
Abstract
Members of the large family of WRKY transcription factors are involved in a wide range of developmental and physiological processes, most particularly in the plant response to biotic and abiotic stress. Here, an analysis of the soybean genome sequence allowed the identification of the full complement of 188 soybean WRKY genes. Phylogenetic analysis revealed that soybean WRKY genes were classified into three major groups (I, II, III), with the second group further categorized into five subgroups (IIa–IIe). The soybean WRKYs from each group shared similar gene structures and motif compositions. The location of the GmWRKYs was dispersed over all 20 soybean chromosomes. The whole genome duplication appeared to have contributed significantly to the expansion of the family. Expression analysis by RNA-seq indicated that in soybean root, 66 of the genes responded rapidly and transiently to the imposition of salt stress, all but one being up-regulated. While in aerial part, 49 GmWRKYs responded, all but two being down-regulated. RT-qPCR analysis showed that in the whole soybean plant, 66 GmWRKYs exhibited distinct expression patterns in response to salt stress, of which 12 showed no significant change, 35 were decreased, while 19 were induced. The data present here provide critical clues for further functional studies of WRKY gene in soybean salt tolerance.
Similar content being viewed by others


Background
Soybean (Glycine max) is a global cash crop. Apart from its major contribution to human and animal nutrition, the seed provides a feedstock for biodiesel production and represents a significant raw material for a number of pharmaceutical and industrial processes (Phang et al. 2008; Wang et al. 2010). In recent years, the demand for soybean is increasing rapidly, so it attracted more and more attention to improve soybean agronomic traits, such as stress tolerance. Soybean productivity is greatly compromised by soil salt. However, during the long period of evolution, soybean has evolved complex strategies to survive salt stress. These strategies are originated from the changes of various aspects, such as the genome, gene expression, metabolism and physiology (Phang et al. 2008). To present, functionally reported salt tolerance related genes in soybean are mainly categorized into several classes, including ion transporter coding genes (e.g. GmHKT1, GmSALT3, GmNHX1, GmCAX1 and GmCHX1) (Chen et al. 2011; Guan et al. 2014; Li et al. 2006; Luo et al. 2005a; Qi et al. 2014), transcription factors (TFs) (e.g. GmNAC11/-20/-29, GmDREB1, GmMYB76/-92/-174/-177, GmbZIP44/-62/-78, GmWRKY27 and GmERF7) (Hao et al. 2011; Jin et al. 2010; Liao et al. 2008a, b; Wang et al. 2015; Zhai et al. 2013) and others genes (e.g. glutathione S-transferase gene GsGST, late embryogenesis abundant gene GmLEA, calcineurin B-like protein coding gene GmCBL1 and flavone synthase gene GmFNSII) (Ji et al. 2010; Lan et al. 2005; Li et al. 2012; Phang et al. 2008; Wang et al. 2011; Yan et al. 2014; Zhou et al. 2010).
The product of a TF gene binds to a specific cis-regulatory sequence(s) in the promoter of its target gene. The WRKYs are among the largest class of plant TFs, and their promoter target (the W-box) has the sequence (T)(T)TGAC(C/T) (Rushton et al. 2010). WRKY TFs are recognized by the presence of a conserved DNA-binding region composed of about 60 residues (the “WRKY” domain), which harbors the WRKYGQK heptapeptide followed by a C2H2 or C2HC zinc finger motif (Rushton et al. 2010). In some cases, the heptapeptide can take the form WRKYGKK or WRKYGEK (Rushton et al. 2010). WRKY TFs have been classified into three main groups: those possessing two heptapeptides are clustered into group I; both group I and II members harbor one C2H2 type zinc finger motif, while the group III members feature a C2HC one. The large size of group II has been addressed by its division into five subgroups (IIa, IIb, IIc, IId and IIe), based on peptide sequence (Eulgem et al. 2000; Rushton et al. 2010).
Since the first reports of WRKY TFs (dePater et al. 1996; Ishiguro and Nakamura 1994; Rushton et al. 1995, 1996), considerable progress has been made in revealing the function of WRKY TFs. They are elucidated to be involved in various developmental and physiological processes (Rushton et al. 2010), such as seed development (Lagace and Matton 2004; Luo et al. 2005b), seed dormancy and germination (Xie et al. 2005, 2007; Zentella et al. 2007), senescence (Miao et al. 2004; Robatzek and Somssich 2002; Ulker et al. 2007) trichome morphogenesis (Johnson et al. 2002), metabolic pathways (Kato et al. 2007; Sun et al. 2003) and plant development (Cai et al. 2014; Devaiah et al. 2007; Guo et al. 2015; Yu et al. 2013, 2016). The particularly prominent roles of WRKY in plant appear to be the modulation of response to biotic and abiotic stresses (Chen et al. 2012; Eulgem and Somssich 2007; Pandey and Somssich 2009). In the root of the model plant Arabidopsis, 18 WRKY genes have been shown to be induced by exposure to salt stress (Jiang and Deyholos 2006); In rice, ten WRKY genes (of 13 analyzed) respond differentially to a range of abiotic stress treatment (Qiu et al. 2004), while in Brachypodium distachyon, over 60 % of a set of 86 WRKY genes assayed were up-regulated by heat and cold stress and over 50 % were down-regulated by salt, drought and/or oxidation stress (Wen et al. 2014). Among the soybean WRKY TFs, 25 out of 64 have been shown to be differentially expressed in response to at least one abiotic stress treatment (Zhou et al. 2008).
