
Abstract
Capecitabine, a fluoropyrimidine prodrug, has been a frequently chosen ligand for the last one and half decades to inhibit thymidylate synthase (TYMS) for treatment of colorectal cancer. TYMS is a key enzyme for de novo synthesis of deoxythymidine monophosphate and subsequent synthesis of DNA. Recent years have also seen the trait of modifying ligands using halogens and trifluoromethyl (–CF3) group to ensure enhanced drug performance. In this study, in silico modification of capecitabine with Cl, Br, I atoms and –CF3 group has been performed. Density functional theory has been employed to optimize the drug molecules and elucidate their thermodynamic and electrical properties such as Gibbs free energy, enthalpy, electronic energy, dipole moment and frontier orbital features (HOMO–LUMO gap, hardness and softness). Flexible and rigid molecular docking have been implemented between drugs and the receptor TYMS. Both inter- and intra-molecular non-covalent interactions involving the amino acid residues of TYMS and the drug molecules are explored in details. The drugs were superimposed on the resolved crystal structure (at 1.9 Å) of ZD1694/dUMP/TYMS system to shed light on similarity of the binding of capecitabine, and its modifiers, to that of ZD1694. Together, these results may provide more insights prior to synthesizing halogen-directed derivatives of capecitabine for anticancer treatment.
Similar content being viewed by others
Background
Thymidylate synthase (TYMS) is a homodimeric protein with identical subunits (molecular weight for each unit ~35 kDa) and one of the most conserved protein entities in nature (Chu et al. 1991; Phan et al. 2001). It has long been well-known as a drug target, notably of 5-fluoro uracil (5-FU) and the anti-folate raltitrexed, for treatments of colorectal cancer (Chu et al. 1991; Phan et al. 2001; Costi et al. 2005). Both subunits of TS contribute to each of the two active sites; however, it shows “half-of-the-site activity” i.e. conformational switching between active and inactive states, thus only one subunit being active at a time (Świniarska et al. 2010; Luo et al. 2011). It is a key enzyme in folate metabolic pathway—thereby an essential precursor for biosynthesis of DNA, RNA and protein (Arooj et al. 2013; Hardy et al. 1987). TYMS catalyses the de novo pathway for the production of deoxythymidine monophosphate (dTMP), one of the three nucleotides which form thymine (a nucleic acid in DNA) and dihydrofolate from deoxyuridine monophosphate (dUMP) and 5,10-methylenetetrahydrofolate (mTHF) (Chu et al. 1991; Peters et al. 2002; Salo-Ahen and Wade 2011). The process involves a cycle, where dietary folate is reduced to dihydrofolate and then to tetrahydrofolate (THF) by dihydrofolate reductase while D2NADPH supplying necessary hydrogen. Serine transhydroxymethylase converts THF to mTHF. Meanwhile, dUMP binds to a receptor site of TYMS—prompting a configurational change to create a binding site for mTHF. Transfer of a methyl group to the uridine ring results in the formation of dihydrofolate and dTMP (Danenberg and Danenberg 1978; Santi et al. 1974). Because of its pivotal role in synthesis of DNA, TYMS remains one of the important target proteins in cancer chemotherapy.
