
Abstract
An increasing number of studies have investigated the association between SLCO1B1 −521T>C and −388A>G polymorphisms and the risk of statin-induced adverse drug reactions (ADRs), but the results have been inconsistent. This meta-analysis was performed to gain more insight into the relationship. PubMed, Embase, Cochrane Library and Web of Science were searched for relevant articles published before March 5th, 2015. The quality of included studies was evaluated by the Newcastle-Ottawa Quality scale. Pooled effect estimates (odds ratios [ORs] or hazard ratios [HRs) and corresponding 95 % confidence intervals (CIs) were calculated to assess the association in overall and subgroup analyses for various genetic models. Begg’s rank correlation test and Egger’s linear regression test were used to examine the publication bias. A total of nine cohort and four case–control studies involving 11, 246 statin users, of whom 2, 355 developing ADRs were included in the analysis. Combined analysis revealed a significant association between the SLCO1B1−521T>C polymorphism and increased risk for ADRs caused by various statins, but the synthesis heterogeneity was generally large (dominant model: pooled effect estimate = 1.85, 95 % CI 1.20–2.85, P = 0.005; I 2 = 80.70 %, Pheterogeneity < 0.001). Subgroup analysis by statin type showed that the ADRs risk was significantly elevated among simvastatin users (dominant model: pooled effect estimate = 3.43, 95 % CI 1.80–6.52, P = 0.001; I 2 = 59.60 %, Pheterogeneity = 0.060), but not among atorvastatin users. No significant relationship was found between the −388A>G polymorphism and ADRs caused by various statins (dominant model: pooled effect estimate = 0.94, 95 % CI 0.79–1.13, P = 0.526; I 2 = 40.10 %, Pheterogeneity = 0.196). The meta-analysis suggests that SLCO1B1 −521T>C polymorphism may be a risk factor for statin-induced ADRs, especially in simvastatin therapy. Conversely, there may be no significant association for −388A>G polymorphism.
Similar content being viewed by others


Background
Statins or the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors have become the most important pharmaceutical intervention for the primary and secondary prevention of cardiovascular diseases by lowering blood concentrations of low density lipoprotein cholesterol (LDL-C) (Baigent et al. 2005; Stewart 2013). In general, statins are safe and well tolerated, but there are still 25–50 % of patients with coronary artery disease noncompliant after 1 year’s medication mainly because of adverse drug reactions (ADRs) (Ho et al. 2008). The musculoskeletal problem is the most common intolerance, symptoms of which range from mild clinical myalgias, “incipient” myopathy, “definite” myopathy to fatal rhabdomyolysis (Feng et al. 2012; Ghatak et al. 2010; Link et al. 2008; Thompson et al. 2006). Though the rate of statin-induced myopathy is low, the absolute number of patients with statin-related muscle symptoms may be substantial due to the high prevalence of cardiovascular diseases and the wide use of statin drugs (Wilke et al. 2005a, b). In addition, many patients discontinue statin therapy for some mild myopathic symptoms, which increases their potential risk of cardiovascular events to some extent (Wilke et al. 2012). Therefore, understanding the mechanism of statin-induced ADRs has great significance to reduce statin toxicity and optimize patients’ adherence.
Previous studies showed that a series of clinical factors may contribute to the risk of developing muscle toxicity during statin therapy, including older age (though existing controversy) (Schech et al. 2007; Graham et al. 2004), low body mass index, female gender (de Keyser et al. 2014), higher dose (de Lemos et al. 2004; McClure et al. 2007), metabolic comorbidities (i.e., hypothyroidism), intense physical exercise (Meador and Huey 2010), interactions with other drugs such as fibrates, ciclosporin, protease inhibitors, macrolide antibiotics, and amio darone (Wilke et al. 2012; Graham et al. 2004; Niemi et al. 2011). In addition, genetic factors also play a vital role. It was reported that variability in statin metabolizing enzyme genes (CYP3A4/5, CYP2C8/9, CYP2D6, UGT1), membrane transporter genes (SLCO1B1, ABCB1, ABCG2) and pharmacodynamic related genes (CPT2, COQ2, RYR2) could influence the development and severity of statin-associated muscle toxicity (Feng et al. 2012; Ishikawa et al. 2004; Wilke et al. 2005a, b; Frudakis et al. 2007; Fiegenbaum et al. 2005; Vladutiu et al. 2006; Vladutiu 2008; Oh et al. 2007; Marciante et al. 2011). Among them, the SLCO1B1 gene is widely investigated. The SLCO1B1 gene locates on the chromosome 12 (Chr 12p12.2) occupying 109 kb, which encodes the organic anion transporting polypeptide 1B1 (OATP1B1). This is an influx transporter expressed on the sinusoidal membrane of human hepatocytes and facilitates the liver uptake of most statins (atorvastatin, rosuvastatin, pravastatin, simvastatin, and lovastatin) (Pasanen et al. 2007; König et al. 2006; Pasanen et al. 2006; Chen et al. 2008). Only a few are known to have functional effects in spite of many single-nucleotide polymorphisms (SNPs) have been identified in SLCO1B1. Previous studies mostly focused on the variants −521T>C (rs4149056) and −388A>G (rs2306283). It is reported that patients with the −521C minor allele had reduced hepatic uptake and increased blood concentration of various statins, which might increase the ADRs risk (Pasanen et al. 2006, 2007; Kameyama et al. 2005; Niemi et al. 2004). The role of −388A>G in the transporting activity of OATP1B1 varies in different studies (Niemi et al. 2011). In recent years, an increasing number of studies began to explore the role of SLCO1B1 polymorphisms in statin-induced ADRs. However, the results remain inconsistent and with limited power (Link et al. 2008; de Keyser et al. 2014; Marciante et al. 2011; Voora et al. 2009; Danik et al. 2013; Donnelly et al. 2011; Carr et al. 2013; Santos et al. 2012; Brunham et al. 2012).
