
Abstract
Background
The Azadirachta indica (neem) tree is a source of a wide number of natural products, including the potent biopesticide azadirachtin. In spite of its widespread applications in agriculture and medicine, the molecular aspects of the biosynthesis of neem terpenoids remain largely unexplored. The current report describes the draft genome and four transcriptomes of A. indica and attempts to contextualise the sequence information in terms of its molecular phylogeny, transcript expression and terpenoid biosynthesis pathways. A. indica is the first member of the family Meliaceae to be sequenced using next generation sequencing approach.
Results
The genome and transcriptomes of A. indica were sequenced using multiple sequencing platforms and libraries. The A. indica genome is AT-rich, bears few repetitive DNA elements and comprises about 20,000 genes. The molecular phylogenetic analyses grouped A. indica together with Citrus sinensis from the Rutaceae family validating its conventional taxonomic classification. Comparative transcript expression analysis showed either exclusive or enhanced expression of known genes involved in neem terpenoid biosynthesis pathways compared to other sequenced angiosperms. Genome and transcriptome analyses in A. indica led to the identification of repeat elements, nucleotide composition and expression profiles of genes in various organs.
Conclusions
This study on A. indica genome and transcriptomes will provide a model for characterization of metabolic pathways involved in synthesis of bioactive compounds, comparative evolutionary studies among various Meliaceae family members and help annotate their genomes. A better understanding of molecular pathways involved in the azadirachtin synthesis in A. indica will pave ways for bulk production of environment friendly biopesticides.
Similar content being viewed by others

Background
Azadirachta indica A. Juss (neem) is an evergreen tree native to the Indian subcontinent[1] belonging to the family Meliaceae and order Rutales. The neem tree is extensively studied for its natural products. Neem oil and its limonoids such as azadirachtin, nimbin, salannin among others are of substantial economic value due to their wide array of applications in agriculture[2], healthcare[3] and soil conservation[4]. Azadirachtin, isolated half a century ago, has been intensively studied and its commercial formulations have been found to be toxic against a large range of insect species, whilst retaining very low mammalian toxicity[5]. Recent success in the total synthesis of azadirachtin[6] has renewed interest towards its commercial exploitation. Neem-derived azadirachtin and other limonoids are also used as anti-proliferative[7], cytotoxic[8–10], larvicidal[11, 12], and anti-inflammatory[7, 13] agents, suggesting the need for better understanding of molecular pathways involved in their synthesis.
In spite of the varied uses of azadirachtin and other neem-derived limonoids, a modern agro-chemical and/or pharmaceutical program focusing on understanding their molecular mechanism(s) of action is yet to be established. A better understanding of the biology of differentiation of secretory cells known to harbor azadirachtin and other triterpenoids[14] may permit development of varieties with higher percentage of these cells in the cotyledons. This information may also be exploited in stalling or delaying further differentiation of these cells to permit greater accumulation of the terpenoids of interest. The elucidation of complete pathways leading to terpenoid biosynthesis and expression of genes involved in such pathways in A. indica will pave ways towards development of newer terpenoid-based biotechnological applications.
De novo sequencing and assembly of the transcriptome from A. indica fruit was reported previously[15]. This report extends the earlier study to cover the whole genome and transcriptomes from root, leaf, stem and flower of neem. Comparative genomics among the Meliaceae family members will be enabled and, cheaper and environment friendly biopesticides may result using results presented in this study.
Results
Genome assembly and similarity with other plant genomes
Sequencing libraries generated using multiple platforms and chemistries were tested to assemble the neem genome and transcriptomes (Table1, Table S1 in Additional file1). Various combinations of sequencing libraries were used to obtain the best scaffold N50s and N90s using SOAPdenovo[16]. The scheme for A. indica genome assembly is given in Figure1a. The de novo assembly of A. indica genome produced a scaffold N50 length of 452,028 bp with corresponding scaffold N90 length of 56,222 bp, contig N50 length of 740 bp and contig N90 length of 172 bp. The genome size of neem was determined to be 364 Mbp using the kmer frequency plot (Figure S1 in Additional file2) representing 95% of the total published genome size[17]. Chargaff's symmetry rule was applied for assessing the quality of the genome assembly[18–21] by comparing the symmetry of 4-mers between the A. indica scaffolds and the Arabidopsis thaliana (A. thaliana) genome. There was a tighter distribution of symmetry in A. indica at around 0.5 (Figure S2 in Additional file2) compared to the A. thaliana suggesting good assembly results. Genome scaffolds (>N50) mapping with Citrus sinensis (C. sinensis) and Citrus clementina (C. clementina) showed conserved macrosynteny between neem and citrus (Figure S3 in Additional file2) but not with chromosomes from A thaliana, Oryza sativa (O sativa), Theobroma cacao (T cacao), Vitis vinifera (V vinifera), Ricinus communis (R communis) and Sorghum bicolor (S. bicolor) (Figure S3 in Additional file2). In A. indica, 385 and 435 scaffolds (>N50) out of a total of 534 scaffolds (>N50) mapped to C. sinensis and C. clementina genome scaffolds (>N50), respectively, with an Expect value of zero. Phylogenetic bootstrap analysis[22] corroborated the taxonomic closeness of A. indica to C. sinensis, as indicated by 100% bootstrap value using rbcL and rbcS (Figure2a). Phylogenetic analysis within the Meliaceae family grouped A. indica with Owenia and Melia sp. with a 100% bootstrap value (Figure2b). Like A. indica, Melia azadirach is known to bear bioactive limonoids[23] suggesting a common evolutionary trajectory with regard to synthesis of these compounds.

