK2 genes in 19 patients with large PNH clones.
Results
We found one patient with a novel somatic nonsense mutation in TET2 in multiple hematopoietic lineages, which was detectable upon repeat testing. This patient has had severe thromboses and has relatively higher peripheral blood counts compared with the other patients—but does not have other features of a myeloproliferative neoplasm.
Conclusions
We conclude that mutations in TET2 may contribute to clonal expansion in exceptional cases of PNH.
Similar content being viewed by others


Background
Paroxysmal nocturnal hemoglobinuria (PNH) is characterized by complement mediated hemolysis, immune mediated marrow failure, and an expansion in the marrow of a stem cell with an acquired somatic mutation in PIG-A [1]. This gene is essential for the biosynthesis of glycosylphosphatidylinositol (GPI), and circulating cells derived from the PNH clone are missing all GPI-linked proteins, including the complement inhibitors CD55 and CD59 [2]. The lack of these proteins sensitizes the red cell to complement mediated lysis. Platelets derived from the mutant stem cell clone have the same surface defect as red cells, but here the effect of uninhibited complement may primarily lead to a state of activation, explaining the marked hypercoagulable state seen in this disorder [3,4,5,6]. Other contributing factors may involve a decrease in fibrinolysis due to abnormal post-translational modification of the GPI-linked uPAR receptor [7], increased thrombin generation on platelet-derived microparticles [8,9,10], and defects related to the GPI-linked tissue factor pathway inhibitor [11].
Normal individuals harbor occult circulating blood cells with PIG-A mutations [12], and in mice, Pig-A disruption is not sufficient to drive clonal expansion [13], suggesting that clonal expansion depends on additional mechanisms. The immune escape model [14] posits that the PIG-A gene mutation represents the “first hit”, and aplastic anemia (AA)—which selects for GPI (−) stem cells—represents the necessary “second hit”. In support of this model, the GPI-anchor can fit into the groove of the HLA-like molecule CD1d [15], there is recent evidence that GPI itself may be the auto-antigen [16], and lymphocyte cultures can be raised to selectively kill GPI (+) cells [17]. Furthermore, the immune escape model is supported by the demonstration of oligoclonal T cell expansions [18] and an HLA DR-15/16 [19] association. Although not all patients with PNH develop AA, many have a subclinical form of stem cell loss [20].
There are, however, features of PNH that cannot be so readily explained by the immune escape model, such as the observation that rare patients will develop acute leukemia [21], advanced myelodysplasia [22], or features of a myeloproliferative neoplasm (MPN) [23]. Therefore, there is long-standing interest in identifying second genetic hits. In 24% of patients with PNH, we have found an abnormal karyotype [22], but this frequently regresses, despite persistence of the PNH clone. Two exceptional patients have been reported with an abnormality involving chromosome 12 [24], leading to over-expression of HMGA2 (on chromosome 12q14.3), which results in a myeloproliferative syndrome when overexpressed in mice [25]. We have reported that activating JAK2V617F mutations represent a second genetic hit seen occasionally in PNH [26], and this phenomenon probably accounts for the case reports from the 1970’s of patients with a positive HAM test and an MPN [23].
We have now hypothesized that, as for JAK2, mutations in TET2 could also represent a second genetic hit. TET2 encodes an enzyme of 2002 amino acids that is involved in the conversion of methylcytosine to hydroxymethylcytosine, using α-ketoglutarate as a co-factor [27,28,29]. This is likely to induce changes in gene expression patterns as a consequence of altered cytosine methylation, leading to a proliferation of myeloid cells: indeed, homozygous or heterozygous Tet2 inactivation in mice results in an advantage for stem cells in competitive reconstitution experiments [30, 31]. Mono-allelic mutations in TET2 have been found in about 12% of MPNs, about 20% of cases of MDS, as well as in CMMoL, AML, and mastocytosis [32]. Because TET2 functions as a haploinsufficient tumor suppressor gene, it has an unusual combination of features: a broad spectrum of somatic inactivating mutations are pathogenic, and only one allele needs to be mutated. Indeed, PIG-A, because it is X-linked, represents the other most prominent example of a gene with both of these features. We therefore investigated whether TET2 mutations would be found as a second genetic hit in patients with PNH.
