
Abstract
Azadirachta indica (neem) is the only source of azadirachtin, which is known for its insecticide activity. Melia azedarach is a related species of A. indica, widely distributed in the south of China. In this study, the leaf transcriptomes of these two Meliaceae plants were sequenced. More than 40 million clean reads were generated from each library. About 80 % of A. indica reads were mapped to the neem genome, while 93 % of M. azedarach reads were mapped to its assembled transcripts and unigenes dateset. After mapping and assembly, 225,972 transcripts and 91,607 unigenes of M. azedarach were obtained and 1179 new genes of A. indica were detected. Comparative analysis of the annotated differentially expressed genes (DEG) showed that all six DEGs involved in terpenoid backbone biosynthesis were up-regulated in A. indica. Chemical analysis of the two plants revealed A. indica leaves contained 2.45 % total terpenoid and nearly 20–50 µg azadirachtin per gram, whereas azadirachtin was not detected in M. azedarach and total terpenoid content was reached 1.67 %. These results give us a better insight into the transcriptomes differences between A. indica and M. azedarach, and help us to understand the terpenoid biosynthesis pathway in vivo.
Similar content being viewed by others
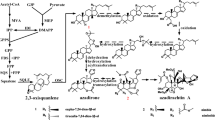


Background
Azadirachta indica A. Juss (neem tree) and Melia azedarach Linn. are two species in the Meliaceae family, that have a close relationship in phylogenetic systematics. However, in chemical analysis of different tissues in the two species, azadirachtin (a kind of triterpene) was found in nearly all parts of A. indica, whereas no azadirachtin or its derivatives were found in M. azedarach (Tan and Luo 2011). Azadirachtin is the most important activated compound in the neem tree, having effective biological functions and huge commercial value (Atawodi and Atawodi 2009). Azadirachtin is an efficient environmentally friendly plant-derived pesticide, that interfere with insect growth and development (Qiao et al. 2014). A study on the biology and mortality of rice leaffolder larvae treated with neem extract showed azadirachtin was a potent pesticide and caused almost 100 % larval mortality at a 1 ppm concentration (Senthil Nathan et al. 2006). Because of its broad spectrum toxicity to insects, azadirachtin has been registered as a pesticide in many countries. NeemAzal, a kind of azadirachtin-based commercial insecticide, was shown to have a strong inhibitory effect on Rhyzopertha dominica, Sitophilus oryzae and Tribolium confusum (Athanassiou et al. 2005). Besides insecticidal activity, neem tree extracts also have many pharmaceutical functions, such as anticancer, antimicrobial, anti-inflammatory and antidiabetic activities (Thoh et al. 2010; Soares et al. 2014). However, despite plentiful information on the usefulness of the neem tree, there have been few molecular studies on this plant, especially about the biosynthesis of azadirachtin in vivo. Fortunately, the whole A. indica genome and five transcriptomes (including stem, leaf, flower, root and fruit) have been sequenced (Krishnan et al. 2011, 2012), which has established a solid foundation for molecular biological research on the neem tree. Though the mechanism of azadirachtin biosynthesis is unknown, a lot of research has focused on the synthesis of azadirachtin, including chemosynthesis (Veitch et al. 2007), hairy root culture (Srivastava and Srivastava 2013), callus culture (Rodrigues et al. 2014) and cell line culture in vitro (Singh and Chaturvedi 2013).
Using the omics strategy to study the metabolic pathways which active phytomedicinals are produced has become a hotspot of secondary metabolite research in recent years (Misra 2014), and lies at the intersection of chemistry, biology, mathematics and computer science. Using this method, researchers sequenced the root transcripts of American ginseng and found that one CYP450 and four UDP-glycosyltransferases were most likely involved in ginsenoside biosynthesis (Sun et al. 2010). Based on their different terpenoid products, in this research we comparatively analyzed the leaf transcriptomes of A. indica and M. azedarach by specific RNA-seq, and screen for genes related to azadirachtin biosynthesis. This study will help us to understand the formation of azadirachtin in vivo, and also provides potential targets for regulation with existing research strategies to harvest greater amounts of environment-friendly biopesticides.