A meta-analysis of seven studies investigating association of statin myopathy with the SLCO1B1−521T>C was published in 2013 (Carr et al. 2013). However, there were three otherwise eligible articles (de Keyser et al. 2014; Danik et al. 2013; Santos et al. 2012) not included in the analysis, which would influence the validity of the pooled estimates. Also, the publication did not present the exact literature selection criteria or the extracted data of its included original studies, thus it was difficult to check some pivotal information. We performed this meta-analysis to provide a more comprehensive estimation of the association between the two SNPs, SLCO1B1 −521T>C and −388A>G polymorphisms and statin-induced various ADRs not only myopathy, under various genetic models. The finding of a significant correlation may become useful in prestatin treatment screening in order to predict the chance of the development of adverse effects.
Methods
Search strategy
Four databases were electronically searched to retrieve studies on association between statin-induced ADRs and polymorphisms of the SLCO1B1 gene until 5 March 2015, including PubMed, ISI web of knowledge, Embase, and the Cochrane Library. Searching terms were: “HMG-CoA reductase inhibitor” or “statin” or “simvastatin” or “lovastatin” or “fluvastatin” or “atorvastatin” or “pravastatin” or “rosuvastatin” or “cerivastatin” or “mevastatin”, combined with “SLCO1B1”. In addition, references of the retrieved publications were checked for relevant studies.
Inclusion and exclusion criteria
Titles and abstracts of all retrieved publications were screened. Then the full-text screening was conducted by two researchers (Jiang and Tang) independently. Any discordance was subsequently resolved through discussion or a third party (Zhang). Studies were considered eligible if they met all of the following criteria: (1) It investigated the association between −521T>C or −388A>G polymorphisms in the SLCO1B1 gene and the risk of statin-induced ADRs; (2) It provided effect estimates (OR, RR, or HR) and their corresponding 95 % confidence intervals (95 % CIs) or allele or genotype frequencies for calculating the effect estimates; (3) The publication language was English. Studies consistent with any of the following conditions were excluded: (1) Reviews, case reports, comments, letters, news, editorials; (2) In vitro or animal trials; (3) Studies lacking information necessary or usable data for the analysis; (4) For overlapping and republished studies, only the most recent or the largest population was included.
Data extraction
Data were extracted independently by two reviewers (Jiang and Tang). Disagreements were solved by discussion, and a third party (Zhang) was involved when necessary. The collected information included: first author, year of publication, country where study was conducted, ethnicity, study design, definition of ADRs, characteristics of participants, statin type, dose and regimen, sample size, polymorphism region, allele or genotype frequencies in patients with or without ADRs during the treatment of statins, crude and adjusted effect estimates (ORs, RRs, HRs) and their corresponding 95 % CIs as reported, any multivariate analyses adjustment factors, genotyping method, and information about Hardy–Weinberg Equilibrium (HWE).
Quality assessment of included studies
Newcastle-Ottawa Quality scale (NOS) (Crowther et al. 2010; Cota et al. 2013) was developed for quality assessment. A “star system” (range 0–9 stars) was used for the present analysis to evaluate each included study on the following: selection of the study groups, between-group comparability, and the ascertainment of either the exposure for case–control studies or the outcome for cohort studies. A study can be awarded a maximum of one star for each numbered item within the Selection and Exposure or Outcome categories. A maximum of two stars can be assigned for Comparability. With respect to the follow-up period sufficient for outcomes to occur, the minimum follow-up of the exposed group was set at 1 year, given that the risk of an adverse drug reaction is greater during the first year of therapy, and decreases afterwards (de Keyser et al. 2014).
Statistical analysis
The effect estimates that were extracted, if available, or de novo calculated from available data, were crude and adjusted ORs and HRs. We pooled OR estimates and HR estimates for each study together. If both the crude and the adjusted effect estimates were available for the same outcome, we incorporated the adjusted one into the analysis. Pooled effect estimates were calculated for all of the following genetic models: the allele contrast, the homozygote comparison, the heterozygote comparison, the dominant model, the recessive model, and the additive model respectively. Heterogeneity among included studies was assessed by Chi square-based Q test and I 2 test (Higgins and Thompson 2002). If the data showed no heterogeneity (P > 0.10, I 2 < 50 %), the Mantel–Haenszel fixed effect model was used (Mantel and Haenszel 1959), otherwise the DerSimonian-Laird random effect model was used (DerSimonian and Laird 1986). In addition, stratification analysis was conducted by the type of statin. If the original research did not provide information about HWE, we calculated it using an online HWE calculation tool (Rodriguez et al. 2009). Sensitivity analysis was carried out to investigate the influence of a single study on the overall risk by omitting one study each time and was conducted based on leave-one-out sensitivity procedure. Publication bias was assessed by the Begg’s rank correlation test and Egger’s linear regression test if the number of included studies in the meta-analysis was more than two (Begg and Mazumdar 1994; Egger et al. 1997). Data were analyzed using the STATA 12.0 (Stata Statistical Software, College Station, TX, USA, www.stata.com) software. A P value of 0.05 for any test or model was considered to be statistically significant.