Capecitabine (N 4-pentyloxycarbonyl-5-deoxy-5-fluorocytidine) is a fluoropyrimidine-based novel oral prodrug designed by Miwa et al. This drug received the approval by FDA (US Food and Drug Administration) in 1998 and has been in use as a ligand for TYMS inhibition. A prodrugis an inactive chemical derivative of an active drug molecule which, after being administered specifically into the target cells or tumors, is converted to the desired form and ensures improved bioavailability. When delivered orally, capecitabine is absorbed through the intestinal wall of cells and converted to 5′-deoxy-5-fluorouridinein a three-step sequential enzymatic pathway, which includes the last-step tumor selective reaction catalyzed by the tumor-associated angiogenic factor thymidine phosphorylase—eventually leading to transformation into 5-fluorouracil (5-FU). 5-FU has been known as the mainstream antifolate that inhibits TYMS. Entering the cell, 5-FU is converted to 5-fluorodeoxyuridine monophosphate (FdUMP) which, mutually with the methyl donor-reduced folate, forms a covalent stable ternary complex with TYMS, thus preventing thymidine synthesis in cells. In addition, its alteration into fluorouridine triphosphate (FUTP) and fluorodeoxyuridine triphosphate (FdUTP) disrupts the function of RNA and DNA in tumor cells respectively (Stella et al. 1985; Papamichael 2000; Etienne et al. 2004; Longley et al. 2003; Park et al. 2002; Miwa et al. 1998; Ishikawa et al. 1998). The drawback of direct administration of 5-FU due to primary and secondary resistance and its potential toxicity to normal, non-tumored cells, however, has been a matter of concern. 5-FU undergoes rapid metabolic change by dihydropyrimidine dehydrogenase (DPD) in the mucosa of the gastrointestinal tract and the liver—limiting its oral bioavailability. In addition, adverse side-effects during its application—diarrhea, nausea and cardiovascular complexities for instance, have been reported (Mader et al. 1998; Saif 2009; Abou and Fadl 2009). Contrariwise, capecitabine is well-tolerated and enhances drug concentration at the tumor site, avoids complication associated with venous access and reduces cytotoxicity. Infact, stage III trials for treatment of colorectal cancer using capecitabine have been undertaken and shown markedly superior results, compared to 5-FU, with improved safety profiles (Miwa et al. 1998; Scheithauer et al. 2003; Schmoll and Arnold 2006).
In governing the mechanisms responsible for diverse ligand–protein complex systems such as the one mentioned above, non-covalent intermolecular forces play a pivotal role. Elucidation and quantification of non-covalent forces are central to pharmaceutical drug-design and lead optimization. Important non-covalent interactions include hydrogen bonding, electrostatic interactions, stacking interactions (cation–pi, anion–pi or pi–pi), van der Walls forces, and hydrophobic interactions (Meyer et al. 2003; Cerný and Hobza 2007; Dougherty 2007; Wheeler and Bloom 2014; Wheeler 2011; Varma et al. 2010). Recent years have witnessed the importance of non-covalent interactions involving halogen atoms, otherwise termed as halogen bonding, due to their intriguing chemical features. Halogen bonds have been identified in many of biological low-mass compounds and in complexes of biomolecules with halogenated ligands. Halogen atoms can either function as electrophilic species or as nucleophilic Lewis bases and display anisotropic charge distribution along the region of a C–X bond with an equatorial distribution of negative and positive charges—thereby creating what is called a σ-hole. A σ-hole maintains a diminished electron density site and entices an electronegative site of another molecule to interact. Such cases occur, especially those with larger atomic radii (chlorine, bromine and iodine) (Lu et al. 2009, 2012; Sirimulla et al. 2013). On the other hand, drug candidates containing fluorine show increased metabolic stability and membrane permeation, hence use of them has become commonplace. The small-size and very high electronegativity of fluorine attribute to its eccentric behaviors, which in turn contribute to its versatile interactions in biomolecular receptor-ligand moieties (Hagmann 2008; Smart 2001; Zhou et al. 2009).
This in silico study focuses on quantum mechanical and molecular docking analysis of thymidylate synthase (TYMS) against capecitabine and its halogenated derivatives, modified by trifluoromethane (–CF3), Cl, Br and I, to investigate compelling non-covalent interactions in order to attain a deeper understanding and interpret the core characteristics of such receptor-ligand synergy. In-silico approach, computer-aided drug design (CADD) in other words, has brought about a major revolution to facilitate the design and discovery of novel therapeutic solutions (Kapetanovic 2008). CADD has been utilized in diverse ways—choosing the most exact ligands and their target proteins from digital repositories such as DrugBank (Wishart et al. 2006), PubChem (Wang et al. 2009) or PDB (Berman et al. 2002), using softwares adept in sophisticated quantum mechanical calculations and 3-D computer graphics to model and/or manipulate potential inhibitors, carrying out molecular docking in order to foresight energetically favorable binding sites and interactions among the lead-candidates and target molecules with acute physicochemical and pharmaceutical details (Koutsoukas et al. 2011; Kapetanovic 2008; Tang et al. 2006; Song et al. 2009; Joseph-Mccarthy 1999). Discovery and marketing of a drug on average require around 7–12 years of research and cost from US$800 million to US $1.2 billion; however, CADD has been successful in minimizing the lengthy trial-and-error research cycle and gigantic costs involving drug discovery (Kapetanovic 2008; Tang et al. 2006). In this work, thermo-chemical and molecular orbital calculation for the drug candidates were performed. Each of the ligand molecules was subjected to flexible and rigid docking to elucidate binding energies, binding sites and characteristics of various non-bonding interactions among those complex receptor–ligand environments. In addition, the results have been compared to the previously determined crystal structure of ZD1694–dUMP–TYMS complex at 1.9 A resolution to identify the degree of superposition of the presently observed systems to that of a resolved one (Phan et al. 2001).