Methods
Plant materials
Seeds of A. indica were obtained from Yuanmou Desert Ecosystem Research Station, Yuanmou County, Yunnan Province, China, while seeds of M. azedarach were collected from Dong’an Forest Park, Chaohu County, Anhui Province, China. The two Meliaceae plants were identifed by Yanping Zhang (Research Institute of Resources Insects of the Chinese Academy of Forestry, China) and Yiming Hu (Anhui Academy of Forestry, China), respectively. Sampling of plant materials did not affect the local ecology and was performed with permission from local administrative departments. After seed germination, the plants were grown in a greenhouse at 28 °C with a 16 L:8 D photoperiod. Leaves of A. indica and M. azedarach were collected from the plants and immediately frozen in liquid nitrogen and stored at −80 °C until analysis.
Azadirachtin and total terpenoid quantitative analysis in leaves
One gram of cryopreserved leaf powder was weighted into a 5-mL centrifuge tube, 3 mL methanol was added for extraction, and the tube was mixed using a homogenizer (Fluko, Essen, Germany) for 2 min. The tube was then centrifuged at 4000 rpm for 3 min and the supernatant was removed to a 10-mL volumetric flask. The extraction was repeated three times, and the supernatants were combined and dried using a Termovap Sample Concentrator (Organomation, Berlin, MA, USA). The concentrated sample was purified with an ENVI-Carb SPE (Supelco, Bellefonte, PA, USA) according to the manufacturer’s instructions and then concentrated to 1 mL. Last, a Waters model 2695 high performance liquid chromatography (HPLC) apparatus equipped with a model 2996 photodiode array detector (PAD) (Waters, Milford, MA, USA), was used to detect the azadirachtin content in each sample; the detection parameters were as follows: X Terra® RP18 column (4.6 × 250 mm, 5 µm), 10 μL sample size, mobile phase of methanol–water (6:4), 1 mL/min velocity, 210–360 nm detection wavelength. Total terpenoid content assay was performed with Ghorai’s methods (Ghorai et al. 2012).
RNA extraction and strand-specific library construction
Total RNAs were isolated using an EASYspin Plus Complex Plant RNA Kit (Aidlab, Beijing, China), and then unwanted cytoplasmic, mitochondrial, and chloroplast ribosomal RNAs were removed using Ribo-Zero™ rRNA Removal Kits (Illumina, San Diego, CA, USA). The quality of the collected RNAs was initially estimated with a Nanodrop 8000 UV–Vis Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and then the integrity of the RNA samples was precision detected with an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) according to the user’s guide. Strand-specific cDNA libraries of the two Meliaceae plants were constructed for transcriptome sequencing using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA) according to the directions. First, purified mRNAs were randomly broken into shorter fragments, and random primers were added by hybridization. Next, first-strand cDNA was synthesized using the fragments as templates, and the second-strand cDNA was synthesized using dUTP instead of dTTP for labelling. The fragments were then purified, end-repaired, dA-tailed and ligated with adapters. Finally, the second-strand was selectively removed using the USER enzyme (NEB) while the first-strand was left for the PCR amplification.
Sequencing and quality control of the data
Based on sequencing by synthesis technologies, the test qualified libraries were then sequenced using an Illumina Hiseq™ 2500 with 125 bp pair-end reads at the Biomarker Technologies Company in Beijing, China. Via base calling, huge numbers of raw reads were acquired. The quality scores of the bases (Q-values), reflecting the probability of mismatched bases, and the base distribution was determined to evaluate sequencing quality. Finally, reads with adaptors, reads with unknown nucleotides larger than 10 % and low quality reads in which the percentage of bases with Q-values <10 was more than 50 % were removed, leaving only the clean reads. The datasets for each sample were deposited in the Short Read Archive (SRA) database of the National Institutes of Health (NIH).