Results
Literature search
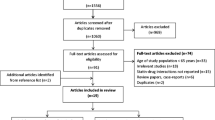
A total of 676 records were yielded by the search strategy from four electronic databases. Among these, 35 publications met the inclusion criteria after title and abstract screening. Based on full-text inspection, we excluded another 25 publications because 11 of them lacked information necessary or usable data for the analysis (Dlouha et al. 2013; Dmitry et al. 2013; Mafalda et al. 2013; Pranculis and Kucinskas 2013; Hopewell et al. 2012; Khan et al. 2011; Johansen et al. 2010; Tamraz et al. 2013; Toms et al. 2010; Kamatani and Mushiroda 2011; Kroemer 2010); seven articles were reviews (three articles) (Patel et al. 2014; de Keyser et al. 2012a, b; Dendramis 2011), comments (two articles) (Wright et al. 2011; Nakamura 2008), news (one article) (Dolgin 2013) or letters (one article) (Puccetti et al. 2010); four articles reported overlapping data for the same study (de Keyser et al. 2012a, b; Toms et al. 2009; Danik et al. 2012; Voora et al. 2008); two studies were published in Russian (Sychev et al. 2013; Petrov et al. 2013) and one study was an in vitro study (Furihata et al. 2009). Among the 10 publications included, one (de Keyser et al. 2014) described four independent studies. Of these, two investigated simvastatin-related ADRs and the remaining two investigated atorvastatin-related ADRs. Thus, 13 studies from 10 publications (Link et al. 2008; de Keyser et al. 2014; Marciante et al. 2011; Voora et al. 2009; Danik et al. 2013; Donnelly et al. 2011; Carr et al. 2013; Santos et al. 2012; Brunham et al. 2012; Linde et al. 2010) were included in the meta-analysis. Details of the study selection process are presented in Fig. 1.

Flow chart of study selection. n Number of studies
Study characteristics
Characteristics of the eligible studies are summarized in Table 1. All studies were published between 2008 and 2014, with a total number of 11, 246 subjects, of whom 2, 355 developing ADRs. Five studies were conducted in the Netherlands, three in the UK, two in the US, one in Brazil, one in the US and Canada and one in 26 countries. There were 11 studies entirely or predominantly involving Caucasian participants and two not reporting the ethnicity. Nine were cohort studies and four were case–control studies. Definitions of statin-induced ADRs, doses and treatment periods were heterogeneous among these studies. There were four hospital-based and nine population-based studies. Among the included 13 studies, three focused on simvastatin, three on atorvastatin, one on rosuvastatin, one on cerivastatin, two on mixed statins and three on both mixed statins and specific statin. Seven studies provided genotype frequencies which helped us to calculate the crude ORs under various genetic models. Nine studies reported adjusted effect estimates in some genetic models.
Quality of included studies
Rating of the quality of the included studies according to the NOS is presented in Table 2. The quality scores ranged from 5 to 9. The case definition was adequate in all of the included case–control studies, but the non-response rate between the case and control groups was equal in only one study (Brunham et al. 2012). For all cohort studies, 4 stars were assigned to Selection category. The follow-up time for exposed participants was not reported in the study by Linde et al. (2010) and not long enough (<1 year) for the outcome to occur in the study by Voora et al. (2008). Nearly all included cohort studies did not mention the loss to follow-up.
Meta-analysis results
All of the 13 included studies explored the association between −521T>C polymorphism of the SLCO1B1 gene and statin-induced ADRs. The results of meta-analysis are presented in Table 3. A significant increased risk of ADRs induced by different types of statins was found for the C vs. T: pooled effect estimate = 1.99, 95 % CI 1.20–3.29, P = 0.007); the homozygote comparison (CC vs. TT: pooled effect estimate = 2.21, 95 % CI: 1.41–3.47, P = 0.001); the dominant model (TC/CC vs. TT: pooled effect estimate = 1.85, 95 % CI 1.20–2.85, P = 0.005); the recessive model (CC vs. TT/TC: pooled effect estimate = 2.76, 95 % CI 1.73–4.39, P < 0.001) and the additive model (per C allele: pooled effect estimate = 1.76, 95 % CI 1.25–2.50, P = 0.001), but not for the heterozygote comparison (TC vs. TT: pooled effect estimate = 1.26, 95 % CI 0.96–1.65, P = 0.091). Figure 2 shows the pooled effect estimate and 95 % CI for any statin induced ADRs in the dominant model.

Association between SLCO1B1 −521T>C polymorphism and risk of adverse drug reactions caused by any statin. Dominant genetic model (TC/CC vs. TT). The ES is odds ratio (OR) or hazard ratio (HR); the size of the square is proportional to the weight of each study; horizontal lines represent the 95 % CI
In the stratified analysis by statin type (Table 3), the synthesis of studies investigating simvastatin pointed to a statistically significant increased risk for the allele contrast model (C vs. T: pooled effect estimate = 3.00, 95 % CI 1.39–6.48, P = 0.005), the homozygote comparison (CC vs. TT: pooled effect estimate = 3.62, 95 % CI 1.33–9.83, P = 0.012), the dominant model (TC/CC vs. TT: pooled effect estimate = 3.43, 95 % CI 1.80–6.52, P < 0.001) (Fig. 3), the recessive model (CC vs. TT/TC: pooled effect estimate = 5.98, 95 % CI 2.53–14.13, P < 0.001) and the additive model (per C allele: pooled effect estimate = 2.87, 95 % CI 1.67–4.94, P < 0.001). Whereas in contrast, the combined effect estimates for atorvastatin-induced ADRs were far from being statistically significant in any of the six genetic models. The association between the −521T>C polymorphism and atorvastatin-induced ADRs in the dominant model is shown in Fig. 4.