Schematic representation of various analytical tools used in A. indica genome and transcriptome assembly, annotation and expression analysis.

Molecular phylogeny of A. indica with plants of other family (a) and within the Meliaceae family (b). Individual rbcL and rbcS alignments (*.phy) were concatenated and bootstrapped using 100 replicates (a) through 'seqboot' in PHYLIP version 3.69[22]. For intra-family phylogeny, rbcL sequences were bootstrapped (b). Phylogenies for each replicate was reconstructed using the 'dnaml' option in Phylip, after choosing either Chlamydomonas reinhardtii (a) or Citrus sinensis (b) as outgroups. The consensus phylogeny with bootstrap values highlighting the branch support was plotted using 'consense' and 'drawgram' options in Phylip.
Repeat analyses
Repeat Modeler (RepeatModeler Open-1.0, 2008–2010)[24] and Repeat Masker (RepeatMasker Open-3.0, 1996–2010)[25] were used to determine the de novo repeats and to perform homology-based repeat analysis respectively among A. indica genome repeats. For repeat analysis, we used consensus repeat libraries (of R.communis[26], Glycine max (G.max)[27] and S. bicolor[28]), in-built plant specific libraries, Repbase[29] and the de novo neem repeat library constructed using Repeat Modeler. In addition, LTR-finder[30], TransposonPSI[31] and MITE-hunter[32] were used to study repeats in the A. indica genome. The neem genome was found to harbour fewer repeat elements compared to other sequenced eudicots with a total non-redundant repeat content of 13.03%. We further classified the repeats into two categories, the interspersed elements (11.21%) comprising of retrotransposons (3.72%) and DNA transposons (0.75%), and the tandem repeats (3.58%) comprising of the low complexity repeats (3.17%), simple repeats (0.05%), and the satellite repeats (0.38%) (Table S2 in Additional file1). The detected interspersed repeat sequences identified using different tools (with similarity of at least 80% and more than 200 nucleotides long) were clustered using Vmatch[33], giving rise to a consensus neem repeat element library of length 23,920,944 bp (~6.59% of the genome).
We studied the insertion age distributions among the LTR-retrotransposons in A. indica made by the Kimura 2-parameter distance matrix[34] and inferred that the oldest insertion event in neem dated back to 13.35 million years (Figure3). The frequency of insertion events peaks around 1 million years ago with smaller secondary peaks observed at ~6 million and ~11 million years ago respectively.

The frequency distribution of LTR-retrotransposon insertion ages in A. indica .
De novo transcriptome assembly
The scheme for transcriptome assembly and annotation is provided in Figure1b. The transcriptomes of different organs were independently assembled using Inchworm (a part of the de novo transcriptome assembler Trinity[35]). Trinity performs better in comparison to other de novo transcriptome assembly programs for a wide range of parameters like genome complexity, read coverage and spliced isoforms[35, 36] and hence was a natural choice for A. indica transcriptome assembly. Chimeric transcripts observed during Inchworm assembly[37] were filtered using PASA (Program to Assemble Spliced Alignments)[38]. The PASA-filtered transcripts were then anchored to the genome scaffolds by assembling overlapped transcript alignments into maximal alignment assemblies in order to remove multiple spliced isoforms.
Transcript annotation and gene prediction analysis
The transcriptome assemblies were annotated using the KEGG Automatic Annotation Server (KAAS)[39]. In addition to the KAAS, the assemblies were serially annotated using; MegaBLAST[40] against the non-redundant nucleotide database, BlastX against the non-redundant protein database, MegaBlast against the RefSeqRNA, Expressed Sequence Tag (EST), Transcriptome Shotgun Assembly (TSA) databases[40] and AutoFACT[41] using uniref90 and uniref100[42], kegg[43], cog[44] and nr[40] databases (Table S3 in Additional file1). In total, 380,629 common and 1,374 unique KEGG annotations (KO terms) across all organs (Figure S4 in Additional file2) were observed. Annotated/predicted genes in A. indica were compared with a non-redundant database containing green plants gene identifier (gi) lists using homology-based searches. As shown in Figure4, a higher percentage of overlap was found between A. indica and V. vinifera, a plant known to express a host of terpernoids[45]. As expected, genes common between A. indica and other species compared (A. thaliana, O. sativa, V. vinifera and in four sequenced organs of A. indica) had the highest number of KEGG annotation associated with them (Figure4). The transcripts from all organs were further assigned Gene Ontology (GO) terms using Blast2GO[46] (Figure S5 in Additional file2 and Table S4 in Additional file1).

Gene annotations and comparison across species. Predicted genes in A. indica were used for homology-based search against the nr database restricting the hits to the green plant gene identifiers. Similar analyses were performed using the annotations in A. thaliana, V. vinifera and O. sativa. The overlapping gi list were used to draw the Venn diagram.