Reads assembly, mapping and new gene detection
The A. indica reference genome (364M) and gene set were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA_000439995.3_AzaInd2.1), and also could be browsed online from the official website of the Ganit Labs, Bio-IT Centre, Institute of Bioinformatics and Applied Biotechnology, (http://115.119.161.46:96/cgi-bin/gb2/gbrowse/neemV2/) using Gbrowse 2.0. After the neem genome was prepared, clean reads from the A. indica libraries (Y1–3) were aligned to the reference genome using TopHat2 (Kim et al. 2013). Briefly, the mapping process was divided into two steps. First, the reads were aligned against the neem genome, and then split into smaller segments, which were aligned to the genome. After that, the Cufflinks software was used to assemble the mapped reads (Trapnell et al. 2010), which were compared with the original genome annotation information to screen for new genes.
For the M. azedarach libraries (K1–3), because there was little genome information for M. azedarach. it was necessary to assemble the clean reads before annotation. Based on the known related species: Citrus Clementina (301M), Citrus sinensis (328M) and A. indica (364M), the genome size of M. azedarach was likely between 300 to 400M. The assembler program Trinity (Grabherr et al. 2011), which is better than other de novo transcriptome assembly programs in many respects, was used for this process. Briefly, Trinity first extends the clean reads set in k-mer space and breaks ties. Next, it overlaps linear sequences by overlaps of k-1 to build graph components. Last, it builds a De Bruijn graph and compacts it to get the transcripts and unigenes. After reads assembly was finished, the clean reads were mapped to transcripts and unigenes sets using Bowtie2 (Langmead et al. 2009).
Functional annotation of new genes and unigenes
Before further bioinformatics analysis, it was necessary to test the quality of the transcriptome libraries. Normally, detecting the distribution of inserted segments in the genes or unigenes, the length profile of the inserted fragments and the saturation curve map, which are common methods to assess the quality of libraries. For functional annotation, the new genes and unigenes were aligned to five public databases: Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Eukaryotic Orthologous Groups (KOG), Swiss-Prot and Non-redundant Protein (Nr) databases.
Quantitative analysis of gene expression
After using Bowtie to align the clean reads and unigenes set, RNA-seq by expectation maximization (RSEM) was used to accurately quantify the expression levels of transcripts from the RNA-seq data (Li and Dewey 2011). Fragments per kilobase of transcript per million fragments mapped (FPKM) values were used to reflect the expression levels of transcripts or genes.
In this study, to compare the differentially expressed genes (DEG) in the two Meliaceae plants, we needed to screen for orthologous genes was needed. OrthoMCL was used to analyze the homologous proteins among the known protein sequences in A. indica and predicted protein sequences in M. azedarach (Li et al. 2003). After that, according to methods established by Brawand et al. (2011), a corresponding degree of scaling was used to normalize the gene expression levels in the different species (Brawand et al. 2011). The fold changes of gene expression were assessed by the log2 ratio (FPKM-Y/FPKM-K).
Results and discussion
Sequence analysis and assembly
To obtain a comprehensive overview of the differences in terpenoid biosynthesis between the two Meliaceae plants, three cDNA libraries from A. indica and three from M. azedarach were constructed. For convenient analysis of the RNA-seq data, the three A. indica libraries were named Y1–Y3 and the M. azedarach libraries were named K1–K3. The six libraries were sequenced using the Illumina Hiseq™ 2500 sequencing platform. After removing the adapter and low quality reads, 46,203,176, 43,956,038 and 56,227,214 clean reads were acquired from Y1 to Y3, respectively (Table 1), while 51,341,436, 45,384,504 and 41,737,868 clean reads were obtained from K1 to K3, respectively (Table 2). The number of reads from each library was ten-fold higher than in the previous sequencing (Krishnan et al. 2012).
Though A. indica and M. azedarach are related species, there were great differences between the two Meliaceae plants at the transcriptome level. Initially, we planned to align the reads of three M. azedarach libraries to the neem genome database, but the low mapping ratio (<20 %) made it necessary to perform de novo assembly. Using the assembler Trinity (Grabherr et al. 2011), 225,972 transcripts and 91,607 unigenes were acquired, with corresponding N50 lengths of 2628 and 1321 bp, respectively (Table 3).
Mapping of reads to the A. indica genome dataset, and M. azedarach transcripts and unigenes
To identify the corresponding genes of the sequences in each library, the clean reads were mapped to the A. indica genome. The mapping results showed that more than 80 % of reads from each library were matched to the reference genome while about 50 % were uniquely matched. Based on the reference genome, the cufflinks software was used to splice the mapped reads of the A. indica libraries (Y) and 53,381 genes were acquired. After the mapped reads were assembled, 1179 new genes were screened out by comparison with the original neem genome annotation information.