Association between SLCO1B1 −521T>C polymorphism and risk of adverse drug reactions caused by simvastatin. Dominant genetic model (TC/CC vs. TT). The ES is odds ratio (OR); the size of the square is proportional to the weight of each study; horizontal lines represent the 95 % CI

Association between SLCO1B1 −521T>C polymorphism and risk of adverse drug reactions caused by atorvastatin. Dominant genetic model (TC/CC vs. TT). The ES is odds ratio (OR) or hazard ratio (HR); the size of the square is proportional to the weight of each study; horizontal lines represent the 95 % CI
Three of the included studies explored the relationship between the −388A>G polymorphism of the SLCO1B1 gene and statin-induced ADRs. Due to the limited available data from the original studies, we only performed meta-analysis under the dominant model and the additive model based on two studies respectively. Both of the results yielded a decreased risk for statin-related ADRs among −388G allele carriers, although the pooled effect estimates did not reach statistical significance (AG/GG vs. AA: pooled effect estimate = 0.94, 95 % CI 0.79–1.13, P = 0.526; the additive model: pooled effect estimate = 0.91, 95 % CI 0.81–1.02, P = 0.114) (Table 3).
Sensitivity analysis
For −521T>C polymorphism and risk of ADRs induced by any statin, the conclusion of insignificant association for the heterozygote comparison was materially altered by removing the study by de Keyser et al. (2014) with the overall effect estimates changing to 1.36 (95 % CI 1.02–1.84, P = 0.038) and 1.34 (95 % CI: 1.01–1.79, P = 0.044) respectively. In addition, the result changed and became insignificant when we excluded the study by Carr et al. (2013) in the homozygote comparison for −521T>C polymorphism and simvastatin-related ADRs (pooled effect estimate = 3.17, 95 % CI 0.96–10.53, P = 0.059). The results were not materially altered for other analysis except for the above situations.
Publication bias
The Begg’s rank correlation test and Egger’s linear regression test indicated no evidence of publication bias among studies testing −521T>C and −388A>G polymorphisms of the SLCO1B1 gene and the risk of ADRs induced by various statins or specific statin (P > 0.05 for all of the genetic models) except in the analysis of −521T>C polymorphism and ADRs induced by any statin in the heterozygote comparison (Begg’s, P = 0.049; Egger’s, P = 0.046) and the additive model (Begg’s, P = 0.386; Egger’s, P = 0.025) (Table 3).
Discussion
ADRs give rise to the discontinuation of statin treatment (Simons et al. 1996). Recent studies showed that genetic variants in the SLCO1B1 gene modified the risk of statin-induced myopathy, but the results were controversial. The meta-analysis aimed to retrieve all published relative articles to identify the associations between the two functional variants −521T>C and −388A>G of the SLCO1B1 gene and the risk of ADRs during statin therapy. We included articles about any ADR and the dose decrease, discontinuation or switches to other cholesterol-lowering drugs related to statin toxicity as indicators of ADRs.
The results of the meta-analysis demonstrated that the minor allele of the −521C significantly increased the risk of ADRs caused by various statins, which is consistent with that of one previous meta-analysis (Carr et al. 2013). However, the synthesis heterogeneity was generally large under the different genetic models except for the recessive model. So we could not draw a strong conclusion that the association between the rs4149056 polymorphism and the ADRs induced by statins was a class effect. Upon closer review of each selected original study, it was found that results according to different statin types were inconsistent. Thus, we conducted a stratification analysis to explore that.
After stratification analysis by the statin type, significant associations between the −521T>C polymorphism and ADRs among simvastatin users for the allele contrast, the homozygote comparison, the dominant model, the recessive model and the additive model were observed, while no significant effect was found among patients treated with atrovastatin in any of the six genetic models. The results were in line with those of one previous meta-analysis by Carr et al. (2013), in which patients who carried at least one minor allele showed a more than three-fold higher risk for simvastatin-induced side effects compared with the reference TT genotype, whereas in contrast, the relationship did not reach statistical significance within atorvastatin users. The results from the original studies indicated that the −521C minor allele was not a risk factor for rosuvastatin-induced myalgia (HR = 0.95, 95 % CI 0.79–1.15 per allele, P = 0.59) (Danik et al. 2013) or pravastatin-induced composite adverse events (TC/CC vs. TT: OR = 1.03, 95 % CI 0.41–2.57, P = 0.948) (Voora et al. 2009). On the contrary, an additional copy of the minor allele was associated with the risk of cerivastatin-associated rhabdomyolysis (OR = 2.45, 95 % CI 1.61–3.75, P = 3.11E−05) (Marciante et al. 2011). However, as there was only one study investigating cerivastatin, rosuvastatin and pravastatin respectively, we did not perform a meta-analysis to provide evidence for their relationship.