Computational methods
Optimization of drugs using quantum mechanical calculations
Quantum mechanical calculations were performed using Gaussian 09 program-suit (Gaussian 09 Revision 2009). The structures of capecitabine was fully optimized utilizing density functional theory employing B3LYP/MidiX level of theory. The basis set is initially developed from the Huzinaga MIDI basis and more compatible for molecules with halogen atoms (Easton et al. 1996). The initial structure of capecitabine was manipulated replacing fluorine with –CF3, Cl, Br and I; the modified compounds, labeled from D1–D4 sequentially (Figs. 1, 2), were then optimized using the same level of theory. Internal electronic energy, enthalpy, Gibbs free energy and dipole moment were investigated for each of the molecules. Energetics data of a system exploiting Gaussian 09 are produced by solving traditional thermodynamic and quantum mechanical equations. Contributions from each of translational, rotational, vibrational and electronic motions are considered to estimate the thermodynamic parameters of a system. Sum of electronic and thermal energies in a molecule is represented by the following equation (Gaussian 09 Revision 2009)
where εo is the total electronic energy for a given system and E tot is the correction to internal thermal energy when E tot = E t + E r + E v + E e. Similarly,
where H corr and G corr are the thermal corrections for enthalpy and Gibbs free energy for the given system respectively.

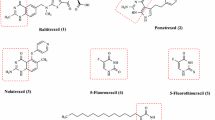
Optimized structure of capecitabine and its halogenated derivatives (D1) generated at B3LYP/MidiX level of theory

Optimized structure of halogenated derivatives (D2, D3 and D4) generated at B3LYP/MidiX level of theory
Frontier molecular orbital calculations were also conducted using the same level of theory. Considering the correlation of ionization potential (I) with HOMO and electron affinity (E) with LUMO according to Koopmans theorem, hardness (η) and softness (S) of the drugs were calculated according to the following equations (Pearson 1986)
Preparation of protein
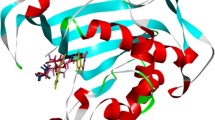
Crystal structure of thymidylate synthase (TYMS) was collected from Protein Data Bank (PDB, ID: 1HVY; Chain A) (Fig. 3). Prior to docking, heteroatoms, lipids and water molecules were removed from the crystal structure using PyMol (version 1.3) (DeLano 2002). Geometry and energy minimization of the crystal structure were carried out with Swiss-PDBViewer (version 4.1.0) employing GROMOS96 force field (Guex and Peitsch 1997; Scott et al. 1999). The ligand and protein structures were saved as .pdb files.

Crystal structure of thymidylate synthase (TYMS)
Binding site and docking
The active binding pocket of TYMS was predicted using CASTp (Dundas et al. 2006), which found the largest pocket area and volume to be 1347.7 Å2 and 1976.3 Å3 respectively (Additional file 1: Figure S1 provides with some of the most favorable pocket area and volume alongside the amino acid residue chains. The colored region in the protein structure represented largest pocket area and volume). These information were used to generate the grid boxes during molecular docking.