The A. indica RNA-Seq reads were aligned to the genomes or genome scaffolds of multiple plant species. The alignments of A. indica RNA-Seq reads to C. sinensis and C. clementina genome scaffolds by TopHat[47] and CuffLinks[48] suggest sequence similarity between neem and citrus (Table S5 in Additional file1). This is in agreement with the results obtained from our phylogenetic analysis (Figure2a). We built training sets using C. sinensis and C. clementina gene models and used GlimmerHMM[49] to subsequently predict genes in A. indica. Additionally, we used the A. thaliana training sets to represent a distantly related species for gene prediction analysis.
GlimmerHMM[49] predicted greater number of gene models in A. indica using C. sinensis (34,624) and C. clementina (34,737) training sets compared to A.thaliana (23,397). The predicted genes were then serially annotated using Megablast[40] and TblastX[40] (with Expect value of 10-10) resulting in 22,760 and 22,840 annotations with C. sinensis and C. clementina respectively. Statistics for PASA[38] predicted mapped transcripts, gene structures and assembly is provided in Table S6 in Additional file1. The overlap between the GlimmerHMM and PASA predicted gene models was marginally higher using the C. clementina training set compared to the C. sinensis training set (Figure S6 in Additional file2). The genes predicted by GlimmerHMM using C. sinensis training set and training set candidates predicted by PASA identified 10,667, 10,965, 12,209 and 11,181 genes expressed in the root, leaf, stem and flower, respectively (Figure S6c in Additional file2).
Nucleotide composition, transcript expression and gene structure analysis
The nucleotide composition analyses in the transcriptome from all organs suggested different Adenine (A), Thymine (T), and Guanine (G) Cytosine (C) content distribution. The A + T content of most transcripts ranged between 20-40% while for G + C there were two distinct populations of transcripts, one with 0-20% and the other with 20-40% (Figure S7 in Additional file2).
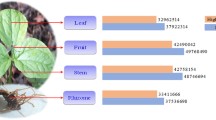
Fragments per transcript kilobase per million fragment (FPKM) values derived from Trinity transcriptome assembly pipeline were used for expression analysis in different organs of neem (Figure1c). The FPKM values for transcripts with overlapping peaks at the same level of expression in all organs were plotted (Figure S8 in Additional file2). The frequency histograms plotted after binning the transcripts suggested maximum density around ~40% GC with 5.0-5.5 log2 (FPKM) values (Figure S9 in Additional file2). In order to avoid erroneous expression analysis resulting from genes with multiple isoforms, the PASA-assembly pipeline were used to remove them and infer relative abundance of transcripts in different organs (Figure5).

Organ-specific expression for un-annotated transcripts. KEGG annotations obtained for transcripts from all organs were parsed to retain only the un-annotated ones. PASA assemblies, establishing correspondence between transcripts and common genomic loci were obtained for the same. The top 300 highly expressed transcripts for each organ were chosen, filtered for redundancies, to yield a total of 888 unique assemblies and plotted.
Post-annotation, GC content comparisons were made between the genome versus transcriptome, introns versus exons and first introns versus first exons (Figure6). The raw genomic reads were relatively GC-poor and the exons were relatively GC-rich (Figure6). The genes those were inferred to accumulate to higher abundances in neem harbored longer introns when compared to the ones in A. thaliana, O. sativa and V. vinifera (Figure7). Although, there was variation in the exon size, similar correlation between the cumulative exon length and expression was observed.

GC density profiles. The %GC compositions are plotted as density curves, (a). for the raw sequencing reads from the neem genome and pooled transcriptome from all organs and (b) for all exons, all introns, first exon and first intron from the gene structures assembled using PASA, corresponding to the pooled organ transcriptome.

Correlation between expression and gene structure. (a) The average intron and exon sizes of the ten most and least expressed genes bearing KEGG Orthology assignments, from A. indica, A.thaliana, O.sativa and V.vinifera are plotted separately for the low and high expression level categories. The error bars indicate standard deviation in the intron and exon sizes respectively. These sizes were obtained from gene structures provided by PASA assemblies mapping these genes to corresponding genome scaffolds. (b) The gene structures depicting the organization and sizes of exons (boxes) and introns (lines) are represented using the web tool StrDraw, for the above mentioned low (left panel) and high (right panel) transcript expression categories in all species.
Comparative analysis of transcript expression
In neem, transcript abundances (TAs) were compared with their orthologs from other species after inter-species normalization of the expression values with elongation factor 1-alpha (EF1A; NCBI gene ID: 836161). Among the 88 genes that were differentially abundant in neem, transcripts from three genes (GGPS, malZ and polygalacturonase) were expressed at a higher level in A. indica compared to the other three species studied (Table S7 in Additional file1). GGPS (geranylgeranyl diphosphate synthase type II, NCBI gene ID: 816377) is involved in terpenoid backbone synthesis, malZ (alpha-glucosidase, NCBI gene ID: 823737) in galactose metabolism and polygalacturonase (NCBI gene ID: 820815) in starch and sucrose metabolism. Unlike GGPS, no other consensus gene was over-expressed to the same magnitude (<10-fold) in neem when compared to the other three species studied. A complete list of the genes and their relative TAs in species is provided in Table S7 in Additional file1.