There remains uncertainty about the biological mechanisms underlying the statin-associated myopathy but statin concentrations in the blood was one possible reason (Stewart 2013). A non-synonymous coding SNP, rs4149056 in the SLCO1B1 gene encodes a valine-to-alanine substitution that leads to a less active form of the OATP1B1 transporter and hence increases the plasma drug concentration (Kameyama et al. 2005; Tirona et al. 2001; Nozawa et al. 2002; Iwai et al. 2004). A previous study showed that in transient expression systems of HEK293 and HeLa cells using statins as substrates, the transporting activities of those expressing SLCO1B1 −521C allele decreased significantly (Kameyama et al. 2005). Furthermore, there are differences in the effects of SLCO1B1 −521T>C polymorphism on the pharmacokenetics of various statins. In patients with homozygous minor allele genotype, the observed plasma areas under the curve of active simvastatin acid, pitavastatin, atorvastatin, pravastatin, and rosuvastatin have been 221, 162–191, 144, 57–130, and 62–117 % higher respectively than that in patients with the wild-type genotype (Wilke et al. 2012). This may partially account for the disparate roles of rs4149056 variant in the development of ADRs during different types of statin therapy. Several case–control (Link et al. 2008; de Keyser et al. 2014; Carr et al. 2013) and cohort studies (de Keyser et al. 2014) as well as our meta-analysis confirmed the positive correlation between −521C minor allele and simvastatin-induced adverse effects. Link et al. (2008) observed that the OR for myopathy of 4.5 (95 % CI 2.6–7.7, P = 2 × 10−9) per copy of the C allele in patients taking 80 mg of simvastatin daily in the SEARCH study was approximately 2 times of that in those taking 40 mg daily within the Heart Protection Study (OR = 2.6, 95 % CI 1.3–5.0, P = 0.004). Besides, Carr et al. (2013) discovered in the stratification analysis for dose (a cut-off point of 40 mg), the significant increased risk associated with rs4149056 variant was only found in patients receiving ≥ 40 mg/day simvastatin. These provided evidence for dose-genotype interaction in simvastatin treatment. Consequently, the Clinical Pharmacoenomics Implementation Consortium (CPIP) Guideline for SLCO1B1 and Simvastatin-Induced Myopathy (2012) (Wilke et al. 2012) recommends that prescribing physicians should be alerted to the FDA advice on avoiding high-dose simvastatin. In our analyses, all of extracted or de novo effect estimates for atrovastatin-related ADRs showed a consistent and no significant association. While in one of the included studies by de Keyser et al. (2014), an association between the −521T>C polymorphism and dose decrease or switching to another cholesterol-lowering drug in the highest-dose category (> 20 mg) was found (the adjusted HR = 3.26, 95 % CI: 1.47–7.25, P = 0.004), however, we extracted the HR for whole population, which showed no significance, other than the outcome of stratification analysis. Therefore, we could anticipate that a significant higher risk would appear if patients are prescribed to high dose atorvastatin. Puccetti et al. (2010) conducted a case–control study to explore specific genetic and/or environmental factors to statin intolerance and they found a significant association between the C allele of rs4149056 SNP in the SLCO1B1 and myopathy in atorvastatin-treated patients (OR = 2.7, 95 % CI 1.3–4.9, P < 0.001). Since the genetic model under which the reported OR had been calculated was not available, we did not incorporate this study into our meta-analysis, which may influence our results.
The −388A>G is another common variant in the SLCO1B1 gene, which is in strong linkage disequilibrium with the −521T>C SNP (Link et al. 2008). Unlike rs4149056, rs2306283 SNP enhances liver uptake of pravastatin bringing about a reduced area under the curve for plasma pravastatin concentration (Niemi et al. 2004). The synthesis of studies on −388A>G polymorphism yielded a non-statistically significant protective effect on ADRs caused by any statin. But the results should be interpreted with caution because there were only two available studies incorporated to the meta-analyses. Therefore, more well-designed large sample size original studies are needed to identify the association.
When we performed the meta-analyses on association between SLCO1B1 −521T>C and ADRs caused by various statins, high heterogeneity was present in all genetic models except for the recessive model. The disparate definitions of ADRs, study design, characteristics of participants (i.e., age, sex, BMI, health status), statin type, dose and duration of treatment, and some other factors maybe the potential explanation. Unfortunately, we could only conduct the subgroup analysis for statin type but not other factors owing to the limited available data and small number of original studies. After stratification analyses, the I 2 values were decreased to zero in the atorvastatin group but for the simvastatin the heterogeneity remained almost the same. In order to further explore the source of heterogeneity and to investigate the influence of individual study to the overall results, sensitivity analysis was performed. When we removed studies by de Keyser et al. (2014) respectively, the pooled effect estimates changed to be statistically significant in the heterozygote comparison. Both of the two studies reported insignificant reduced risk of dose decrease or switches in TC carriers, which may take place by chance, and might contributed more to the overall insignificant result. For simvastatin users, no significant association between −521T>C polymorphism and ADRs was found with the exclusion of one study by Carr et al. (2013) for the homozygote contrast. Possible explanation was that in this study, borderline statistically significant differences between cases and controls in terms of previous history of type 2 diabetes (P = 0.046), asthma (P = 0.080), and hypertension (P = 0.087) were determined, which might be confounders making the calculated OR biased consequently influencing the pooled result.
Several limitations in our study may affect the results. Firstly, it is noteworthy that the definitions of statin-induced ADRs varied among individual studies, which contributed to the presence of heterogeneity to a large extent. Further studies using uniform definitions are required to reach more definitive conclusions. Secondly, the crude and adjusted effect estimates were combined together due to the limited available data from the original studies. Moreover, the adjusting factors for each effect estimate were not completely consistent. As such, some potential confounding risk factors may be introduced to influence our pooled results, such as the differences in statin therapy including dose and duration of treatment. It is indicated that methodological improvement in the individual studies should be focused. Thirdly, the data from original studies were insufficient. We could not perform further stratification analyses to explore dose-, gender-, age-, BMI- and comedication-gene interactions. Additionally, the limited number of studies on −388A>G polymorphism lowered our power and the results of meta-analyses should be interpreted with caution. Fourthly, we only included articles published in English in four databases, relevant articles published in other databases and in other languages and unpublished studies may have been missed. What’s more, as the number of included studies was small, the power of detecting the publication bias in Begg’s and Egger’s test was low. Finally, the original studies were entirely or predominantly based on Caucasians, thus additional researches in other ethnicities are needed to generalize the findings.