Base on the availability of the complete soybean genome sequence and several databases (PlantTFDB, SoyDB, SoyTFKB, NCBI and Phytozome), a previous study reported a genome-wide characterization of the WRKY family in soybean and a functional analysis of some genes involved in response to Phakopsora pachyrhizi (Bencke-Malato et al. 2014). However, in Phytozome (release v10), the new assembly (v2.0) replaces the Glyma1 assembly. The new database corrects several issues in pseudomolecule reconstruction in the Glyma1 assembly. According to the new database and a recent RNA-Seq result (Belamkar et al. 2014), Song et al. identified 176 GmWRKYs and analyzed their expression files in different tissues and in response to drought and salt stress (Song et al. 2016). However, beside Phytozome, there are many other soybean genome sequence databases. Furthermore, several other transcriptome experiment data sets of soybean under abiotic stress are provided by NCBI website. Here, integrating more databases and RNA-Seq results (Belamkar et al. 2014; Wei et al. 2015), we made a new genome-wide identification of soybean WRKYs and compared their response to salt stress in different tissues. In addition, we analyzed expression profiles of 66 GmWRKYs by quantitative RT-PCR (RT-qPCR). Our findings provide new clues for further investigation of WRKY gene in soybean salt tolerance.
Methods
Retrieval of GmWRKY sequences
A set of 185 GmWRKY sequences was recovered from Phytozome (http://phytozome.jgi.doe.gov/pz/portal.html, release 10.2) using the keyword PF03106 as a search term, along with three further GmWRKY sequences from the NCBI database (http://www.ncbi.nlm.nih.gov). The presence of a WRKY domain(s) in all 188 GmWRKYs was confirmed by running the SMART program (http://smart.embl-heidelberg.de) (Letunic et al. 2015). These GmWRKY genes were further checked in PlantTFDB (http://planttfdb.cbi.pku.edu.cn/, release 3.0) (Jin et al. 2014) and SoyTFKB (http://www.igece.org/Soybean_TF/).
Multiple sequence alignment and phylogenetic analysis
A multiple alignment of the WRKY sequences was performed using the ClustalW program implemented in MEGA v6.06 software package (http://www.megasoftware.net/) (Tamura et al. 2013). The sequences were also subjected to a phylogenetic analysis using the neighbor-joining method; the resulting tree was based on 1000 bootstrap replicates, the p-distance model and pairwise deletion.
Gene structure and conserved motifs analysis
The exon–intron structure of each gene was derived by comparing its coding sequence with the corresponding genomic DNA sequence, using the GSDS program (http://gsds.cbi.pku.edu.cn/) (Hu et al. 2015). The online program MEME v4.10.1 (http://meme-suite.org/tools/meme) was used to identify the conserved motifs present; the relevant parameters were: number of repetitions = any; maximum number of motifs = 16; optimum width of each motif = 6–70 residues.
Genomic location and gene duplication
Each WRKY gene was positioned in the genome by reference to the full genome sequence. The gene duplications in GmWRKY genes were identified based on the investigations described in previous study (Schmutz et al. 2010), and Circos software was used to provide a graphical representation of the position of homeologous chromosome segments (Krzywinski et al. 2009).
RNA-seq analysis
A transcriptomic analysis was based on archival RNA-seq data collected from a set of salt stress experiments, mounted on the NCBI GEO database (Belamkar et al. 2014). Experiments GSM1377923, -24 and -25 represented three independent replicates of plants sampled before any exposure to salt; GSM1377935, -36 and -37 related to plants sampled after a 1 h exposure to the stress; GSM1377938, -39 and -40 after a 6 h exposure; and GSM1377941, -42 and -43 after a 12 h exposure. The other transcriptomic analysis was also based on archival RNA-seq data derived from the NCBI GEO database (Wei et al. 2015), experiment GSE57960 was related to plants sampled after a 12 h exposure to salt stress, the aerial part of plants was used for sequencing. The reads per kilobase of exon model per million mapped reads (RPKM) algorithm was used for normalization and mean normalized values were used for the analysis. The transcription response was given in the form of fold changes relative to the 0 h control. Cluster v3.0 software (University of Tokyo, Human Genome Center) was used to perform hierarchical clustering, which was visualized using Java TreeView software (Saldanha 2004). The relevant parameters were: similarity measurement: correlation (uncentered); linkage method: average linkage method.
Plant materials
Seed of cv. Williams 82 was germinated on a sheet of moist filter paper, and the seedlings were grown under a regime of 28/20 °C, 14 h photoperiod, light intensity 800 μmol m−2 s−1 and relative humidity 55 %. Two weeks old seedlings were exposed to 200 mM NaCl for either 0, 2, 6 or 24 h, after which the whole seedling was harvested, snap-frozen in liquid nitrogen and stored at −80 °C.
RT-qPCR analysis
Total RNA was extracted from the frozen plant material using the TRIzol reagent (Invitrogen, USA) following to the manufacturer’s instructions. The resulting RNA was treated with RNase-free DNaseI (Promega, USA) to remove genomic DNA contamination, and the cDNA First strand was synthesized with 3 μg total RNA by TransScript One-step gDNA Removal and cDNA synthesis SuperMix (TransGen, China) following the manufacturer’s protocol. The subsequent RT-qPCRs and data analysis were performed using a Bio-Rad Real-Time PCR detection system (Bio-Rad) based on the SYBR Green I master mix, as reported previously (Bustin et al. 2009; Seo et al. 2009). According to a previous study, GmELF1b was most stably expressed under salt stress, so it was used as a reference gene (Le et al. 2012). All reactions were carried out in triplicate, using samples harvested from independent plants. The relevant primer sequences are given in Additional file 1: Table S1.