Terpenoid biosynthesis pathways
The genes related to quinone, terpenoid and terpenoid-backbone synthesis pathways were identified by KEGG’s KAAS automatic pathway annotation pipeline[39]. GGPS (NCBI gene ID: 816377), COQ6 (NCBI gene ID: 822006) and CLA1 (NCBI gene ID: 827230) were among the top 5 differentially expressed genes in neem leaf compared to the other organs (Figure8). Eight genes (TPS21, NCBI gene ID: 832461; lytB/ispH, NCBI gene ID: 829585; ispE, NCBI gene ID: 817234; GGPS, NCBI gene ID: 816377; +neomenthol dehydrogenase, NCBI gene ID: 825294; FDPS, NCBI gene ID: 827430; FDFT1, NCBI gene ID: 829616 and SQLE, NCBI gene ID: 816814) involved in the synthesis of sesquiterpenes and triterpenes leading to azadirachtin-A were over-expressed in neem compared to A. thaliana, O. sativa, C. sinensis and V. vinifera, (Figure8c). The length of introns in the terpenoid and steroid biosynthesis gene families in A. indica were greater compared to the ones in A. thaliana, O. sativa, C. sinensis and V. vinifera (Figure S10 in Additional file2).

Genes involved in the terpenoid biosynthesis pathways. Organ-wide transcript abundance heat-maps were drawn for top (a) and bottom (b) five transcripts in each organ. The clustering and heat-map analyses were done using R. Differential transcript abundance were calculated between terpenoid pathway related genes expressed in A. indica leaf to those in leaves of A. thaliana, C. sinensis and O. sativa, and V. vinifera, after normalizing the transcript abundance of all genes to that of elongation factor 1-alpha (EF1A). The differential indices were further log2-transformed and represented as cumulative bar histograms for all species (c).
Discussion
The current study describes the draft genome and transcriptomes from root, stem, leaf and flower. Both neem and citrus belong to the Order Rutales and our phylogenetic studies reaffirmed their taxonomic closeness. Additionally, phylogenetic studies grouped A. indica with the Melia species, one that is also known to harbour bioactive compounds (Figure2b) suggesting a common evolutionary process with regard to synthesis of these compounds in Meliaceae. Repeat analysis showed low repeat content in A. indica genome compared with other sequenced angiosperms. This could have been due to the presence of xenobiotic terpenoids specific to the plant, which might have been a major impediment for horizontal gene transfer[50]. This impediment in horizontal gene transfer could have skewed the conventional host-pathogen interactions countering the accumulation of repeats in A. indica compared to other contemporary angiosperms. Insertion times of LTR elements in neem suggest a major wave of retro-transposition about 1million years ago (Figure3), similar to the observation made in S. bicolor[28]. However, in A. indica the oldest insertion event date back much further than S. bicolor (13.35 million years in neem against 3 million years in Sorghum, Figure3). Monocot genomes have higher GC content than dicots, which is reflected in an average difference in codon usage between them[51, 52]. Interestingly, like O. sativa[53] but unlike A. thaliana and others[54], the A. indica genome shows a bimodal distribution of GC content (Figure S11 in Additional file2). Domesticated crop genomes with higher GC content are likely to have evolved by a relaxation of a natural selection process against higher nitrogen use in DNA due to the use of nitrogen-rich fertilizers[55]. A. indica, a hardy plant, is not domesticated and can grow in poor and degraded soil including in semi-arid conditions with tolerance for high temperature[56]. Additionally, there is no history of any usage of nitrogen rich fertilizers to grow A. indica. This might have played a role in shaping the AT-rich nucleotide composition of A. indica genome.
Transcript abundances of genes in A. indica correlated well with their functions in respective organs; for example, root, leaf, stem and flower showed the presence of highly expressed ones involved in ion homeostasis, photosynthesis, ion transport and ATPase activity, respectively. Unlike the genome, A. indica transcriptomes exhibited unimodal GC distribution roughly overlapping with the higher GC peak of the genome (Figure S11 in Additional file2). The distribution of A + T in A. indica transcriptomes mapped closer to the A. thaliana transcriptome than to the ones in O. sativa or V. vinifera (Figure S11 in Additional file2). The lengths and the GC content of the first exons and first introns are reported to be associated with functional elements in other large genomes[57]. The GC content of the first exons and first introns in A. indica were similar to that of other exons and introns in the genome (Figure7). Highly expressed primary transcripts of A. indica bear longer intron structures suggesting their possible functional role in expression and conforming with the results shown in other plants[58] and yeast[59].
Sequencing and analysis of the genome and transcriptomes of neem holds significance due to the plant’s utility in agriculture and medicine. The use of botanical products as pest control and deterrence has acquired greater significance with the shifting trends in mainstream agriculture towards sustainable and organic farming. Neem provides a suitable option for developing such eco-friendly and sustainable pesticides[56]. Currently a very small percentage of farmers use neem-based products as a substitute to synthetic pesticides as the general awareness of such practices remains limited. Azadirachtin, derived from neem seed kernels, has been proved to be a potent and effective pesticide. Total chemical synthesis of azadirachtin is challenging owing to the low yield of synthesis and remains a bottleneck for commercial bulk manufacturing. Secondary metabolites synthesized by terpene synthases are the major components of essential oils and are of great economic value. In plants, synthesis of terpenes is compartmentalized into monoterpenes and diterpenes (as well as carotenoids and chlorophylls) that are produced via the 1-deoxy-d-xylulose-5-phosphate (DXP) pathway in the plastids while the sesquiterpenes and triterpenes are made in the cytosol via the mevalonate pathway[60]. The enzymes involved in the synthesis of azadirachtin have not been studied in detail. The pathway hierarchy in neem-derived tetranortriterpenoid azadirachtin-A primarily requires the synthesis of a terpenoid backbone followed by steroid and triterpenoid biosynthesis. We found that the genes in the terpenoids and steroid biosynthesis family (TPS21, lytB, ispH, ispE, GGPS, +neomenthol dehydrogenase, FDPS, FDFT1 and SQLE) were more abundant in neem compared to A. thaliana, O. sativa, C. sinensis and V. vinifera.