In conclusion, our meta-analysis suggests that SLCO1B1 −521T>C polymorphism may be significantly associated with increased risk of statin-induced adverse reactions, especially in the simvastatin therapy. Conversely, the −521C minor allele might not modify the risk of atorvastatin-associated adverse effects. Besides, there may be no significant association between −388A>G polymorphism and statin-related adverse reactions. However, the finding should be interpreted with caution because of the small number of studies and small sample sizes. Well-designed epidemiological studies with large sample size in the treatment of various statins among different ethnicities should be carried out to confirm these associations and interaction between dose-, sex-, BMI-, extensive physical exercise-, comedication-gene should also be further investigated.
References
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R (2005) Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90, 056 participants in 14 randomised trials of statins. Lancet 366:1267–1278
Begg CB, Mazumdar M (1994) Operating characteristics of a rank correlation test for publication bias. Biometrics 50:1088–1101
Brunham LR, Lansberg PJ, Zhang L, Miao F, Carter C, Hovingh GK, Visscher H, Jukema JW, Stalenhoef AF, Ross CJ, Carleton BC, Kastelein JJ, Hayden MR (2012) Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenomics J 12:233–237
Carr DF, O’Meara H, Jorgensen AL, Campbell J, Hobbs M, McCann G, van Staa T, Pirmohamed M (2013) SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink. Clin Pharmacol Ther 94:695–701
Chen C, Stock JL, Liu X, Shi J, Van Deusen JW, DiMattia DA, Dullea RG, de Morais SM (2008) Utility of a novel Oatp1b2 knockout mouse model for evaluating the role of Oatp1b2 in the hepatic uptake of model compounds. Drug Metab Dispos 36:1840–1845
Cota GF, de Sousa MR, Fereguetti TO, Rabello A (2013) Efficacy of anti-leishmania therapy in visceral leishmaniasis among HIV infected patients: a systematic review with indirect comparison. PLoS Negl Trop Dis 7:e2195
Crowther M, Lim W, Crowther MA (2010) Systematic review and meta-analysis methodology. Blood 116:3140–3146
Danik JS, MacFadyen J, Nyberg F, Ridker P (2012) Lack of association between polymorphisms in the SLC01B1 gene and clinical myalgia following rosuvastatin therapy. J Am Coll Cardiol 59:E1621
Danik JS, Chasman DI, MacFadyen JG, Nyberg F, Barratt BJ, Ridker PM (2013) Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am Heart J 165:1008–1014
de Keyser CE, Becker ML, Maitland-Van Der Zee A-H, Uitterlinden AG, Hofman A, Visser LE, Stricker BH (2012a) The SLCO1B1 C.521T>C polymorphism and the risk of adverse reactions during simvastatin and atorvastatin therapy. Pharmacoepidemiol Drug Saf 21:184
de Keyser CE, Stricker BH, Becker ML (2012b) Research highlights. Pharmacogenomics 13:15–16
de Keyser CE, Peters BJ, Becker ML, Visser LE, Uitterlinden AG, Klungel OH, Verstuyft C, Hofman A, Maitland-van der Zee AH, Stricker BH (2014) The SLCO1B1 c.521T>C polymorphism is associated with dose decrease or switching during statin therapy in the Rotterdam Study. Pharmacogenet Genomics 24:43–51
de Lemos JA, Blazing MA, Wiviott SD, Lewis EF, Fox KA, White HD, Rouleau JL, Pedersen TR, Gardner LH, Mukherjee R, Ramsey KE, Palmisano J, Bilheimer DW, Pfeffer MA, Califf RM, Braunwald E (2004) Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 292:1307–1316
Dendramis G (2011) Interindividual differences in the response to statin therapy and gene polymorphisms related to myopathy during statin therapy. G Ital Cardiol (Rome) 12:182–185
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–188
Dlouha D, Hubacek J, Adamkova V, Snejderlova M, Ceska R, Vrablik M (2013) Association between common variants in the SLCO1B1 gene and statin-induced myopathy. Cardiology (Switzerland) 126:75
Dmitry S, Grigory S, Andrey G, Tatyana B (2013) The frequency of allele genotypes of gene SLCO1B1null5 in Russian patients suffering from hyperlipidemia. Drug Metabol Drug Interact 28:A23
Dolgin E (2013) Pharmacogenetic tests yield bonus benefit: better drug adherence. Nat Med 19:1354–1355
Donnelly LA, Doney AS, Tavendale R, Lang CC, Pearson ER, Colhoun HM, McCarthy MI, Hattersley AT, Morris AD, Palmer CN (2011) Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a go-DARTS study. Clin Pharmacol Ther 89:210–216
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315:629–634
Feng Q, Wilke RA, Baye TM (2012) Individualized risk for statin-induced myopathy: current knowledge, emerging challenges and potential solutions. Pharmacogenomics 13:579–594
Fiegenbaum M, da Silveira FR, Van der Sand CR, Van der Sand LC, Ferreira ME, Pires RC, Hutz MH (2005) The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin Pharmacol Ther 78:551–558
Frudakis TN, Thomas MJ, Ginjupalli SN, Handelin B, Gabriel R, Gomez HJ (2007) CYP2D6*4 polymorphism is associated with statin-induced muscle effects. Pharmacogenet Genomics 17:695–707