Methods
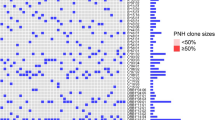
Patients diagnosed with PNH were recruited to an IRB-approved protocol after providing written informed consent. Diagnostic flow cytometry for PNH was performed by staining whole blood with FITC conjugated anti-CD59 for red cells, PE-conjugated anti-CD24 and Alexa-488 conjugated to FLAER for granulocytes and identification by FSC/SSC (on a log–log scale for red cells). For DNA extraction, granulocytes were isolated from whole blood by sedimentation in 6% hetastarch, centrifugation over ficoll, followed by osmotic lysis of red cells. We have included in this analysis only patients with more than 75% PNH granulocytes.
Extracted DNA from granulocytes was subjected to whole genome amplification, followed by bi-directional sequencing using a dye terminator approach using previously published primers [33]. To isolate separate GPI (+) and GPI (−) lymphocyte populations as well as monocyte populations, buffy coat cells were incubated with FLAER-Alexa 488 (which directly binds to GPI) and anti-CD33-PE, followed by sorting on a DakoCyomation MoFlo instrument. To isolate nucleated red cells, cells from the buffy coat layer were sorted based on their expression of glycophorin A and based on their FSC/SSC properties using the red cell settings.
Results
Upon sequencing of the TET2 gene in our cohort of patients, we identified the presence of several previously reported SNPs. In 11 out of 19 patients, we identified the c.5284A>G; p.I1762V variant, with an allele frequency of 34% compared with 22% in the NCBI dbSNP database. In 6 of the patients, we identified the c.5162T>G; p.L1721W variant, with an allele frequency of 15.8%, compared with 9.2% in the database. In 3 patients, we identified the c.1088C>T; p.P363L variant, with an allele frequency of 7.9%, compared with 3% in the database. None of these differences were statistically significant. One remarkable patient (patient 14), however, was heterozygous for all three of these SNPs– and was also heterozygous for a nonsense mutation, c.2697T>A; pY899X, which has not been previously reported (Table 1).
A set of repeat sequencing reactions confirmed the presence of the 2697T>A mutation in granulocyte samples taken 8 months and then again 16 months after the initial sample (Fig. 1). The mutation was not found in either the sorted GPI (+) or GPI (−) lymphocytes, essentially ruling out a germline mutation. The TET2 mutation was found in nearly a 1:1 ratio amongst the sorted monocytes, and was found in a lower ratio amongst sorted nucleated red cells and granulocytes.

Electropherograms demonstrate the presence of the 2697T>A (Y899X) mutation in blood cells from patient 14. a Sequences amplified from granulocytes from the initial blood sample. b–f Sequences amplified from cells from the third sample obtained 16 months after the initial blood sample: b sorted glycophorin A-positive nucleated red cells; c sorted CD33-positive Monocytes; d Granulocytes; e sorted FLAER-positive lymphocytes; f sorted FLAER-negative lymphocytes. The arrow indicates the peak at the 2697 position. The highest proportion of mutant sequences was seen amongst the monocytes
In this patient, almost 100% of the granulocytes and red cells display the PNH phenotype. Using a method that we have recently developed (manuscript in preparation)—we have determined that nearly 100% of her platelets are GPI-negative, as well as approximately 30% of the lymphocytes. There was a history of severe thrombotic complications (involving the middle hepatic vein, portal vein, extremity DVT and PE, and a spontaneous hemorrhagic CVA that may have been secondary to a thrombotic event). This patient has a persistently high reticulocyte count and is unusual in that there were elevated blood counts compared with the other patients in the cohort (WBC of 7.7 vs median of 3.7, platelet count of 321 vs median of 131). Of the WBC’s, 4% were nucleated red cells. Interestingly, this patient’s absolute monocyte count ranged from 2 to 4 times above the upper limit of normal. PNH was first confirmed by flow cytometry in 1999, in the setting of hemolysis and a history of having had thrombosis. This patient had been transfusion dependent with an LDH 9 times the upper limit of normal prior to starting eculizumab in 2008. This patient has not had new thromboses in at least the past 18 years while on warfarin. Based on the timeline of symptoms, it is likely that this patient has had PNH since the 1980’s, without having ever had aplastic anemia. While there are no other features of a myeloproliferative neoplasm, the natural history of this patient’s presentation may have been altered due to a splenectomy for the presumptive diagnosis of ITP prior to the diagnosis of PNH.