Results and discussion
Identification of WRKY genes in soybean
The WRKYs represent one of the largest families of plant TFs. The acquisition of full genome sequences has simplified the enumeration of WRKY copy number, so that it is now clear that there are 81 WRKY copies in tomato (Huang et al. 2012), 55 in cucumber (Ling et al. 2011), 104 in poplar (He et al. 2012), 59 in grapevine (Wang et al. 2014), 116 in cotton (Dou et al. 2014) and 119 in maize (Wei et al. 2012). In a previous study, 182 putative WRKY gene models were identified (Bencke-Malato et al. 2014). In recent, phytozome updated the soybean assembly; the new assembly (v2.0) replaced the Glyma1 assembly. Therefore, we performed a comprehensive analysis of soybean WRKY sequences obtained from Phytozome, PlantTFDB and NCBI, and finally identified a total of 185 non-redundant putative WRKY genes. Compared with previous 182 WRKY genes, six genes GmWRKY49 (Glyma.05G203900), GmWRKY53 (Glyma.06G061900), GmWRKY72 (Glyma.07G161100), GmWRKY108 (Glyma.10G171000), GmWRKY130 (Glyma.14G085500) and GmWRKY131 (Glyma.14G100100) are novel ones, while three previous genes GmWRKY17 (Glyma06g06530), GmWRKY38 (Glyma18g48460) and GmWRKY132 (Glyma14g11440) are considered obsolete according to the current version of the annotated genome. However, the three obsolete genes have been retained for further study. Thus, a total of 188 annotations of GmWRKYs were presented in this study (Table 1). All the 188 retrieved sequences were proved to contain WRKY domains using SMART analysis.
In previous genome-wide studies, the commonly accepted nomenclature for WRKY members was based on their location order on chromosomes (Dou et al. 2014; Ling et al. 2011). Identically, in the present study, GmWRKYs was designated from GmWRKY1 to GmWRKY188 based on their exact physical position from the top to the bottom on the soybean chromosomes 1–20 (Table 1). For genes producing more than one transcript, only the primary sequence was named. This nomenclature system was different from previous study (Bencke-Malato et al. 2014). A full comparison of currently known WRKY genes is given in Additional file 2: Table S2.
In silico mapping revealed that the WRKY genes were distributed over all 20 soybean chromosomes. Chromosome 7 harbored the highest number of GmWRKY genes (16, 8.51 %), while chromosome 11, 12 and 20 harbored only three (1.60 %). The largest WRKY product was encoded by GmWRKY46 (1355 residues), and the shortest was GmWRKY131 (68 residues) (Fig. 1; Table 1).

The distribution of WRKY genes on the 20 chromosomes of soybean
Classification of WRKY genes in soybean
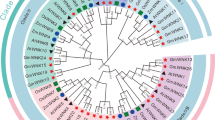
As described previously, WRKY family is typically categorized into three main groups defined by the number of WRKY domains present and the configuration of their zinc finger (Rushton et al. 2010). The 188 soybean WRKY genes were also categorized into the three main groups (Additional file 3: Fig. S1). The group I members numbered 32 (GmWRKY65 and -72 harbored a single N-terminal WRKY domain); there were 130 sequences assigned to group II, sub-divided into subgroup IIa (14 members), IIb (33 members), IIc (42 members), IId (21 members) and IIe (20 members); the remaining 26 sequences belonged to group III (Table 1; Fig. 1). In Arabidopsis and poplar, group I houses the largest number of WRKYs, while in rice, group III is the largest (He et al. 2012). However, the largest group in soybean is group II, implying that this group had experienced more gene duplications during the evolutionary course.
Although most of the sequences harbored the well conserved WRKYGQK motif, variants were present in 24 of the sequences: WRKYGKK in 11, WRKYGEK in three, WKKYGQK in two, and WRKYGKR, WRKYEDK, WIKYGQK and WHQYGLK each in one. Strikingly, A WRKYGQK-like stretch was lacking in GmWRKY20, -49, -105 and -130, while the group I members WRKY65 and -72 had both lost their N terminal WRKYGQK-like stretch (Table 1; Additional file 3: Fig. S1). The largest number of variants belonged to group IIc, 11 out of 24. The WRKYGQK sequence was highly conserved in subgroups IIb, IId and IIe, as well as in the C terminal WRKY domain of group I members (Table 1; Additional file 3: Fig. S1). This is consistent with the implication that this group experienced more gene duplications. There was also some variation in the zinc finger motif (including its complete absence) in 11 of the sequences (WRKY6, -42, -52, -65, -72, -79, -94, -106, -112, -139 and -165) (Additional file 3: Fig. S1).
Gene duplication of soybean WRKY genes
Duplication events contribute not only to functional redundancy, but also generate functional novelty (Moore and Purugganan 2005). The modern soybean genome has undergone two whole genome duplication (WGD) events, the first, associated with the evolution of the legume clade occurred ~59 million years ago (Lavin et al. 2005), while the second, which was responsible for the creation of the Glycine genus, occurred ~13 million years ago (Schmutz et al. 2010). To investigate whether the expansion of GmWRKY genes had primarily happened during both WGD events, we mapped the GmWRKYs to the duplicated blocks (Fig. 2). Consistent with previous study (Schmutz et al. 2010), the blocks between chromosomes involved more than just two chromosomes. Of 188 GmWRKYs, 180 (95.7 %) genes were located in the blocks (the exceptions were GmWRKY46, -63, -65, -72, -94, -105, -106, and -139) (Fig. 2), indicating that WGD was the primary reason for the expansion of GmWRKYs (Fig. 2).