Furihata T, Satoh N, Ohishi T, Ugajin M, Kameyama Y, Morimoto K, Matsumoto S, Yamashita K, Kobayashi K, Chiba K (2009) Functional analysis of a mutation in the SLCO1B1 gene (c.1628T>G) identified in a Japanese patient with pravastatin-induced myopathy. Pharmacogenomics J 9:185–193
Ghatak A, Faheem O, Thompson PD (2010) The genetics of statin-induced myopathy. Atherosclerosis 210:337–343
Graham DJ, Staffa JA, Shatin D, Andrade SE, Schech SD, La Grenade L, Gurwitz JH, Chan KA, Goodman MJ, Platt R (2004) Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 292:2585–2590
Higgins JP, Thompson SG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21:1539–1558
Ho PM, Magid DJ, Shetterly SM, Olson KL, Maddox TM, Peterson PN, Masoudi FA, Rumsfeld JS (2008) Medication nonadherence is associated with a broad range of adverse outcomes in patients with coronary artery disease. Am Heart J 155:772–779
Hopewell JC, Offer A, Parish S, Haynes R, Li J, Jiang L, Lathrop M, Armitage J, Collins R (2012) Environmental and genetic risk factors for myopathy in Chinese participants from HPS2-THRIVE. Eur Heart J 33:445
Ishikawa C, Ozaki H, Nakajima T, Ishii T, Kanai S, Anjo S, Shirai K, Inoue I (2004) A frameshift variant of CYP2C8 was identified in a patient who suffered from rhabdomyolysis after administration of cerivastatin. J Hum Genet 49:582–585
Iwai M, Suzuki H, Ieiri I, Otsubo K, Sugiyama Y (2004) Functional analysis of single nucleotide polymorphisms of hepatic organic anion transporter OATP1B1 (OATP-C). Pharmacogenetics 14:749–757
Johansen C, Phillips MS, Dube M-P, Wang J, Lin T, Ban MR, Kennedy BA, Tardif J-C, Hegele RA (2010) SLCO1B1 is not associated with statin-induced myopathy in patients from a tertiary referral lipid clinic. Arterioscler Thromb Vasc Biol 29:e23
Kamatani N, Mushiroda T (2011) Use of the data from genome-wide association study (GWAS) for pharmacogenomics and new drug development: focusing on transporter genes. J Pharmacol Sci 115:42P
Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K (2005) Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15 + C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics 15:513–522
Khan AA, Ding K, Khader S, Kullo IJ (2011) Association of a polymorphism in SLCO1B1 with statin-induced myalgias, myositis and myopathy: an electronic medical record based pharmacogenetic study. J Am Coll Cardiol 57:E1160
König J, Seithel A, Gradhand U, Fromm MF (2006) Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol 372:432–443
Kroemer HK (2010) Cardiovascular drug transport: influence of genetics and disease. Drug Metab Rev 42:7–8
Linde R, Peng L, Desai M, Feldman D (2010) The role of vitamin D and SLCO1B1*5 gene polymorphism in statin-associated myalgias. Dermatoendocrinol 2:77–84
Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R (2008) SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 359:789–799
Mafalda M, Leticia B, Claudia CB, Luisa M-V (2013) Characterization of the functional genetic variants relevant to statin response in the Azores Islands population (Portugal). Drug Metabol Drug Interact 28:A37
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22:719–748
Marciante KD, Durda JP, Heckbert SR, Lumley T, Rice K, McKnight B, Totah RA, Tamraz B, Kroetz DL, Fukushima H, Kaspera R, Bis JC, Glazer NL, Li G, Austin TR, Taylor KD, Rotter JI, Jaquish CE, Kwok PY, Tracy RP, Psaty BM (2011) Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet Genomics 21:280–288
McClure DL, Valuck RJ, Glanz M, Murphy JR, Hokanson JE (2007) Statin and statin-fibrate use was significantly associated with increased myositis risk in a managed care population. J Clin Epidemiol 60:812–818
Meador BM, Huey KA (2010) Statin-associated myopathy and its exacerbation with exercise. Muscle Nerve 42:469–479
Nakamura Y (2008) Pharmacogenomics and drug toxicity. N Engl J Med 359:856–858
Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, Backman JT, Kerb R, Schwab M, Neuvonen PJ, Eichelbaum M, Kivisto KT (2004) High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics 14:429–440
Niemi M, Pasanen MK, Neuvonen PJ (2011) Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 63:157–181
Nozawa T, Nakajima M, Tamai I, Noda K, Nezu J, Sai Y, Tsuji A, Yokoi T (2002) Genetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): allele frequencies in the Japanese population and functional analysis. J Pharmacol Exp Ther 302:804–813
Oh J, Ban MR, Miskie BA, Pollex RL, Hegele RA (2007) Genetic determinants of statin intolerance. Lipids Health Dis 6:7
Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M (2006) SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics 16:873–879
Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M (2007) Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther 82:726–733
Patel J, Abd T, Blumenthal RS, Nasir K, Superko HR (2014) Genetics and personalized medicine–a role in statin therapy? Curr Atheroscler Rep 16:384
Petrov VI, Smuseva ON, Solovkina YV (2013) Integrated assessment of statin-associated muscle damage predictors in patients with ischemic heart disease. Ration Pharmacother Cardiol 9:247–250
Pranculis A, Kucinskas V (2013) Pharmacogenomic landscape of the Lithuanian population. Drug Metabol Drug Interact 28:A28–A29