Discussion
In a series of 19 patients with “classic” PNH, we have found a TET2 mutation only in patient 14, and in contrast to a separate series of patients [26], activating JAK2 mutations were not found in any patient here. This confirms that the PNH/MPN overlap syndrome is a rare phenomenon, and we conclude that TET2 mutations are not a common feature of PNH. Patient 14 had an unusual occurrence of 4 different base pair substitutions; while 3 of these are known polymorphisms, the Y899X substitution is very likely to be affecting the enzymatic activity of the TET2 protein, given that it is a truncating mutation, occurring at the 3′ end of exon 3, in a region where chain terminating mutations have been previously reported in patients with myeloid disorders [34].
The fact that the same mutation was seen in granulocytes, monocytes, and nucleated red cells, and was still present 16 months later suggests that the mutation resides in a long-lived stem cell clone with a potential for multilineage hematopoiesis. Although the patient does not have a clinically evident MPN, it is interesting that the WBC is higher than all of the other patients in the cohort, and this patient’s platelet count was the second highest of the 19 patients. Interestingly, this patient’s absolute monocyte count ranged from 2 to 4 times above the upper limit of normal, which is not a typical finding in PNH. The relative elevation in the blood counts and a particular increase in monocytes, however, is very much consistent with findings from mice that are genetically haploinsufficient for Tet2 [31].
In mice, haploinsufficiency for Tet2 also produces an advantage for stem cells based on in vivo competitive repopulation studies [31]. Therefore, we posit that the TET2 mutation identified here conferred a survival advantage, perhaps in the context of an injured marrow. Conversely, given that 5.6% of healthy women with clonal hematopoiesis over the age of 65 have monoallelic inactivating TET2 mutations [35], one might question whether this patient’s TET2 mutation could be simply a part of that process. However, among 96 healthy women under the age of 60 who were known to have clonal hematopoiesis, TET2 mutations were not found [35], and of note, patient 14 was 52 years old at the time of the study. Interestingly, in 2 subsequent studies, T → A transversions, such as seen in patient 14, accounted for only 3 out of 103 of all age-related TET2 mutations [36, 37], and so it seems that this patient’s mutation is distinct from the phenomenon of age-related clonal hematopoiesis. It seems more likely that that this patient’s TET2 mutation provides an advantage for the stem cell clone in a manner that does not completely compete against the normal stem cell pool and which is dependent upon an abnormal stem cell environment—as is thought to be the case for the PNH clone [14, 38].
It is notable that practically 100% of the red cells and granulocytes in patient 14 are GPI-negative, whereas the TET2 mutant allele burden is < 50% in granulocytes and nucleated red cells, approximately 50% in monocytes, and undetectable amongst GPI (+) and GPI (−) lymphocytes. These findings suggest that the TET2-mutated population belongs to a subclone. Notably, the TET2 mutant clone and the PNH population are both large and stable in relative size in this patient, in contrast to TET2 mutations reported previously in patients with bone marrow failure [39,40,41]. The simplest explanation for the findings is that first, a PNH stem cell population expanded, followed by expansion of a TET2 mutant clone, which arose out of a subset of the PNH stem cells. It is possible that the TET2 mutation is not evident among lymphocytes because it is instead driving the stem cell towards monocytic differentiation. The fact that the TET2 mutation was not seen in either the GPI (+) or GPI (−) lymphocyte population strongly suggests that the mutation is not in the germline.