Chromosomal location of the soybean WRKY genes. The illustrated genome-wide chromosome organization caused by whole genome duplication events is accomplished using the Circos software based on the duplication coordinates defined in the current genome assembly v2.0. Segmental duplicated blocks are color coded. Paralogous pairs are connected with lines
Besides WGD, tandem duplication event is the other approach for gene expansion. Precise mapping analysis showed the presence of 14 adjacent genes possibly due to tandem duplication (Fig. 2; Additional file 4: Fig. S2a). These 14 WRKY genes were localized in 6 distinct tandem duplicate gene clusters, with four clusters containing two tandem genes (GmWRKY120/123, GmWRKY121/122, GmWRKY131/132 and GmWRKY151/152) and two clusters possessing three ones (GmWRKY108/109/110 and GmWRKY136/137/138). All the 14 tandem duplicated WRKY genes were mapped onto the duplicated blocks, implying that local duplications occurred earlier than the WGD.
Gene structure and conserved motifs of GmWRKYs
Gene structural diversity may reflect the evolution of multigene families (Hu et al. 2010). In order to look into the structural diversity of GmWRKY genes, we first constructed a phylogenetic tree based on the full-length GmWRKY polypeptide sequences, and they were also categorized into seven subfamiles as above (Additional file 4: Fig. S2a). From the tree, we could find that each clade consists of two to four genes, which well matched the two WGD events and confirmed that the expansion of GmWRKY happened during both WGD events. We then analyzed the exon–intron organization in the coding sequences of each soybean WRKY genes HD-ZIP genes (Additional file 4: Fig. S2b). Previous study showed that most Populus WRKY genes contain two to four introns (He et al. 2012). Similarly, the majority of soybean WRKY members harbored two to four introns. For instance, over 60 % members of subgroups IIc (26/42), IId (17/21), IIe (14/20) and III (23/26) harbored two introns; over 60 % group I members (17/32) harbored four; most members in group IIa harbored three (7/14) or four (5/14) (Additional file 4: Fig. S2b). In contrast, the gene structure appeared to be more variable in groups IIb, the number of introns in this group varied from one to six (Additional file 4: Fig. S2b). In Populus, although there were only eight members in group IIb, the numbers of introns varied from three to six (He et al. 2012). These results indicated that WRKY genes in different species were relatively conserved during the evolution. Furthermore, genes shared similar exon–intron organization within the same subgroup, while they were strikingly distinct in the gene structure among different groups, suggesting that they were not only conserved, but diverged during the evolution.
To better understand the conservation and diversification of WRKY genes in soybean, putative motifs of GmWRKYs were predicted using MEME software and finally 16 distinct motifs were identified (Additional file 4: Fig. S2c). As expected, most of the closely related members in the phylogenetic tree shared common motif compositions, suggesting that the WRKY proteins within the same subfamily might be of similar functions. However, like putative motifs predicted in ZmWRKYs (Gao et al. 2014), the biological significance of most of the putative motifs in GmWRKYs was also unclear because they did not have homologs when searching against Pfam (http://pfam.sanger.ac.uk/search) and SMART (Simple Modular Architecture Research Tool) databases. The same phenomenon also existed in Populus NAC and HD-ZIP proteins (Hu et al. 2010, 2012). According to previous study, WRKY proteins harbor typical WRKY domains and zinc-finger motifs (Eulgem et al. 2000; Rushton et al. 2010). Here, motif 1, 2 and 9 comprised the WRKY domain, motif 3 and 10 were the partial zinc-finger motifs followed motif 2 and 9 (Additional file 5: Table S3). The product size of the group I and subgroup IIb genes was larger than that of members of the other groups (Table 1), consistent with their harboring a greater number of motifs (Additional file 4: Fig. S2c). In contrast, although subgroup IIc possessed the largest number of members, they harbored the least number of motifs (one to three). Even though the C-terminal regions of GmWRKYs were highly divergent, we could also identify several conserved motifs which were present in GmWRKYs from specific subgroups, for example, motifs 3, 5 and 6 in group I, motif 14 in subgroup IIb, and motif 13 in subgroups IIa and IIb (Additional file 4: Fig. S2c). Whether these motifs play functional roles remained to be further elucidated.
Expression profiles of GmWRKYs in response to salt stress
The WRKY gene family is heavily implicated in the plant response to abiotic stress (Chen et al. 2012), as indicated by a number of microarray-based transcriptomic data sets. Several studies have reported the influence of abiotic stress on WRKY genes based on these data sets (Dou et al. 2014; Satapathy et al. 2014; Wei et al. 2012). In soybean (cv. Kefeng No. 1), the response of a set of 64 WRKY genes following the plant’s exposure to salt stress has been described (Zhou et al. 2008), but this number represents only about one-third of the total WRKY genes. In order to give insight to the function of GmWRKYs in plant response to salt tolerance, we analyzed the soybean (cv. Williams 82) gene expression profiles under salt stress (Belamkar et al. 2014). Finally, 66 of the 188 GmWRKY genes were transcriptionally regulated under salt stress (Fig. 3a; Additional file 6: Table S4). 65 genes were up-regulated, with only WRKY71 being down-regulated (Fig. 3a). The response of WRKY was typically quite rapid (Eulgem et al. 2000), most notably in the case of GmWRKY20, -47, -76, -126, -134, -153, -164, which responded by an at least five fold rise in transcript abundance after a 1 h exposure to the stress. In some cases, the response was transient: some examples were the genes WRKY44, -51, -54, -78, -81, -85, -102 and -107, for which transcript abundance peaked after a 6 h exposure and then fell away (Fig. 3a; Additional file 6: Table S4). The most responsive gene (WRKY134) belonged to subgroup IIa, which was increased to ~226 fold after a 6 h exposure (Fig. 3a; Additional file 6: Table S4). By contrast, the expression of GmWRKY71, a member of group IId was down-regulated in response to salt stress (Fig. 3a).