Molecular docking is a method that predicts preferred orientation of a compound bound to a second during the formation of a stable complex and finds its frequent application in in silico pharmaceutical design. Current methods of docking include pose prediction, using docking algorithms and energy-based scoring functions that identify the optimal binding modes of a drug, i.e. energetically most favorable conformations, to the active sites of a target protein. Lower free energy means better ligand–protein binding (Thomsen and Christensen 2006; Gschwend et al. 1996; Kroemer 2007). Two types of docking algorithms are most common: flexible and rigid. In flexible docking, either one or both of the molecules involved in binding are considered as flexible objects, whereas rigid docking imposes conformational restriction on both the ligand and protein, thereby considering them as rigid solid objects (Zhou et al. 2007; Halperin et al. 2002). In the current work, both flexible and rigid docking for each of the ligand–protein entities were performed using Autodock Vina (Trott and Olson 2010). TORSDOF was set for all the ligands followed by the conversion of all rotatable bonds into non-rotatable during rigid docking. While performing flexible docking, ligand molecules were kept flexible and the protein was kept rigid. The grid boxes were constructed in such a manner that it covered the colored volumes corresponding to the binding pockets identified in Additional file 1: Figure S1. For both flexible and rigid docking, the dimensions of grid boxes were chosen as follows: 54.6895, 43.2718 and 54.3972 Å towards X, Y and Z co-ordinates respectively. Detailed analysis of the residues involved in non-covalent interactions between the ligands and protein was explored using Accelyrs Discovery Studio 4.1 (2013) and LigPlot+ (version 1.4.5) (Laskowski and Swindells 2011).
Pharmacokinetic parameters
AdmetSAR online database has been utilized to generate the pharmacokinetic parameters related to drug absorption, metabolism and toxicity for the parent drug and its modifiers (Cheng et al. 2012). SDF (Structure Data File) and SMILES (simplified molecular-input line-entry system) strings were utilized throughout the generation process.
Results and discussion
Electronic and thermodynamic behavior of capecitabine and its derivatives
The current work has employed the equations mentioned in “Optimization of drugs using quantum mechanical calculations” section to produce thermochemical data that predict the energetic availability and flexibility of capecitabine and its halogenated derivatives, as depicted in Table 1. Exchange of F in the initial structure with bioisosteres –CF3, Cl, Br and I saw a gradual increase in the negative value of electronic and thermal energies, enthalpy and free energy, hence suggesting energetically and configurationally more preferable trifluoromethylated, chlorinated, brominated and iodinated molecules. To be specific, Br and I-substituted capecitabine molecules (D3 and D4) showed marked changes in the thermochemical data, in contrast to that of –CF3 and Cl-substituted ones (D1 and D2), suggesting greater atomic radii—halogen substituents allowing the modified drugs become more stable.
Dipole moment is a useful parameter in the study of drug-receptor systems and plays a significant role for the formation of hydrogen bond in biological systems (Lien et al. 1982). The numerical value of dipole moment was highest for capecitabine. As listed in Table 1, high electronegativity of fluorine contributed to the overall high molecular dipole moment in capecitabine—partial charge on fluorine being −0.298 a.u., whereas the three fluorine atoms in –CF3 modified molecule D1 showed even more electronegativity, ranging from −0.305 to −0.310 a.u. but a relatively overall lower dipole moment value. Partial charge for iodine in the iodinated structure D4 was found to be +0.148 a.u. and the value of molecular dipole moment seemed to be relatively higher, indicating relatively higher polarity for D4 (the partial charge maps of capecitabine and its derivatives are provided from Additional file 1: Figure S2).
Frontier molecular orbitals
Frontier orbitals (FO) are general terms used to denote both highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). Frontier orbitals’ energy values are important to determine chemical reactivity and the extent to which a drug interacts with a particular receptor. Even, energy of HOMO along can be a pivotal determinant to find a relationship between a class of drug’s activity and their electronic configuration, as reported by Snyder et al. (Snyder and Merril 1965). The energy gap between HOMO and LUMO predicts of a molecule’s kinetic and chemical stability. Larger FO gap concords with high kinetic stability, but low chemical reactivity, as it is energetically unfavorable for an electron to have it elevated from a low-energy HOMO to a relatively high-lying LUMO (Aihara 1999; Hoque et al. 2013).
Pictorial views of HOMOs and LUMOs of capecitabine and its four derivatives are shown in Additional file 1: Table S1. Frontier orbital energy gap in capeciabine was found to be 0.1778 a.u.; values for the four derivatives ranged from 0.1770 to 0.1889 a.u. (Table 2). The iodinated derivative D4 showed the lowest energy gap among the derivatives, 0.1770 a.u., which is even lower than the initial structure—hence enhanced softness, least hardness and high chemical reactivity. The –CF3 incorporated structure, on the other hand, revealed that it’s energy gap was the most unfavorable one in the group towards chemical reactivity.