An alternative model, proposing that hypermutability could contribute to the generation of somatic mutations in PIG-A in PNH [42, 43] could explain why some patients have several distinct PNH clones [44,45,46], and in some cases, mutations in genes other than PIG-A. Indeed, in PNH and aplastic anemia, in addition to JAK2, TET2 and HMGA2 (mentioned above), mutations in ASXL1, DNMT3a, BCOR, BCORL1, SUZ12, and U2AF1 have been reported [39,40,41]. However, considering that TET2 can be inactivated by a single mutation, and given that haploinsufficiency for TET2 can drive clonal expansion [31], and given that TET2 and PIG-A have a comparable number of codons, we believe that if hypermutability were fundamental to PNH, then TET2 would have been commonly mutated in our cohort of patients as well. Our data, then, is more consistent with experimental models suggesting that the mutation rate is normal in PNH [47,48,49], and we believe that the association with aplastic anemia and the immune escape model explains clonal expansion in most patients. However, recent reports of PNH arising in the setting of a CALR-mutated MPN [50],CML [51] and a relapse of AML associated with TET2 and JAK2 mutations [52] raise the question of whether autoimmunity is necessary in all cases. The elucidation of the autoantigen in aplastic anemia and the identification of the responsible T cell clones may shed light on this question in exceptional cases such as these and the one described here [53].
Conclusions
Based on a large cohort of patients with highly expanded PNH clones, we conclude that mutations in TET2 can contribute to clonal expansion in exceptional cases. Our findings argue against the hypermutability model in most patients with PNH.
Availability of data and materials
The datasets used and/or analused during the current study are available from the corresponding author on reasonable request
Abbreviations
- PNH:
-
paroxysmal nocturnal hemoglobinuria
- GPI:
-
glycosylphosphatidylinositol
- MPN:
-
myeloproliferative neoplasm
- AA:
-
aplastic anemia
References
Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259:1318–20.
Nicholson-Weller A, Spicer D, Austen K. Deficiency of the complement regulatory protein, “Decay-accelerating factor” on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1985;312:1091–7.
Devine DV, Siegel RS, Rosse WF. Interactions of the platelets in paroxysmal nocturnal hemoglobinuria with complement. Relationship to defects in the regulation of complement and to platelet survival in vivo. J Clin Invest. 1987;79(1):131–7.
Dixon RH, Rosse WF. Mechanism of complement-mediated activation of human blood platelets in vitro: comparison of normal and paroxysmal nocturnal hemoglobinuria platelets. J Clin Invest. 1977;59(2):360–8.
Polley M, Nachman R. Human complement in thrombin-mediated platelet function. J Exp Med. 1979;150:633–45.
Zimmerman TS, Kolb WP. Human platelet-initiated formation and uptake of the C5-9 complex of human complement. J Clin Invest. 1976;57(1):203–11.
Ronne E, Pappot H, Grondahl-Hansen J, Hoyer-Hansen G, Plesner T, Hansen NE, et al. The receptor for urokinase plasminogen activator is present in plasma from healthy donors and elevated in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1995;89(3):576–81.
Hugel B, Socie G, Vu T, Toti F, Gluckman E, Freyssinet JM, et al. Elevated levels of circulating procoagulant microparticles in patients with paroxysmal nocturnal hemoglobinuria and aplastic anemia. Blood. 1999;93(10):3451–6.
Sims PJ, Faioni EM, Wiedmer T, Shattil SJ. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem. 1988;263(34):18205–12.
Wiedmer T, Hall SE, Ortel TL, Kane WH, Rosse WF, Sims PJ. Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood. 1993;82(4):1192–6.
Ott I, Miyagi Y, Miyazaki K, Heeb MJ, Mueller BM, Rao LV, et al. Reversible regulation of tissue factor-induced coagulation by glycosyl phosphatidylinositol-anchored tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2000;20(3):874–82.
Araten D, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci USA. 1999;96:5209–14.
Keller P, Payne JL, Tremml G, Greer PA, Gaboli M, Pandolfi PP, et al. FES-Cre targets phosphatidylinositol glycan class A (PIGA) inactivation to hematopoietic stem cells in the bone marrow. J Exp Med. 2001;194:581–90.