The transcription response of the WRKY genes in response to salt stress. Transcript abundance levels have been normalized and hierarchical clustered. Blue colored blocks indicate a decreased and yellow ones an increased level of transcription relative to the control. a The set of 66 genes transcriptionally altered in soybean root by the stress. Genes with remarkable changed expressions are labeled in red. b–d The transcription profiles of genes belonging to subgroups b IIb and c IIc and to d group III. hr number of hours of exposure to the stress. e The set of 49 genes transcriptionally altered in the aerial part of soybean by the stress

RT-qPCR-based transcription profiling of 66 WRKY genes. a Unaffected WRKY genes. b Salt-inhibited WRKY genes. c Salt-inducible WRKY genes. Error bars represent SD (n = 3). (t test, *P < 0.05; **P < 0.01)
In soybean, a notable number of responsive genes belonged to subgroup IIb (18 of 33), although their level of induction by salt treatment was only modest (Fig. 3b). In addition, most of subgroup IIc (14/42) and group III (14/26) members were significantly induced and these genes tended to be dramatically up-regulated after a 6 h exposure (Fig. 3c, d). In Arabidopsis root, the 18 salt induced members belong to group I (4/18), II (11/18) and III (3/18), respectively (Jiang and Deyholos 2006). These data indicated that either in soybean or Arabidopsis, group II members made major contribution in salt response, suggesting that WRKY functions in response to salt stress in different organisms appeared to be conserved during evolution.
The material used in the above RNA-seq was soybean root which was not able to represent other parts. We then analyzed the other RNA-seq data which were derived from the aerial part of soybean plants (cv. SuiNong 28) (Wei et al. 2015). A total of 49 GmWRKY genes were transcriptionally regulated under salt stress (Fig. 3e; Additional file 7: Table S5). 47 genes were down-regulated, with only two (WRKY155 and WRKY183) being up-regulated (Fig. 3e). These results were quite different with the above RNA-seq analysis, indicating that GmWRKY genes showed distinct response profiles in different tissues.
Investigation of GmWRKY gene expressions by RT-qPCR
The transcriptional profiles we analyzed above could provide clues for revealing the function of GmWRKYs in plant response to salt tolerance. However, the material used in RNA-seq was soybean root or aerial part which was not able to represent the entire plant. To shed light on the expression profiles of GmWRKY genes, 2 weeks old soybean seedlings (cv. Williams 82) were exposed to 200 mM NaCl for 0, 2, 6 or 24 h, respectively, and then the total RNA of the whole plant was isolated used for RT-qPCR analysis. 66 GmWRKY genes were tested and exhibited distinct expression patterns in response to salt stress, of which 12 showed no significant change (Fig. 4a), 35 were decreased (Fig. 4b), while 19 were induced (Fig. 4c). GmWRKY38, -120 and -185 were substantially decreased, especially GmWRKY120. In contrast, GmWRKY20, -89, -114 and -142 were remarkably induced. These expression patterns were different with the above RNA-seq analysis, indicating that GmWRKY genes showed distinct response profiles in the whole plants compared to different tissues. The response of WRKY to abiotic stresses was generally rapid and transient (Eulgem et al. 2000). Likely, most of the GmWRKY genes responded rapidly, their expressions were decreased (31/35) or induced (16/19) after only a 2 h exposure (Fig. 4b, c). In addition, the response of GmWRKYs was transient, such as GmWRKY36, -82, -83, -141, -153, -159 and -66 (Fig. 4b, c).
In tomato and cucumber, most WRKYs are up-regulated by salt stress (Huang et al. 2012; Ling et al. 2011). In contrast, the majority of Brachypodium distachyon WRKYs are down-regulated by the stress (Wen et al. 2014). In soybean, most WRKYs were up-regulated in root, while down-regulated in the aerial part by the stress (Fig. 4). Species differences presumably reflected a major degree of functional divergence in the WRKY gene family.
Conclusion
The present study has taken a genome-wide view of the soybean WRKY gene family, and characterized their transcriptional response to salt stress. An analysis of their phylogeny, chromosomal location, gene structure and content of conserved motifs has allowed the genes to be classified into the standard set of groups. The expansion in copy number of the GmWRKYs has occurred largely as a result of the two well recognized ancient whole genome duplication events. To date, only three GmWRKY genes have been functionally investigated (Jiang and Deyholos 2009), leaving unknown the function of the remaining more than 180. The responsiveness to salt stress of about one-third of the GmWRKY complement confirms the potential of gene manipulation within this gene family as means of improving the salt tolerance of important crop species.