Molecular docking: binding energy of the receptor-ligand systems
Table 3 lists the active site free energies of binding for each of the current receptor-ligand systems and shows a comparison between the binding energy values obtained via flexible and rigid docking for each of the entities. The interaction energy for each of the ligand atoms is calculated for all the possible binding sites—discredited through a grid box—of the receptor, resulting in multiple values of binding energies for each system. For instance, flexible and rigid docking for each of the capecitabine-TYMS entities produced total eighteen possible energy values. However, we have chosen the values corresponding to the second pose of the ligands as they tend to superimpose most appropriately on ZD1694—as we shall see in the latter sections. Binding energies for capecitabine-TYMS interactions of −7.1 and −8.8 Kcal mol−1 for flexible and rigid dockings respectively demonstrated significant deviation into energy values. The modified molecules followed the same trend as well. Flexible docking of D1–D4 against TYMS showed binding energy values from −8.0 to −7.4 Kcal mol−1, when rigid docking showed values from −9.5 to −8.2 Kcal mol−1. The trifluoromethylated and chlorinated ligands possessed the two most negative energy values among the group, making them favorable modified derivatives than the other two.
Non-covalent interactions within the receptor–ligand systems
Crystal structure of ZD1694/dUMP/TYMS complex, in the earlier researches of Phan et al. confirmed the fact that a Cα–S and a O–H covalent bonding at Cys195 and His196 of hTYMS are involved during the transformation of dUMP to dTMP (Phan et al. 2001). ZD1694 in the complex acts as a competitive binder against antifolate THF and inhibits the transformation required for DNA synthesis. The local environment of ZD1694/dUMP/TYMS in their work showed a direct N–H–O contact between ZD1694 molecule and Asp218 as well as Gly222. The local environment, with all hydrophobic contacts being taken into consideration, has been regenerated for the purpose of this work—as depicted by Additional file 1: Figure S4. Possible non-covalent interactions analogous to the most negative free energies of binding in capecitabine-TYMS complex have also been shown in Table 4, a thorough checking of which elucidates that flexible docking provides with more interaction sites—compared to that of rigid docking. Moreover, further comparison between Capecitabine/dUMP/TYMS and ZD1694/dUMP/TYMS complexes shows the local environment to be nearly identical in case of flexibly docked molecules. For instance, non-bonding hydrophobic contacts surrounding ZD1694 in the regenerated structure involved the following amino acid residues—Lys77, Phe80, Gly87, Ile108, Gly222, Phe225, Tyr 258 and Met311. On the other hand, flexibly docked capecitabine showed hydrophobic contacts involving Arg78, Val79, Phe80, Ile108, Leu221, Gly222, Phe225 and Met309; interaction sites are found completely different, however, in the rigid-docked molecule. As Fig. 4 would suggest, ZD1694 and flexibly docked capecitabine are well superimposed on each other-taking into account that the geometry of the latter here resembles only one of the nine poses—when the rigidly docked drug is significantly away. The case is same for the modified derivatives where rigidly docked ligands are observed not to superimpose on the crystal structure of ZD1694. Some more evidences will be found in Additional file 1: Figure S5, where the non-bonding interactions involving rigid ligands have been shown. We, therefore, shall mostly focus on the non-bonding interactions involving flexibly docked ligands.

Superposition of a Capecitabine and b D1 (trifluoromethylated ligand) on the ZD1694/dUMP/TYMS crystal structure resovlved at 1.9 Å. Green, red and purple molecules indicate ZD1694, flexibly docked and rigidly docked structures respectively
The nature of the non-bonding interactions varied from conventional hydrogen bonding to stacking interactions, donor–donor and weak van der Walls interactions. For instance, in capecitabine/dUMP/TYMS, F–O and F–H–C halogen bonding at Leu221 and Gly222 occurred. Rigid docking, however, predicted such interaction at Leu212. Here it is worth mentioning that despite reports suggested that halogen atoms with partial positive charges take part in halogen bonding more frequently, electronegative fluorine atoms in these cases were observed to form such interaction (Politzer et al. 2007). Phe225 was found to donate its π-electrons cloud towards the alkyl chain and the carbon attached to F of the drug, thus forming multiple π-stacking interactions. Here the phenyl residue shifts away from the ligand compared to that of ZD1694-Phe225 interactions—as the distance increases to 4.71 from 4.50 Å. A sulphar interaction occurs at Met309 with a bond distance 4.88 Å. Rigid docking, however, shows no interaction at the peripherical amino acid residues to Met311. An alkyl–alkyl weak interaction, however, occurs at Leu198—which is close to the Cys195 and His196. As observed comparing the interaction listed in Table 4, key interactions between TYMS side chains and ZD1694 are mostly, if not entirely, preserved in the flexibly docked capecitabine. The preservation continues in the modified structures as well as we shall see in the upcoming discussions.