Rotoli B, Luzzatto L. Paroxysmal nocturnal hemoglobinuria. Baillieres Clin Haematol. 1989;2(1):113–38.
Joyce S, Woods A, Yewdell J, Bennink J, Silva AD, Boesteanu A, et al. Natural ligand of mouse CD1d1: cellular glycosylphosphatidylinositol. Science. 1998;279(5356):1541–4.
Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, Richichi B, et al. Glycosylphosphatidylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(14):2753–61.
vanBijnen S, Withaar M, Preijers F, van der Meer A, de Witte T, Muus P, et al. T cells expressing the activating NK-cell receptors KIR2DS4, NKG2C and NKG2D are elevated in paroxysmal nocturnal hemoglobinuria and cytotoxic toward hematopoietic progenitor cell lines. Exp Hematol. 2011;39:751–62.
Karadimitris A, Manavalan JS, Thaler HT, Notaro R, Araten DJ, Nafa K, et al. Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood. 2000;96(7):2613–20.
Maciejewski J, Follmann D, Nakamura R, Saunthararajah Y, Rivera C, Simonis T, et al. Increased frequency of HLA-DR2 in patients with paroxysmal nocturnal hemoglobinuria and the PNH/aplastic anemia syndrome. Blood. 2001;98:3513–9.
Elebute M, Rizzo S, Tooze J, Marsh J, Gordon-Smith E, Gibson F. Evaluation of the haemopoietic reservoir in de novo haemolytic paroxysmal nocturnal hemoglobinuria. Br J Haematol. 2003;123:552–60.
Ware R, Hall S, Rosse W. Paroxysmal nocturnal hemoglobinuria with onset in childhood and adolescence. N Engl J Med. 1991;325:991–6.
Araten D, Swirsky D, Karadimitris A, Notaro R, Nafa K, Bessler M, et al. Cytogenetic and morphologic abnormalities in paroxysmal nocturnal hemoglobinuria. Br J Haematol. 2001;115:360–8.
Luzzatto L, Familusi J, Williams C, Junaid T, Rotoli B, Altinito F. The PNH abnormality in myeloproliferative disorders: association of PNH and acute myelosis in two children. Haematologica. 1979;64:13–30.
Inoue N, Izu-Sarumaru T, Murakami Y, Endo Y, Nishimura J, Kurokawa K, et al. Molecular basis of clonal expansion in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2006;108:4232–6.
Ikeda K, Mason P, Bessler M. 3′ UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferrring a clonal growth advantage at the level of HSC in mice. Blood. 2011;117:5860–9.
Sugimori C, Padron E, Caceres G, Shain K, Sokol L, Zhang L, et al. Paroxysmal nocturnal hemoglobinuria and concurrent JAK2V617F mutation. Blood Cancer J. 2012;2:e63.
Ko M, Huang Y, Jankowska A, Pape U, Tahiliani M, Bandukwala H, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43.
Tahiliani M, Koh K, Shen Y, Pastor W, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5.
He Y, Li B, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7.
Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(2):25–38.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Mullighan C. TET2 mutations in myelodysplasia and myeloid malignancies. Nat Genet. 2009;41(7):766–7.
Patel J, Gönen M, Figueroa M, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89.
Abdel-Wahan O, Mullally A, Hedvat C, Carcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alternations in myeloid malignancies. Blood. 2009;114(1):144–7.
Busque L, Patel J, Figueroa M, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–81.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman P, Mar B, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Genovese G, Kahler A, Handsaker R, Lindberg J, Rose S, Bakhoum S, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Araten DJ, Bessler M, McKenzie S, Castro-Malaspina H, Childs BH, Boulad F, et al. Dynamics of hematopoiesis in paroxysmal nocturnal hemoglobinuria (PNH): no evidence for intrinsic growth advantage of PNH clones. Leukemia. 2002;16(11):2243–8.