Abbreviations
- NCBI:
-
National Center for Biotechnology Information
- PlantTFDB:
-
Plant Transcription Factor Database
- SoyDB:
-
Database of Soybean Transcription Factors
- SoyTFKB:
-
Soybean Transcription Factor Knowledge Base
- SMART:
-
Simple Modular Architecture Research Tool
- GSDS:
-
Gene Structure Display Server
- MEME:
-
Multiple Em for Motif Elicitation
References
Belamkar V, Weeks NT, Bharti AK, Farmer AD, Graham MA, Cannon SB (2014) Comprehensive characterization and RNA-Seq profiling of the HD-Zip transcription factor family in soybean (Glycine max) during dehydration and salt stress. BMC Genom 15:950
Bencke-Malato M, Cabreira C, Wiebke-Strohm B, Bucker-Neto L, Mancini E, Osorio MB, Homrich MS, Turchetto-Zolet AC, De Carvalho MCCG, Stolf R, Weber RLM, Westergaard G, Castagnaro AP, Abdelnoor RV, Marcelino-Guimaraes FC, Margis-Pinheiro M, Bodanese-Zanettini MH (2014) Genome-wide annotation of the soybean WRKY family and functional characterization of genes involved in response to Phakopsora pachyrhizi infection. BMC Plant Biol 14:236
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Cai YH, Chen XJ, Xie K, Xing QK, Wu YW, Li J, Du CH, Sun ZX, Guo ZJ (2014) Dlf1, a WRKY transcription factor, is involved in the control of flowering time and plant height in rice. PLoS ONE 9:e102529
Chen H, He H, Yu D (2011) Overexpression of a novel soybean gene modulating Na+ and K+ transport enhances salt tolerance in transgenic tobacco plants. Physiol Plantarum 141:11–18
Chen LG, Song Y, Li SJ, Zhang LP, Zou CS, Yu DQ (2012) The role of WRKY transcription factors in plant abiotic stresses. BBA-Gene Regul Mech 1819:120–128
dePater S, Greco V, Pham K, Memelink J, Kijne J (1996) Characterization of a zinc-dependent transcriptional activator from Arabidopsis. Nucleic Acids Res 24:4624–4631
Devaiah BN, Karthikeyan AS, Raghothama KG (2007) WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol 143:1789–1801
Dou LL, Zhang XH, Pang CY, Song MZ, Wei HL, Fan SL, Yu SX (2014) Genome-wide analysis of the WRKY gene family in cotton. Mol Genet Genomics 289:1103–1121
Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10:366–371
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Gao J, Peng H, He XJ, Luo M, Chen Z, Lin HJ, Ding HP, Pan GT, Zhang ZM (2014) Molecular phylogenetic characterization and analysis of the WRKY transcription factor family responsive to Rhizoctonia solani in maize. Maydica 59:32–41
Guan RX, Qu Y, Guo Y, Yu LL, Liu Y, Jiang JH, Chen JG, Ren YL, Liu GY, Tian L, Jin LG, Liu ZX, Hong HL, Chang RZ, Gilliham M, Qiu LJ (2014) Salinity tolerance in soybean is modulated by natural variation in GmSALT3. Plant J 80:937–950
Guo DS, Zhang JZ, Wang XL, Han X, Wei BY, Wang JQ, Li BX, Yu H, Huang QP, Gu HY, Qu LJ, Qin GJ (2015) The WRKY transcription factor WRKY71/EXB1 controls shoot branching by transcriptionally regulating RAX genes in Arabidopsis. Plant Cell 27:3112–3127
Hao YJ, Wei W, Song QX, Chen HW, Zhang YQ, Wang F, Zou HF, Lei G, Tian AG, Zhang WK, Ma B, Zhang JS, Chen SY (2011) Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J 68:302–313
He HS, Dong Q, Shao YH, Jiang HY, Zhu SW, Cheng BJ, Xiang Y (2012) Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep 31:1199–1217
Hu RB, Qi G, Kong YZ, Kong DJ, Gao QA, Zhou GK (2010) Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol 10:145
Hu RB, Chi X, Chai G, Kong Y, He G, Wang X, Shi D, Zhang D, Zhou G (2012) Genome-wide identification, evolutionary expansion, and expression profile of homeodomain-leucine zipper gene family in poplar (Populus trichocarpa). PLoS One 7(2):e31149
Hu B, Jin JP, Guo AY, Zhang H, Luo JC, Gao G (2015) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31:1296–1297
Huang SX, Gao YF, Liu JK, Peng XL, Niu XL, Fei ZJ, Cao SQ, Liu YS (2012) Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol Genet Genomics 287:495–513
Ishiguro S, Nakamura K (1994) Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol Gen Genet 244:563–571
Ji W, Zhu YM, Li Y, Yang LA, Zhao XW, Cai H, Bai X (2010) Over-expression of a glutathione S-transferase gene, GsGST, from wild soybean (Glycine soja) enhances drought and salt tolerance in transgenic tobacco. Biotechnol Lett 32:1173–1179
Jiang YQ, Deyholos MK (2006) Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol 6:25
Jiang YQ, Deyholos MK (2009) Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol 69:91–105
Jin TC, Chang Q, Li WF, Yin DX, Li ZJ, Wang DL, Liu B, Liu LX (2010) Stress-inducible expression of GmDREB1 conferred salt tolerance in transgenic alfalfa. Plant Cell Tiss Org 100:219–227
Jin JP, Zhang H, Kong L, Gao G, Luo JC (2014) PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res 42:D1182–D1187
Johnson CS, Kolevski B, Smyth DR (2002) TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell 14:1359–1375
Kato N, Dubouzet E, Kokabu Y, Yoshida S, Taniguchi Y, Dubouzet JG, Yazaki K, Sato F (2007) Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol 48:8–18
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Lagace M, Matton DP (2004) Characterization of a WRKY transcription factor expressed in late torpedo-stage embryos of Solanum chacoense. Planta 219:185–189
Lan Y, Cai D, Zheng YZ (2005) Expression in Escherichia coli of three different soybean late embryogenesis abundant (LEA) genes to investigate enhanced stress tolerance. J Integr Plant Biol 47:613–621