In order to test the quality of the assembly, the clean reads were mapped to the unigenes and transcripts dataset. The results are shown in Table 2; more than 93 % of clean reads were mapped. Next, the mapped reads were used to detect the saturation of genes in each library; the saturation curve is shown in Additional file 1: Figure S1.
Analysis of differential genes expression in the leaves of the two Meliaceae plants
For comparative analysis of the DEGs in the libraries of the two speices, protein homology analysis was performed. Using the Orthomcl software (version 2.0.9), 3867 orthologous genes were identified in the two species (Additional file 2: Table S1). Comparison of the expression of orthologous genes showed that the majority of genes were expressed at different levels in the two species. In total, 2478 genes showed more than two-fold expression changes (log2(Fold Change)| ≥ 1); of these, 1388 genes were up-regulated and 1090 were down-regulated (Additional file 3: Table S2). Notably, among the 2478 DEGs, 352 genes showed more than 210-fold changes in expression level, including 71 up-regulated and 281 down-regulated genes. The distribution of fold-changes in DEG numbers between the A. indica (Y) and M. azedarach (K) libraries is shown in Fig. 1.

The distribution of fold-changes in differentially expressed gene numbers
Functional annotation of all genes (A. indica), unigenes (M. azedarach) and DEGs
For the further study, all genes (53,381) of A. indica including new genes (1179), the unigenes (91,607) of M. azedarach and the 2478 DEGs were aligned with the GO, KEGG, KOG, Swiss-Prot and Nr databases. The number of annotated genes in each database is listed in Table 4, and detailed annotation information for the DEGs, new genes and unigenes is shown in Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4.
From the GO annotation, 16,901 (A. indica), 15,649 (M. azedarach) and 1346 (DEGs) annotated genes were categorized into three main groups. For cellular components, genes associated with cell parts and organelles were the most highly represented, while genes related to catalytic activity and binding represented the largest proportion of genes with molecular functions. For biological processes, the most represented GO term was metabolic process, followed by cellular process and single-organism process. More information on the functional categorization of genes in A. indica, the unigenes in M. azedarach and the DEGs is shown in Additional file 6: Figure S2.
Using KEGG annotation, 517 new genes in A. indica, 21,238 M. azedarach genes and 763 DEGs were mapped to different KEGG pathways. The type classification of DEGs from the KEGG annotation results is shown in Fig. 2. Genes related to metabolism represented the largest proportion of DEGs, especially purine metabolism. Plant hormone signal transduction was the second largest category in the classification (Fig. 2). In relation to terpenoid synthesis, 135 unigenes participated in terpenoid backbone biosynthesis, 32 in sesquiterpenoid and triterpenoid biosynthesis, 29 in monoterpenoid biosynthesis and 50 in diterpenoid biosynthesis in M. azedarach, while 106, 52, 34 and 84 genes were involved in the corresponding biosynthetic processes in A. indica, respectively. Notably, only one new gene (new_gene 6030) was found to participate in terpenoid biosynthesis and all six DEGs involved in terpenoid backbone biosynthesis were up-regulated in A. indica (Fig. 3). It is likely that more metabolic flux is transfered into terpenoid synthesis in A. indica.

The type classification of DEGs with KEGG annotation results

DEGs involved in terpenoid backbone biosynthesis
The homologous species distribution from Nr annotation is shown in Fig. 4. The majority of genes of in A. indica and M. azedarach were most similar to homologous genes in C. sinensis (74.15 %) and C. clementina (68.12 %), which are Rutaceae plants. Meliaceae and Rutaceae both belong to Rutineae taxonomically, having a close evolutionary relationship.