This study may help close the gap between traditional knowledge and current practices in the agrochemical industry. The progressive increase in the cost of petroleum-based starting materials has led to a surge in the price of synthetic pesticides scourging the meager profit margins presented to the farmers in developing countries. This study may provide a cost-effective alternative by aiding biotechnological research efforts in enhancing disease resistance in plants.
Conclusions
The de novo sequencing and analyses of the draft genome and organ-specific transcriptomes of neem plant, Azadirachta indica is reported. A. indica is the first Meliaceae family member to be sequenced. The neem genome bears fewer repetitive elements compared to other sequenced higher plants. It has about 20,000 genes with an average transcript length of 1.69 kbp. A. indica’s evolutionary closeness to Citrus species was verified by both molecular phylogenetic analyses and sequence similarity. Transcript expression and the exon-intron junction architecture of underlying genes involved in the terpenoid biosynthesis pathways suggested relative abundance of enzymes involved in the azadirachtin synthesis in neem. Genes involved in the terpenoid biosynthesis pathways in neem bear longer introns compared to the same genes in A. thaliana, O. sativa, V. vinifera and C. sinensis.
Methods
Sample collection, identification, nucleic acids extraction and quality control
Different parts of Azadirachta indica A. Juss were collected from a locally grown tree. The genus and species was confirmed at the South Regional Centre, Botanical Survey of India, Tamil Nadu Agricultural University Campus, Coimbatore, Tamil Nadu, India using the herbarium of the twigs bearing flowers and fruits. All parts of the plant (root, leaf, stem, and flower) were collected and immediately flash frozen in liquid nitrogen and stored at -80°C until further use. Total genomic DNA was extracted using Qiagen plant genomic DNA extraction kit. Total RNA was extracted using Plant Total RNA extraction kit (Bioteke, China). The quality, quantity of genomic DNA and integrity of total RNA were accessed using Nanodrop, Qubit methods and Agilent Bioanalyzer RNA 6000 Nano chip respectively (Text S1 in Additional file3) before using the DNA and RNA in making sequencing libraries.
Sequencing library preparation
Solexa sequencing-by-synthesis
Short-insert paired-end sequencing libraries were prepared using Illumina (San Diego, California, USA) TruSeq library preparation kit with one modification. The library was not amplified post-adapter ligation to minimize amplification-related bias and the un-amplified, adapter-ligated library was directly used to generate clusters on cBOT instrument following the manufacturer’s recommendation. Long-insert mate pair sequencing libraries were prepared using multiple inserts (10 kbp, 3 kbp and 1.5 kbp) using Illumina mate pair library preparation kit. RNA-seq sequencing libraries from all the four organs were prepared using Illumina TruSeq-RNA library prep kit following the manufacturers instructions. Details of the methods including the QC of samples and sequencing libraries are provided in the Text S1 in Additional file3.
Pyro-sequencing using IonTorrent Personal Genome Machine (PGM)
Genomic DNA of neem was sheared to 200 bp using Covaris (Woburn, Massachusetts, USA) to make libraries using IonTorrent PGM protocol. Sequencing was performed following the manufacturer’s instructions. Details of the methods including the QC of libraries are provided in the Text S1 in Additional file3.
Cloning of neem genomic DNA
Genomic DNA (2-11 kbp) was cloned into pJAZZ-OC linear cloning vector using BigEasy v2.0 kit from Lucigen (Middleton, Wisconsin, USA) following manufacturer’s specifications. Following cloning, positive colonies were picked and sub-cultured. Plasmid DNA was isolated and checked for inserts by restriction digestion (Text S1 in Additional file3). Positive clones were selected and used for capillary sequencing.
Capillary sequencing
Neem genomic DNA clones were used for capillary sequencing using BigDye Terminator kit using the manufacturer’s instructions. Positive clones were selected and used for sequencing in either forward or reverse direction using M13 primers using the Applied Biosystems 3500 instrument (Text S1 in Additional file3).
De novo genome and transcriptome assembly
Two sets of Illumina short insert (150 bp and 350 bp) paired-end libraries and three long insert (1.5 kbp, 3 kbp and 10 kbp) mate-pair libraries, capillary Sanger sequencing reads generated from cloned neem genomic DNA and pyro-sequencing reads generated with IonTorrent Personal Genome Machine (Table S1 in Additional file1) were used in assembling the neem genome with SOAPdenovo[16]. We assembled the neem transcriptomes from all organs using the raw RNA-seq paired reads from four organs: root, leaf, stem and flower using the genome-independent transcriptome assembler Trinity[35].
Phylogenetic analyses
The concatenated alignment of either the rbcL and rbcS genes of 24 plant species including neem or rbcL gene alone for Meliaceae family members were used to generate a bootstrapped phylogeny using Phylip v3.69[22]. For reporting the phylogenetic analysis results, we followed the “Minimal Information About a Phylogenetic Analysis” (MIAPA) standard[61]. Detail attributes and the actual files are provided in Additional files4,5 and6.