Kulasekararaj A, Jiang J, Smith A, Mohamedali A, Mian S, Gandhi S, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplasia syndrome. Blood. 2014;124:2698–704.
Shen W, Clemente M, Hosono N, Yoshida K, Przychodzen B, Yoshizato T, et al. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Investig. 2014;124(10):4529–38.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–74.
Horikawa K, Kawaguchi T, Ishihara S, Nagakura S, Hidaka M, Kagimoto T, et al. Frequent detection of T cells with mutations of the hypoxanthine-guanine phosphoribosyl transferase gene in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2002;99(1):24–9.
Purow DB, Howard TA, Marcus SJ, Rosse WF, Ware RE. Genetic instability and the etiology of somatic PIG-A mutations in paroxysmal nocturnal hemoglobinuria. Blood Cells Mol Dis. 1999;25(2):81–91.
Endo M, Ware RE, Vreeke TM, Singh SP, Howard TA, Tomita A, et al. Molecular basis of the heterogeneity of expression of glycosyl phosphatidylinositol anchored proteins in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87(6):2546–57.
Nafa K, Bessler M, Deeg HJ, Luzzatto L. New somatic mutation in the PIG-A gene emerges at relapse of paroxysmal nocturnal hemoglobinuria. Blood. 1998;92(9):3422–7.
Nishimura J, Inoue N, Wada H, Ueda E, Pramoonjago P, Hirota T, et al. A patient with paroxysmal nocturnal hemoglobinuria bearing four independent PIG-A mutant clones. Blood. 1997;89(9):3470–6.
Araten D, Zamechek L, Halverson G. No evidence of hypermutability in red cells from patients with paroxysmal nocturnal hemoglobinuria using the XK gene. Haematologica. 2014;99:e142–4.
Araten DJ, Luzzatto L. The mutation rate in PIG-A is normal in patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2006;108(2):734–6.
Chen G, Zeng W, Green S, Young NS. Frequent HPRT mutations in paroxysmal nocturnal haemoglobinuria reflect T cell clonal expansion, not genomic instability. Br J Haematol. 2004;125(3):383–91.
Fraiman YS, Cuka N, Batista D, Vuica-Ross M, Moliterno AR. Development of paroxysmal nocturnal hemoglobinuria in CALR-positive myeloproliferative neoplasm. J Blood Med. 2016;7:107–10.
Tominaga R, Katagiri T, Kataoka K, Kataoka K, Wee RK, Maeda A, et al. Paroxysmal nocturnal hemoglobinuria induced by the occurrence of BCR-ABL in a PIGA mutant hematopoietic progenitor cell. Leukemia. 2016;30(5):1208–10.
Santagostino A, Lombardi L, Dine G, Hirsch P, Misra SC. Paroxysmal nocturnal hemoglobinuria with a distinct molecular signature diagnosed ten years after allogenic bone marrow transplantation for acute myeloid leukemia. Case Rep Hematol. 2019;2019:3.
Gargiulo L, Zaimoku Y, Scappini B, Maruyama H, Ohumi R, Luzzatto L, et al. Auto-reactive glycosyl-phosphatidyl-inositol (GPI) specific T cells are present in most patients with idiopathic aplastic anemia (IAA). Blood. 2015;126:2409.
Acknowledgements
We are indebted to Peter Lopez and the staff of the NYU School of Medicine Flow Cytometry Core Facility.
Funding
This work was supported by The PNH Research and Support Foundation Award from the Aplastic Anemia & MDS International Foundation.
Author information
Authors and Affiliations
Contributions
CL, AB, and LBZ performed experiments, DJA wrote the manuscript, and SI, CL, IA, and DJA conceived of the experiments and analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Patients provided written informed consent using protocols that were approved by the IRBs at the New York University School of Medicine and the New York VA Medical Center.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lobry, C., Bains, A., Zamechek, L.B. et al. Analysis of TET2 mutations in paroxysmal nocturnal hemoglobinuria (PNH). Exp Hematol Oncol 8, 17 (2019). https://doi.org/10.1186/s40164-019-0142-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-019-0142-0