Lavin M, Herendeen PS, Wojciechowski MF (2005) Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54:575–594
Le DT, Aldrich DL, Valliyodan B, Watanabe Y, Van Ha C, Nishiyama R, Guttikonda SK, Quach TN, Gutierrez-Gonzalez JJ, Tran LSP, Nguyen HT (2012) Evaluation of candidate reference genes for normalization of quantitative RT-PCR in soybean tissues under various abiotic stress conditions. PLoS One 7(9):e46487
Letunic I, Doerks T, Bork P (2015) SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43:D257–D260
Li WYF, Wong FL, Tsai SN, Phang TH, Shao GH, Lam HM (2006) Tonoplast-located GmCLC1 and GmNHX1 from soybean enhance NaCl tolerance in transgenic bright yellow (BY)-2 cells. Plant, Cell Environ 29:1122–1137
Li ZY, Xu ZS, He GY, Yang GX, Chen M, Li LC, Ma YZ (2012) Overexpression of soybean GmCBL1 enhances abiotic stress tolerance and promotes hypocotyl elongation in Arabidopsis. Biochem Biophys Res Commun 427:731–736
Liao Y, Zou HF, Wang HW, Zhang WK, Ma B, Zhang JS, Chen SY (2008a) Soybean GmMYB76, GmMYB92, and GmMYB177 genes confer stress tolerance in transgenic Arabidopsis plants. Cell Res 18:1047–1060
Liao Y, Zou HF, Wei W, Hao YJ, Tian AG, Huang J, Liu YF, Zhang JS, Chen SY (2008b) Soybean GmbZIP44, GmbZIP62 and GmbZIP78 genes function as negative regulator of ABA signaling and confer salt and freezing tolerance in transgenic Arabidopsis. Planta 228:225–240
Ling J, Jiang WJ, Zhang Y, Yu HJ, Mao ZC, Gu XF, Huang SW, Xie BY (2011) Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genom 12:471
Luo GZ, Wang HW, Huang J, Tian AG, Wang YJ, Zhang JS, Chen SY (2005a) A putative plasma membrane cation/proton antiporter from soybean confers salt tolerance in Arabidopsis. Plant Mol Biol 59:809–820
Luo M, Dennis ES, Berger F, Peacock WJ, Chaudhury A (2005b) MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc Natl Acad Sci USA 102:17531–17536
Miao Y, Laun T, Zimmermann P, Zentgraf U (2004) Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol Biol 55:853–867
Moore RC, Purugganan MD (2005) The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol 8:122–128
Pandey SP, Somssich IE (2009) The role of WRKY transcription factors in plant immunity. Plant Physiol 150:1648–1655
Phang TH, Shao G, Lam HM (2008) Salt tolerance in soybean. J Integr Plant Biol 50:1196–1212
Qi XP, Li MW, Xie M, Liu X, Ni M, Shao GH, Song C, Yim AKY, Tao Y, Wong FL, Isobe S, Wong CF, Wong KS, Xu CY, Li CQ, Wang Y, Guan R, Sun FM, Fan GY, Xiao ZX, Zhou F, Phang TH, Liu X, Tong SW, Chan TF, Yiu SM, Tabata S, Wang J, Xu X, Lam HM (2014) Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat Commun 5:4340
Qiu YP, Jing SJ, Fu J, Li L, Yu DQ (2004) Cloning and analysis of expression profile of 13 WRKY genes in rice. Chin Sci Bull 49:2159–2168
Robatzek S, Somssich IE (2002) Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes Dev 16:1139–1149
Rushton PJ, Macdonald H, Huttly AK, Lazarus CM, Hooley R (1995) Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of α-Amy2 genes. Plant Mol Biol 29:691–702
Rushton PJ, Torres JT, Parniske M, Wernert P, Hahlbrock K, Somssich IE (1996) Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J 15:5690–5700
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258
Saldanha AJ (2004) Java Treeview-extensible visualization of microarray data. Bioinformatics 20:3246–3248
Satapathy L, Singh D, Ranjan P, Kumar D, Kumar M, Prabhu KV, Mukhopadhyay K (2014) Transcriptome-wide analysis of WRKY transcription factors in wheat and their leaf rust responsive expression profiling. Mol Genet Genomics 289:1289–1306
Schmutz J, Cannon SB, Schlueter J, Ma JX, Mitros T, Nelson W, Hyten DL, Song QJ, Thelen JJ, Cheng JL, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu SQ, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du JC, Tian ZX, Zhu LC et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Seo PJ, Xiang FN, Qiao M, Park JY, Lee YN, Kim SG, Lee YH, Park WJ, Park CM (2009) The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol 151:275–289
Song H, Wang P, Hou L, Zhao S, Zhao C, Xia H, Li P, Zhang Y, Bian X, Wang X (2016) Global analysis of WRKY genes and their response to dehydration and salt stress in soybean. Front Plant Sci. doi:10.3389/fpls.2016.00009
Sun CX, Palmqvist S, Olsson H, Boren M, Ahlandsberg S, Jansson C (2003) A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell 15:2076–2092
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Ulker B, Mukhtar MS, Somssich IE (2007) The WRKY70 transcription factor of Arabidopsis influences both the plant senescence and defense signaling pathways. Planta 226:125–137
Wang Z, Libault M, Joshi T, Valliyodan B, Nguyen HT, Xu D, Stacey G, Cheng JL (2010) SoyDB: a knowledge database of soybean transcription factors. BMC Plant Biol 10:14
Wang YX, Suo HC, Zhuang CX, Ma H, Yan XL (2011) Overexpression of the soybean GmWNK1 altered the sensitivity to salt and osmotic stress in Arabidopsis. J Plant Physiol 168:2260–2267
Wang LN, Zhu W, Fang LC, Sun XM, Su LY, Liang ZC, Wang N, Londo JP, Li SH, Xin HP (2014) Genome-wide identification of WRKY family genes and their response to cold stress in Vitis vinifera. BMC Plant Biol 14:103