Repeat identification and analyses
Repeat Modeler (RepeatModeler Open-1.0, 2008–2010)[24] Repeat Masker (RepeatMasker Open-3.0, 1996–2010)[25], LTR_Finder[30], TransposonPSI[31] and MITE-Hunter[32] were used to detect, identify and characterize repeats in the neem genome. Vmatch[33] was used to cluster repeat sequences and build a consensus (>80% similar) neem repeat library.
Calculation of LTR insertion age
5' and 3' LTR sequences of each LTR-retrotransposon, identified by LTR finder[30], were aligned using ClustalW MPI[62]. The distances between 5' and 3' LTR sequences for each alignment were calculated using the Kimura 2-parameter distmat tool from EMBOSS package[63]. The insertion ages were further calculated from these distance values according to the formula[28] T = K / (2r), where T is the insertion age in years, K = Kimura distance value and r is the substitution rate per site per year (taken to be 1.3 × 10-8 as found in O. sativa[64].
Nucleotide composition and expression analyses
The nucleotide composition of the transcripts from the neem organs and genome scaffolds/chromosomes of the neem, A. thaliana, O. sativa and V. vinifera were plotted as frequency histograms and density curves. The fragments per transcript kilobase per million fragments (FPKM) provided by Trinity were log2 transformed and plotted as frequency histograms for each organ, before and after categorizing the transcripts based on their G + C composition.
Transcript annotation, gene prediction and inter-species comparison analyses
The transcripts were serially annotated for each organ using similarity-based BLAST analyses (Text S1 in Additional file3) and functionally classified using BLAST2GO analyses. Gene predictions were performed using GlimmerHMM[49] and PASA[38]. Inter-species gene comparison was performed as described in Text S1 in Additional file3.
Organ-specific transcript expression pathway analyses
The transcripts for individual organs in neem were mapped to KEGG[43, 65–67] pathways using the KAAS server. The KO-assigned transcripts were divided into low and high expression categories. A heat-map of these across the four organs was computed using R. Analyses using KEGG were also carried out for other species, namely A. thaliana, O. sativa, V. vinifera and C. sinensis, focusing on the enzymes mapping to the azadirachtin-A biosynthesis pathway. The differential transcript expression level indices of these enzymes in neem relative to other species were compared. The gene structures for these enzymes, and the structures of the top and bottom ten transcripts ranked according to their expression levels were compared across all species.
KEGG annotations obtained for transcripts from all organs were parsed to retain only the un-annotated ones. PASA assemblies, establishing correspondence between each organ's transcripts and common genomic loci were obtained for the same. The top 300 highly expressed un-annotated transcripts for each organ were chosen, filtered for redundancies, to yield a total of 888 unique assemblies. A heat-map was plotted using the log2 (FPKM) values of these transcripts using R.
Accession number
Genome and transcriptome sequence data described in this report are submitted to the NCBI Short Read Archive database [SRA 053330].
Authors contribution
BP conceived, planned, designed and directed the project; NMK and SP designed the analytical workflow and guided the process; NMK, SP, PJ, PG, RC, and SV analyzed the data, DSA, AKH and BK produced the Solexa Illumina sequencing data. LV, JN produced the capillary sequencing data; NV, KD and KR produced and helped in the generation of pyro-sequencing data, BK and JN performed the neem genomic DNA cloning experiment; NMK, SP and BP wrote the manuscript. All authors read and approved the final manuscript.
References
Schmutterer H: The neem tree : Azadirachta indica A. Juss. and other meliaceous plants : sources of unique natural products for integrated pest management, medicine, industry, and other purposes. Neem Foundation. 2002
Brahmachari G: Neem–an omnipotent plant: a retrospection. Chembiochem : a European journal of chemical biology. 2004, 5 (4): 408-421. 10.1002/cbic.200300749.
Osujih M: Exploration of the frontiers of tradomedical practices: basis for development of alternative medical healthcare services in developing countries. J R Soc Health. 1993, 113 (4): 190-194. 10.1177/146642409311300409.
Saxena RC: In Neem: Today and in the New Millenium. 2004, Dordrecht: Kluwer Academic Publishers
Ley SV: Synthesis and chemistry of the insect antifeedant azadirachtin. Pure & Appl Chem. 1994, 66 (10-11): 4-
Veitch GE, Boyer A, Ley SV: The azadirachtin story. Angew Chem Int Ed Engl. 2008, 47 (49): 9402-9429. 10.1002/anie.200802675.
Schumacher M, Cerella C, Reuter S, Dicato M, Diederich M: Anti-inflammatory, pro-apoptotic, and anti-proliferative effects of a methanolic neem (Azadirachta indica) leaf extract are mediated via modulation of the nuclear factor-kappaB pathway. Genes & nutrition. 2011, 6 (2): 149-160. 10.1007/s12263-010-0194-6.
Harish Kumar G, Chandra Mohan KV, Jagannadha Rao A, Nagini S: Nimbolide a limonoid from Azadirachta indica inhibits proliferation and induces apoptosis of human choriocarcinoma (BeWo) cells. Investigational new drugs. 2009, 27 (3): 246-252. 10.1007/s10637-008-9170-z.