Homologous species distributions in A. indica and M. azedarach
Chemical analysis of the azadirachtin and the total terpenoid content in A. indica and M. azedarach leaves
Using the standard curves (y = 5435.9x − 1501.2, R 2 = 0.9996 for azadirachtin A; y = 3730.0x + 2696.2, R 2 = 0.9995 for azadirachtin B), the total content of azadirachtin was calculated. Accordingly, 24.6, 24.05 and 51.77 mg/kg azadirachtin was detected from samples Y1–3, respectively, while azadirachtin was not detected in the K1–3 samples. Though azadirachtin was undetectable in M. azedarach, limonoids from M. azedarach also showed the activity to inhibit the development of flaviviruses and Mycobacterium tubercolosis (Sanna et al. 2015). Similarly, the total terpenoid content was calculated using the standard curve (y = 0.006x + 0.1064, R 2 = 0.9775). The results showed that there was nearly 12.26 mg terpenoid in 0.5 g A. indica leaves, while 8.33 mg terpenoid in M. azedarach leaves.
Conclusions
In this study, the transcriptome of M. azedarach was sequenced and analyzed for the first time, 225,972 transcripts and 91,607 unigenes were acquired, while 1179 new genes were detected from sequencing of A. indica libraries. Chemical analysis showed azadirachtin was only present in A. indica leaves; no azadirachtin or its derivatives were found in M. azedarach. The total terpenoid content assay showed there were 2.45 % terpenoid in A. indica leaves and 1.67 % terpenoid in M. azedarach leaves, respectively. These results will help us to research genes involved in the synthesis of bioactive compounds, and also associate the gene expression level with the metabolite content, especially terpenoid.
Accession number
The Illumina HiSeq™ 2500 sequencing data from this study have been deposited in the NIH SRA database under the accession numbers: SRR3180937, SRR3181105 and SRR3181166 for A. indica, and SRR3183379, SRR3183380 and SRR3183381 for M. azedarach.
Abbreviations
- SRA:
-
Short Read Archive database
- NIH:
-
National Institutes of Health
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- Nr:
-
Non-redundant Protein databases
- KOG:
-
Eukaryotic Orthologous Groups
- DEG:
-
differentially expressed genes
- FPKM:
-
fragments per kilobase of transcript per million fragments mapped
- RSEM:
-
RNA-seq by expectation maximization
References
Atawodi S, Atawodi J (2009) Azadirachta indica (neem): a plant of multiple biological and pharmacological activities. Phytochem Rev 8:601–620. doi:10.1007/s11101-009-9144-6
Athanassiou CG, Kontodimas DC, Kavallieratos NG, Veroniki MA (2005) Insecticidal effect of NeemAzal against three stored-product beetle species on rye and oats. J Econ Entomol 98:1733–1738. doi:10.1093/jee/98.5.1733
Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, Albert F, Zeller U, Khaitovich P, Grutzner F, Bergmann S, Nielsen R, Paabo S, Kaessmann H (2011) The evolution of gene expression levels in mammalian organs. Nature 478:343–348. doi:10.1038/nature10532
Ghorai N, Chakraborty S, Gucchait S, Saha SK, Biswas S (2012) Estimation of total terpenoids concentration in plant tissues using a monoterpene, linalool as standard reagent. Protoc Exch. doi:10.1038/protex.2012.055
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi:10.1038/nbt.1883
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi:10.1186/gb-2013-14-4-r36
Krishnan NM, Pattnaik S, Deepak SA, Hariharan AK, Gaur P, Chaudhary JR, Vaidyanathan S, Krishna PB, Panda B (2011) De novo sequencing and assembly of Azadirachta indica fruit transcriptome. Curr Sci 101:1553–1561
Krishnan NM, Pattnaik S, Jain P, Gaur P, Choudhary R, Vaidyanathan S, Deepak S, Hariharan AK, Krishna PB, Nair J, Varghese L, Valivarthi NK, Dhas K, Ramaswamy K, Panda B (2012) A draft of the genome and four transcriptomes of a medicinal and pesticidal angiosperm Azadirachta indica. BMC Genomics 13:464. doi:10.1186/1471-2164-13-464
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi:10.1186/gb-2009-10-3-r25
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:1–16. doi:10.1186/1471-2105-12-323
Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi:10.1101/gr.1224503
Misra BB (2014) An updated snapshot of recent advances in transcriptomics and genomics of phytomedicinals. J Postdoc Res 2:1–15. doi:10.14304/surya.jpr.v2n2.1
Qiao J, Zou X, Lai D, Yan Y, Wang Q, Li W, Deng S, Xu H, Gu H (2014) Azadirachtin blocks the calcium channel and modulates the cholinergic miniature synaptic current in the central nervous system of Drosophila. Pest Manag Sci 70:1041–1047. doi:10.1002/ps.3644
Rodrigues M, Festucci-Buselli RA, Silva LC, Otoni WC, Marcelo Festucci-Buselli RA, Silva LC, Otoni WC (2014) Azadirachtin biosynthesis induction in Azadirachta indica A. Juss cotyledonary calli with elicitor agents. Braz Arch Biol Technol 57:155–162. doi:10.1590/S1516-89132014000200001
Sanna G, Madeddu S, Giliberti G, Ntalli NG, Cottiglia F, De Logu A, Agus E, Caboni P (2015) Limonoids from Melia azedarach fruits as inhibitors of flaviviruses and Mycobacterium tubercolosis. PLoS One 10:e0141272. doi:10.1371/journal.pone.0141272
Senthil Nathan S, Kalaivani K, Sehoon K, Murugan K (2006) The toxicity and behavioural effects of neem limonoids on Cnaphalocrocis medinalis (Guenée), the rice leaffolder. Chemosphere 62:1381–1387. doi:10.1016/j.chemosphere.2005.07.051
Singh M, Chaturvedi R (2013) Sustainable production of azadirachtin from differentiated in vitro cell lines of neem (Azadirachta indica). AoB Plants 5:plt034. doi:10.1093/aobpla/plt034
Soares DG, Godin AM, Menezes RR, Nogueira RD, Brito AM, Melo IS, Coura GM, Souza DG, Amaral FA, Paulino TP, Coelho MM, Machado RR (2014) Anti-inflammatory and antinociceptive activities of azadirachtin in mice. Planta Med 80:630–636. doi:10.1055/s-0034-1368507
Srivastava S, Srivastava AK (2013) Production of the biopesticide azadirachtin by hairy root cultivation of Azadirachta indica in liquid-phase bioreactors. Appl Biochem Biotechnol 171:1351–1361. doi:10.1007/s12010-013-0432-7
Sun C, Li Y, Wu Q, Luo H, Sun Y, Song J, Lui EM, Chen S (2010) De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics 11:262. doi:10.1186/1471-2164-11-262
Tan QG, Luo XD (2011) Meliaceous limonoids: chemistry and biological activities. Chem Rev 111:7437–7522. doi:10.1021/cr9004023
Thoh M, Kumar P, Nagarajaram HA, Manna SK (2010) Azadirachtin interacts with the tumor necrosis factor (TNF) binding domain of its receptors and inhibits TNF-induced biological responses. J Biol Chem 285:5888–5895. doi:10.1074/jbc.M109.065847
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi:10.1038/nbt.1621
Veitch GE, Beckmann E, Burke BJ, Boyer A, Maslen SL, Ley SV (2007) Synthesis of azadirachtin: a long but successful journey. Angew Chem Int Ed Engl 46:7629–7632. doi:10.1002/anie.200703027
Authors’ contributions
YW designed, conducted the experiments and wrote the manuscript. XC showed the contribution to analysis the transcriptome data. JS and JW contributed to analysis the chemical components of two Meliaceae plants. HX proofread the manuscript. FT conceived the project. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National 948 Project of China (2014-4-33). All authors also would like to acknowledge the financial support from National forestry public welfare profession scientific research special Project of China (201404601).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
40064_2016_2460_MOESM3_ESM.xlsx
Additional file 3: Table S2. Differentially expressed genes with annotation in the A. indica and M. azedarach libraries.
40064_2016_2460_MOESM6_ESM.pdf
Additional file 6: Fig. S2. Functional categorization of new genes (a) and all genes (b) in A. indica, the unigenes in M. azedarach (c) and the DEGs (d) based on known genes in the GO database.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Y., Chen, X., Wang, J. et al. Comparative analysis of the terpenoid biosynthesis pathway in Azadirachta indica and Melia azedarach by RNA-seq. SpringerPlus 5, 819 (2016). https://doi.org/10.1186/s40064-016-2460-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-2460-6