Puccetti L, Ciani F, Auteri A (2010) Genetic involvement in statins induced myopathy. Preliminary data from an observational case-control study. Atherosclerosis 211:28–29
Rodriguez S, Gaunt TR, Day IN (2009) Hardy-Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol 169:505–514
Santos PC, Gagliardi AC, Miname MH, Chacra AP, Santos RD, Krieger JE, Pereira AC (2012) SLCO1B1 haplotypes are not associated with atorvastatin-induced myalgia in Brazilian patients with familial hypercholesterolemia. Eur J Clin Pharmacol 68:273–279
Schech S, Graham D, Staffa J, Andrade SE, La Grenade L, Burgess M, Blough D, Stergachis A, Chan KA, Platt R, Shatin D (2007) Risk factors for statin-associated rhabdomyolysis. Pharmacoepidemiol Drug Saf 16:352–358
Simons LA, Levis G, Simons J (1996) Apparent discontinuation rates in patients prescribed lipid-lowering drugs. Med J Aust 164:208–211
Stewart A (2013) SLCO1B1 Polymorphisms and statin-induced myopathy. PLoS Curr 5
Sychev DA, Shuev GN, Prokofiev AB (2013) Applied aspects of SLCO1B1 pharmacogenetic testing for predicting of statin-induced myopathy and personalization of statins therapy. Ration Pharmacother Cardiol 9:698–700
Tamraz B, Fukushima H, Wolfe AR, Kaspera R, Totah RA, Floyd JS, Ma B, Chu C, Marciante KD, Heckbert SR, Psaty BM, Kroetz DL, Kwok PY (2013) OATP1B1-related drug-drug and drug-gene interactions as potential risk factors for cerivastatin-induced rhabdomyolysis. Pharmacogenet Genomics 23:355–364
Thompson PD, Clarkson PM, Rosenson RS (2006) An assessment of statin safety by muscle experts. Am J Cardiol 97:69C–76C
Tirona RG, Leake BF, Merino G, Kim RB (2001) Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem 276:35669–35675
Toms TE, Smith JP, Panoulas VF, Douglas KM, Saratzis AN, Kitas GD (2009) Prevalence of risk factors for statin-induced myopathy in rheumatoid arthritis patients: does the SLCO1B1 gene play a role? Rheumatology (Oxford) 48:i83–i84
Toms TE, Smith JP, Panoulas VF, Douglas KM, Saratzis AN, Kitas GD (2010) Prevalence of risk factors for statin-induced myopathy in rheumatoid arthritis patients. Musculoskeletal Care 8:2–9
Vladutiu GD (2008) Genetic predisposition to statin myopathy. Curr Opin Rheumatol 20:648–655
Vladutiu GD, Simmons Z, Isackson PJ, Tarnopolsky M, Peltier WL, Barboi AC, Sripathi N, Wortmann RL, Phillips PS (2006) Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 34:153–162
Voora D, Ali S, Shah SH, Reed CR, Salisbury B, Ginsburg GS (2008) Association of statin-induced musculoskeletal side effects with sex and a hepatic uptake transporter reduced function Allele, SLC01B1*5. Circulation 118:S326
Voora D, Shah SH, Spasojevic I, Ali S, Reed CR, Salisbury BA, Ginsburg GS (2009) The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol 54:1609–1616
Wilke RA, Moore JH, Burmester JK (2005a) Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharmacogenet Genomics 15:415–421
Wilke RA, Reif DM, Moore JH (2005b) Combinatorial pharmacogenetics. Nat Rev Drug Discov 4:911–918
Wilke RA, Ramsey LB, Johnson SG, Maxwell WD, McLeod HL, Voora D, Krauss RM, Roden DM, Feng Q, Cooper-Dehoff RM, Gong L, Klein TE, Wadelius M, Niemi M (2012) The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther 92:112–117
Wright JL, Zhou S, Preobrazhenska O, Marshall C, Sin DD, Laher I, Golbidi S, Churg AM (2011) Statin reverses smoke-induced pulmonary hypertension and prevents emphysema but not airway remodeling. Am J Respir Crit Care Med 183:50–58
Authors’ contributions
All authors contributed in the writing of this paper. JJJ: Literature search, data extraction and production of manuscript, QT: Literature search, data extraction and production of manuscript, JF: Literature search, data extraction and production of figures and manuscript, RD: Final approval of the version to be published, YW: Conception of idea and production of manuscript, YY: Production of manuscript and senior author, XT: Conception of idea and production of manuscript, CD: Result interpretation, made discussion and conclusion, HZ: Senior author for literature search and production of manuscript, YZ: Result interpretation, made discussion and conclusion, FZ: Conception of idea, production of manuscript and senior author. All authors read and approved the final manuscript.
Acknowledgements
We sincerely thank numerous individuals participated in this study. Also, we acknowledge the editors and reviewers for insightful suggestions on this work.
Competing interests
The authors declare that they have no competing interests.
Funding
Support for this study was provided by the Chongqing Social Science Planned Doctor Project (No: 2014BS042).
Author information
Authors and Affiliations
Corresponding author
Additional information
Jiajia Jiang and Qing Tang contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jiang, J., Tang, Q., Feng, J. et al. Association between SLCO1B1 −521T>C and −388A>G polymorphisms and risk of statin-induced adverse drug reactions: A meta-analysis. SpringerPlus 5, 1368 (2016). https://doi.org/10.1186/s40064-016-2912-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-2912-z