Akudugu J, Gade G, Bohm L: Cytotoxicity of azadirachtin A in human glioblastoma cell lines. Life Sci. 2001, 68 (10): 1153-1160. 10.1016/S0024-3205(00)01017-1.
Roy MK, Kobori M, Takenaka M, Nakahara K, Shinmoto H, Isobe S, Tsushida T: Antiproliferative effect on human cancer cell lines after treatment with nimbolide extracted from an edible part of the neem tree (Azadirachta indica). Phytotherapy research : PTR. 2007, 21 (3): 245-250. 10.1002/ptr.2058.
Okumu FO, Knols BG, Fillinger U: Larvicidal effects of a neem (Azadirachta indica) oil formulation on the malaria vector Anopheles gambiae. Malar J. 2007, 6: 63-10.1186/1475-2875-6-63.
Denardi SE, Bechara GH, de Oliveira PR, Camargo Mathias MI: Inhibitory action of neem aqueous extract (Azadirachta indica A. Juss) on the vitellogenesis of Rhipicephalus sanguineus (Latreille, 1806) (Acari: Ixodidae) ticks. Microsc Res Tech. 2011, 74 (10): 889-899. 10.1002/jemt.20973.
Thoh M, Kumar P, Nagarajaram HA, Manna SK: Azadirachtin interacts with the tumor necrosis factor (TNF) binding domain of its receptors and inhibits TNF-induced biological responses. J Biol Chem. 2010, 285 (8): 5888-5895. 10.1074/jbc.M109.065847.
Johnson S, Morgan ED, Peiris CN: Development of the major triterpenoids and oil in the fruit and seeds of neem (Azadirachta indica). Ann Bot. 1996, 78: 6-
Krishnan NM, Pattnaik S, Deepak SA, Hariharan AH, Gaur P, Chaudhary R, Jain P, Vaidyanathan S, Krishna B, Panda B: De novo sequencing and assembly of Azadirachta indica fruit transcriptome. Curr Sci (India). 2011, 101 (12): 1553-1561.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al: De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20 (2): 265-272. 10.1101/gr.097261.109.
Ohri D, Bhargava A, Chatterjee A: Nuclear DNA amounts in 112 species of tropical hardwoods – new estimates. Plant biology. 2004, 6 (5): 555-561. 10.1055/s-2004-821235.
Yamagishi MEB, Hirai RH: Chargaff’s “Grammar of Biology”: New Fractal-like Rules. 2011, arXiv
Sanchez J, Jose MV: Analysis of bilateral inverse symmetry in whole bacterial chromosomes. Biochem Biophys Res Commun. 2002, 299 (1): 126-134. 10.1016/S0006-291X(02)02583-4.
Forsdyke DR: Symmetry observations in long nucleotide sequences: a commentary on the Discovery Note of Qi and Cuticchia. Bioinformatics. 2002, 18 (1): 215-217. 10.1093/bioinformatics/18.1.215.
Prabhu VV: Symmetry observations in long nucleotide sequences. Nucleic Acids Res. 1993, 21 (12): 2797-2800. 10.1093/nar/21.12.2797.
Felsenstein J: PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics. 1989, 5: 164-166.
Tan QG, Luo XD: Meliaceous limonoids: chemistry and biological activities. Chem Rev. 2011, 111 (11): 7437-7522. 10.1021/cr9004023.
Repeat Modeller.http://www.repeatmasker.org,
Repeat Masker.http://www.repeatmasker.org,
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, et al, et al: Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010, 28 (9): 951-956. 10.1038/nbt.1674.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, et al: Genome sequence of the palaeopolyploid soybean. Nature. 2010, 463 (7278): 178-183. 10.1038/nature08670.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al, et al: The Sorghum bicolor genome and the diversification of grasses. Nature. 2009, 457 (7229): 551-556. 10.1038/nature07723.
Jurka J: Repbase update: a database and an electronic journal of repetitive elements. Trends in genetics : TIG. 2000, 16 (9): 418-420. 10.1016/S0168-9525(00)02093-X.
Xu Z, Wang H: LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35: W265-268. 10.1093/nar/gkm286. Web Server issue
TransposonPSI.http://transposonpsi.sourceforge.net,
Han Y, Wessler SR: MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38 (22): e199-10.1093/nar/gkq862.
Vmatch.http://www.vmatch.de,
Kimura M: A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980, 16 (2): 111-120. 10.1007/BF01731581.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29 (7): 644-652. 10.1038/nbt.1883.
Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P: Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinforma. 2011, 12 Suppl 14: S2-
van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, Page JE: The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12 (10): R102-10.1186/gb-2011-12-10-r102.
Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, et al, et al: Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31 (19): 5654-5666. 10.1093/nar/gkg770.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M: KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35: W182-185. 10.1093/nar/gkm321. Web Server issue)
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215 (3): 403-410.
Koski LB, Gray MW, Lang BF, Burger G: AutoFACT: an automatic functional annotation and classification tool. BMC Bioinforma. 2005, 6: 151-10.1186/1471-2105-6-151.
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH: UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007, 23 (10): 1282-1288. 10.1093/bioinformatics/btm098.
Kanehisa M, Goto S: KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28 (1): 27-30. 10.1093/nar/28.1.27.