The value of trifluoromethyl (–CF3) group for its pharmacological activity has been acknowledged since the 50’s of last century. The widespread use in pharmaceutical products has been witnessed in recent years due to its unique chemical and physiological stability and the fact that –CF3 group is lipophilic in nature—an important determinant for increasing the solubility of drugs and letting it penetrate through cell membrane more easily (Yale 1959; Ji et al. 2011). Flexible docking showed that like capecitabine, a fluorine atom (F15) of the –CF3 group formed 2.82 Å long halogen bond with Leu221 and an F–C interaction with Gly222. Another F atom demonstated an anion-π stacking with the delocalized electron cloud of Phe225 (Fig. 5). A π-alkyl interaction at Trp109 was observed here—analogous to the interaction in ZD1694 complex. Lys77 was found to interact at the O of furan ring of the chlorinated derivative D2. Likewise, the halogen atom was found to form stacking and alkyl interactions with variable bond distances at Phe80 and Ile108. A π–π stacking was observed at Phe225 as well (Fig. 5). Nature of the residual environment surrounding the brominated ligand D3 was merely identical to D1 and D2—except Br forming an alkyl interaction at Ile108 as shown by the types of non-bonding interactions and the corresponding bond distances at (D1–D4)-TYMS in Table 5 (Additional file 1: Figure S3 illustrates the non-bonding scenario of D3 and D4 with TYMS). Figure 6 illustrates a more rigorous view of the interactions, including the hydrophic binding sites at the periphery, of capecitabine, D1 and D2 ligands, as these three molecules are closer with respect to the binding energy values to one another.

Non-covalent interactions among a Capecitabine-TYMS, b D1 (trifluoromethylated)-TYMS and c D2 (chlorinated)-TYMS generated by discovery studio

Non-bonding and hydrophobic binding sites (flexible docking) involving a Capecitabine, b –CF3 modified (D1) and c chlorinated derivative (D2) with TYMS
In an article published in 2009, Mairal et al. investigated the potential role of iodine in inhibiting Transthyretin Fibrillogenesis in which they discussed on the binding pockets of Transthyretin (TTR) that could accommodate iodine; using Diflusinal and its derivatives as model compounds, they showed that iodine-inserted binding compounds could become crucial in inhibiting TTR activities for treatment of TTR-related amyloidosis (Mairal et al. 2009). Some other recent researches have also focused on pharmaceutical utilities of iodinated drugs (Barattin et al. 2010; Bois et al. 1998). The thermodynamic, frontier orbital and binding affinity data suggested good energetic availability of compound D4. Same as the D3 derivative, flexible docking produced halogen-alkyl interactions with Ile108 (bond-length 5.18 Å). Partial positive charge of iodine, evidenced by partial charge map of D4 (Additional file 1: Figure S2), might have been a factor behind such interactions. As Table 5 suggests, D4 shows similar types of interactions with the same amino acids compared to its counterparts, with the bond types varying among hydrogen bonding, alkyl interactions and π-stacking.