Wang FF, Chen HW, Li QT, Wei W, Li W, Zhang WK, Ma B, Bi YD, Lai YC, Liu XL, Man WQ, Zhang JS, Chen SY (2015) GmWRKY27 interacts with GmMYB174 to reduce expression of GmNAC29 for stress tolerance in soybean plants. Plant J 83:224–236
Wei KF, Chen J, Chen YF, Wu LJ, Xie DX (2012) Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in Maize. DNA Res 19:153–164
Wei W, Li QT, Chu YN, Reiter RJ, Yu XM, Zhu DH, Zhang WK, Ma B, Lin Q, Zhang JS, Chen SY (2015) Melatonin enhances plant growth and abiotic stress tolerance in soybean plants. J Exp Bot 66:695–707
Wen F, Zhu H, Li P, Jiang M, Mao WQ, Ong C, Chu ZQ (2014) Genome-wide evolutionary characterization and expression analyses of WRKY family genes in Brachypodium distachyon. DNA Res 21:327–339
Xie Z, Zhang ZL, Zou XL, Huang J, Ruas P, Thompson D, Shen QJ (2005) Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol 137:176–189
Xie Z, Zhang ZL, Hanzlik S, Cook E, Shen QXJ (2007) Salicylic acid inhibits gibberellin-induced alpha-amylase expression and seed germination via a pathway involving an abscisic-acid-inducible WRKY gene. Plant Mol Biol 64:293–303
Yan JH, Wang BA, Jiang YN, Cheng LJ, Wu TL (2014) GmFNSII-controlled soybean flavone metabolism responds to abiotic stresses and regulates plant salt tolerance. Plant Cell Physiol 55:74–86
Yu YC, Hu RB, Wang HM, Cao YP, He G, Fu CX, Zhou GK (2013) MlWRKY12, a novel Miscanthus transcription factor, participates in pith secondary cell wall formation and promotes flowering. Plant Sci 212:1–9
Yu YC, Liu ZH, Wang L, Kim SG, Seo PJ, Qiao M, Wang N, Li S, Cao XF, Park CM, Xiang FN (2016) WRKY71 accelerates flowering via the direct activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J 85:96–106
Zentella R, Zhang ZL, Park M, Thomas SG, Endo A, Murase K, Fleet CM, Jikumaru Y, Nambara E, Kamiya Y, Sun TP (2007) Global analysis of DELLA direct targets in early gibberellin signaling in Arabidopsis. Plant Cell 19:3037–3057
Zhai Y, Wang Y, Li Y, Lei T, Yan F, Su L, Li X, Zhao Y, Sun X, Li J, Wang Q (2013) Isolation and molecular characterization of GmERF7, a soybean ethylene-response factor that increases salt stress tolerance in tobacco. Gene 513:174–183
Zhou QY, Tian AG, Zou HF, Xie ZM, Lei G, Huang J, Wang CM, Wang HW, Zhang JS, Chen SY (2008) Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J 6:486–503
Zhou GA, Chang RZ, Qiu LJ (2010) Overexpression of soybean ubiquitin-conjugating enzyme gene GmUBC2 confers enhanced drought and salt tolerance through modulating abiotic stress-responsive gene expression in Arabidopsis. Plant Mol Biol 72:357–367
Authors’ contributions
YY were involved in designing the research. YY and RH collected and analyzed the data. YY and NW performed the experiments. YY and FX wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This research was financially supported by the National High Technology Research and Development Program “863” (Grant No. 2013AA102602-4), National Special Science Research Program of China (Grant No. 2013CB967300), National Natural Science Foundation (Grant Nos. 30970243, 31270328, 31471515), National Key Technology R&D Program of the Ministry of Science and Technology (Grant No. 2011BAI06B01), China Postdoctoral Science Foundation funded project (Grant No. 2014M551893), National Transgenic Project of China (Grant No. 2013ZX08010002-002), Science & Technology Plan of Shandong Province (Grant No. 2013GNC11010), and Research Program for International S&T Cooperation Projects of Shandong Province (Grant No. 2011176). We thank Dr. Robert Koebner (UK) for English editing of the manuscript and Chengming Fan (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences) for his skillful technical assistance in the present study.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
40064_2016_2647_MOESM1_ESM.doc
Additional file 1: Table S1. Primers used in this study.
40064_2016_2647_MOESM2_ESM.doc
Additional file 2: Table S2. A comparison between the WRKY genes identified in the current study and those described previously (Bencke-Malato et al. 2014).
40064_2016_2647_MOESM3_ESM.eps
Additional file 3: Figure S1. Multiple alignment of the conserved WRKY domain sequences.
40064_2016_2647_MOESM4_ESM.eps
Additional file 4: Figure S2. The phylogeny, gene structure and conserved motifs of the soybean WRKY gene family. (a) A multiple sequence alignment of 188 full length polypeptide sequences. The seven groups/subgroups (I, IIa-e and III) are depicted by different colors. (b) Exon–intron structures. (c) The 16 conserved motifs as identified by MEME software. Detailed sequence information given in Additional file 4: Table S3.
40064_2016_2647_MOESM5_ESM.doc
Additional file 5: Table S3. Conserved WRKY motif sequences as predicted by MEME software.
40064_2016_2647_MOESM6_ESM.doc
Additional file 6: Table S4. Normalized transcript levels of 66 GmWRKY genes in root under salinity stress conditions.
40064_2016_2647_MOESM7_ESM.doc
Additional file 7: Table S5. Normalized transcript levels of 49 GmWRKY genes in aerial part under salinity stress conditions.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yu, Y., Wang, N., Hu, R. et al. Genome-wide identification of soybean WRKY transcription factors in response to salt stress. SpringerPlus 5, 920 (2016). https://doi.org/10.1186/s40064-016-2647-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-2647-x