Tatusov RL, Galperin MY, Natale DA, Koonin EV: The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28 (1): 33-36. 10.1093/nar/28.1.33.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, et al: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449 (7161): 463-467. 10.1038/nature06148.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21 (18): 3674-3676. 10.1093/bioinformatics/bti610.
Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 25 (9): 1105-1111. 10.1093/bioinformatics/btp120.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L: Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010, 28 (5): 511-515. 10.1038/nbt.1621.
Majoros WH, Pertea M, Salzberg SL: TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004, 20 (16): 2878-2879. 10.1093/bioinformatics/bth315.
Richardson AO, Palmer JD: Horizontal gene transfer in plants. J Exp Bot. 2007, 58 (1): 1-9.
Fennoy SL, Bailey-Serres J: Synonymous codon usage in Zea mays L. nuclear genes is varied by levels of C and G-ending codons. Nucleic Acids Res. 1993, 21 (23): 5294-5300. 10.1093/nar/21.23.5294.
Carels N, Bernardi G: Two classes of genes in plants. Genetics. 2000, 154 (4): 1819-1825.
Wang HC, Hickey DA: Rapid divergence of codon usage patterns within the rice genome. BMC Evol Biol. 2007, S6-7 Suppl 1
Banerjee T, Gupta SK, Ghosh TC: Compositional transitions between Oryza sativa and Arabidopsis thaliana genes are linked to the functional change of encoded proteins. Plant Sci. 2000, 170 (2): 7-
Acquisti C, Elser JJ, Kumar S: Ecological nitrogen limitation shapes the DNA composition of plant genomes. Mol Biol Evol. 2009, 26 (5): 953-956. 10.1093/molbev/msp038.
Morgan ED: Azadirachtin, a scientific gold mine. Bioorg Med Chem. 2009, 17 (12): 4096-4105. 10.1016/j.bmc.2008.11.081.
Kalari KR, Casavant M, Bair TB, Keen HL, Comeron JM, Casavant TL, Scheetz TE: First exons and introns–a survey of GC content and gene structure in the human genome. In silico biology. 2006, 6 (3): 237-242.
Ren XY, Vorst O, Fiers MW, Stiekema WJ, Nap JP: In plants, highly expressed genes are the least compact. Trends in genetics : TIG. 2006, 22 (10): 528-532. 10.1016/j.tig.2006.08.008.
Vinogradov AE: Intron length and codon usage. J Mol Evol. 2001, 52 (1): 2-5.
Kirby J, Keasling JD: Biosynthesis of plant isoprenoids: perspectives for microbial engineering. Annual review of plant biology. 2009, 60: 335-355. 10.1146/annurev.arplant.043008.091955.
Leebens-Mack J, Vision T, Brenner E, Bowers JE, Cannon S, Clement MJ, Cunningham CW, dePamphilis C, deSalle R, Doyle JJ, et al, et al: Taking the first steps towards a standard for reporting on phylogenies: Minimum Information About a Phylogenetic Analysis (MIAPA). Omics : a journal of integrative biology. 2006, 10 (2): 231-237. 10.1089/omi.2006.10.231.
Thompson JD, Gibson TJ, Higgins DG, et al: Multiple sequence alignment using ClustalW and ClustalX. Current protocols in bioinformatics. Edited by: Baxevanis AD. 2002, Chapter 2:Unit 2 3
Ma J, Bennetzen JL: Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004, 101 (34): 12404-12410. 10.1073/pnas.0403715101.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38 (Database issue): D355-360.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M: The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Database issue): D277-280.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M: KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40 (1): D109-114. 10.1093/nar/gkr988.
Acknowledgements
The study is financed by grants from the Department of Information Technology, Government of India (Ref no:18(4)/2010-E-Infra., 31-03-2010) and the Department of Information Technology, Biotechnology and Science & Technology, Government of Karnataka, India (Ref no:3451-00-090-2-22). We thank Prof. N. Yathindra for encouragement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Authors declare no competing interests.
Neeraja M Krishnan, Swetansu Pattnaik contributed equally to this work.
Electronic supplementary material
12864_2012_4404_MOESM1_ESM.pdf
Additional file 1: Table S1. Description of sequencing read libraries used for genome and transcriptome assembly. Table S2. Classification of repeats in the neem genome. Table S3. Transcript annotations from similarity-based analyses. Table S4. Mapping neem RNA-Seq reads to other plant genomes. Table S5. Transcript mapping statistics using PASA. Table S6. Gene ontology based functional categorization of annotated transcripts. Table S7. Expression levels of genes associated with metabolism in neem compared to other plant species. (PDF 446 KB)
12864_2012_4404_MOESM2_ESM.pdf
Additional file 2: Figure S1. kMer frequency curve. Figure S2. Frequency histograms for symmetric 4-mers. Figure S3. Genome scaffold mapping between neem and other species. Figure S4. Gene ontology based functional categorization of annotated transcripts. Figure S5. Expression density profiles of neem organ transcriptomes. Figure S6. Gene prediction statistics. Figure S7. GC-correlated expression density profiles of neem organ transcriptomes. Figure S8. AT and GC density profiles in genomes and transcriptomes of neem and other plant species. (PDF 4 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Krishnan, N.M., Pattnaik, S., Jain, P. et al. A draft of the genome and four transcriptomes of a medicinal and pesticidal angiosperm Azadirachta indica. BMC Genomics 13, 464 (2012). https://doi.org/10.1186/1471-2164-13-464
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-464