Pharmacokinetic properties of the drugs
Inhibition constant Ki of all the drugs have been calculated using the equilibrium E + I ↔ EI, where E is the enzyme and I is the inhibitor molecule (the reference concentrations for all the entities have been considered 1 mol L−1 for the calculations) and the relationship
where \(\ln {\text{K}}_{\text{b}} = - \Delta {\text{G}}/{\text{RT}}, \, \Delta {\text{G}} = {\text{free}}\;{\text{energy}}\;{\text{of}}\;{\text{binding}}\) and presented in the last row of Table 6. All of the drugs are non-carcinogenic, according to the ADME (absorption, distribution, metabolism, and excretion) analysis and possess a class III acute oral toxicity. The LD50 values also support the level of the acute toxicity, as they reveal good amount of tolerance against oral toxicity. This means that the drugs are relatively safer for oral delivery, and the –CF3 modified drug is the safest in the group—as seen by the LD50 values depicted in Table 6. The drugs are supposed to be absorbed without much complexity as the human intestinal absorption was found positive for all the ligands (Shen et al. 2010). All the drugs are P-glycoprotein non-inhibitor, when the probability is higher for the parent and –CF3 modified ligands. Inhibition of P-glycoprotein affects negatively a drug’s bioavailability and the extent of drug metabolism and intestinal absorption (Broccatelli et al. 2011). The drugs do, however, show a positivity considering blood brain barrier, predicting that the drugs will go through the BBB. Adverse drug–drug interactions and severe cardiac side effects can be avoided with capecitabine and the modified molecules, as the molecules are both CyP450 2C9 and hERG non-inhibitor (Shen et al. 2010; Wang et al. 2012; Cheng et al. 2011).
Conclusion
Halogenation in capecitabine contributed to changes in its physicochemical properties. Thermodynamic calculation demonstrated the stability of capecitabine and its modified derivatives (D1–D4). Free energy and entropy estimation revealed relatively higher negative values, particularly for brominated and iodinated moieties (D3 and D4). The drug molecules were polar in nature confirmed by calculation of dipole moment. Frontier orbital calculations revealed increased hardness for the –CF3 modified drug D1 and increased softness for the iodinated drug D4; moreover, softness values of the drugs, except fluorine, followed an increasing trend with the substitution of halogens of greater atomic radii. The –CF3 modified ligand was found to possess the most negative binding energy value. Molecular docking, both flexible and rigid, exposed the free energies of binding for each ligand–TYMS interaction, when in each case the value obtained by rigid docking was found slightly more negative than that obtained by flexible docking. However, the flexibly docked ligands could be superimposed better on ZD1694 within the crystal structure of TYMS, when rigidly docked molecules showed inconsistency in binding to the proper active sites. Docking identified wide-range non-bonding interactions among the atoms of the drugs and amino acid units of TYMS. Flexible docking, for each ligand–receptor systems, revealed significant halogen interactions as well. Such compilation of information may prove worthwhile for more cost-effective and precise drug-designing and/or manipulation for TYMS inhibition.
References
Abou MH, Fadl E (2009) Cancer of the small intestine 5-fluorouracil—induced cardiotoxicity during chemotherapy for adenocarcinoma of the small bowel. Gastrointest Cancer Res 3:167–170
Accelrys Software Inc. (2013) Discovery studio modeling environment, release 4.0. Accelrys Software Inc., San Diego
Aihara J (1999) Reduced HOMO–LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A 103:7487–7495
Arooj M, Sakkiah S, Cao GP, Lee KW (2013) An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS One 8:e60470
Barattin R, Gerby B, Bourges K, Hardy G, Olivares J, Boutonnat J et al (2010) Iodination increases the activity of verapamil derivatives in reversing PGP multidrug resistance. Anticancer Res 30:2553–2559
Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K et al (2002) The protein data bank. Acta Crystallogr D 58:899–907
Bois F, Beney C, Boumendjel A, Mariotte AM, Conseil G, Di Pietro A (1998) Halogenated chalcones with high-affinity binding to P-glycoprotein: potential modulators of multidrug resistance. J Med Chem 41:4161–4164
Broccatelli F, Carosati E, Neri A, Frosini M et al (2011) A novel approach for predicting P-glycoprotein (ABCB1) inhibition using molecular interaction fields. J Med Chem 54:1740–1751
Cerný J, Hobza P (2007) Non-covalent interactions in biomacromolecules. Phys Chem Chem Phys 9:5291–5303
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105
Cheng F, Yu Y, Shen J, Yang L, Li W, Liu G, Lee PW, Tang W (2011) Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J Chem Inf Model 51:996–1011
Chu E, Koeller DM, Casey JL, Drake JC, Chabner BA, Elwood PC et al (1991) Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc Natl Acad Sci USA 88:8977–8981
Costi MP, Ferrari S, Venturelli A, Calò S, Tondi D, Barlocco D (2005) Thymidylate synthase structure, function and implication in drug discovery. Curr Med Chem 12:2241–2258
Danenberg PV, Danenberg KD (1978) Effect of 5, 10-methylenetetrahydrofolate on the dissociation of 5-fluoro-2′-deoxyuridylate from thymidylate synthetase: evidence for an ordered mechanism. Biochemistry 17:4018–4024
DeLano W (2002) Pymol: an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr
Dougherty DA (2007) Cation-p interactions involving aromatic amino acids. J Nutr 137:1504S–1508S
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34:W116–W118
Easton RE, Giesen DJ, Welch A, Cramer CJ, Truhlar DG (1996) The MIDI! basis set for quantum mechanical calculations of molecular geometries and partial charges. Theor Chim Acta 93:281–301
Etienne M-C, Ilc K, Formento J-L, Laurent-Puig P, Formento P, Cheradame S et al (2004) Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms: relationships with 5-fluorouracil sensitivity. Br J Cancer 90:526–534
Gaussian 09 Revision D.01 (2009) Gaussian Inc., Wallingford CT. http://www.gaussian.com
Gschwend DA, Good AC, Kuntz ID (1996) Molecular docking towards drug discovery. J Mol Recognit 9:175–186
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Hagmann WK (2008) The many roles for fluorine in medicinal chemistry. J Med Chem 51:4359–4368
Halperin I, Ma B, Wolfson H, Nussinov R (2002) Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins Struct Funct Genet 47:409–443
Hardy LW, Finer-Moore JS, Montfort WR, Jones MO, Santi DV, Stroud RM (1987) Atomic structure of thymidylate synthase: target for rational drug design. Science 235:448–455
Hoque MM, Halim MA, Rahman MM, Hossain MI, Khan MW (2013) Synthesis and structural insights of substituted 2-iodoacetanilides and 2-iodoanilines. J Mol Struct 1054–1055:367–374
Ishikawa T, Utoh M, Sawada N, Nishida M, Fukase Y, Sekiguchi F et al (1998) Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem Pharmacol 55:1091–1097
Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S et al (2011) Innate C–H trifluoromethylation of heterocycles. Proc Natl Acad Sci USA 108:14411–14415
Joseph-Mccarthy D (1999) Computational approaches to structure-based ligand design. Pharmacol Ther 84:179–191
Kapetanovic IM (2008) Computer-aided drug discovery and development (CADDD): in silico–chemico–biological approach. Chem Biol Interact 171:165–176
Koutsoukas A, Simms B, Kirchmair J, Bond PJ, Whitmore AV, Zimmer S et al (2011) From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics 74:2554–2574
Kroemer RT (2007) Structure-based drug design: docking and scoring. Curr Protein Pept Sci 8:312–328
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model 51:2778–2786
Lien EJ, Guo ZR, Li RL, Su CT (1982) Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J Pharm Sci 71:641–655
Longley D, Harkin D, Johnston P (2003) 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3:330–338
Lu Y, Liu Y, Xu Z, Li H, Liu H, Zhu W (2012) Halogen bonding for rational drug design and new drug discovery. Expert Opin Drug Discov 7:375–383
Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X et al (2009) Halogen bonding—a novel interaction for rational drug design? J Med Chem 52:2854–2862
Luo B, Repalli J, Yousef AM, Johnson SR, Lebioda L, Berger SH (2011) Human thymidylate synthase with loop 181–197 stabilized in an inactive conformation: ligand interactions, phosphorylation, and inhibition profiles. Protein Sci 20:87–94
Mader RM, Müller M, Steger GG (1998) Resistance to 5-fluorouracil. Gen Pharmacol 31:661–666
Mairal T, Nieto J, Pinto M, Almeida MR, Gales L, Ballesteros A, et al. (2009) Iodine atoms: a new molecular feature for the design of potent transthyretin fibrillogenesis inhibitors. PLoS One 4:e4124
Meyer EA, Castellano RK, Diederich F (2003) Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed 42:1210–1250
Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K et al (1998) Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5 fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer 34:1274–1281
Papamichael D (2000) The use of thymidylate synthase inhibitors in the treatment of advanced colorectal cancer: current status. Stem Cells 18:166–175
Park D, Stoehlmacher J, Zhang W, Tsao-Wei D, Groshen S, Lenz H-J (2002) Thymidylate synthase gene polymorphism predicts response to capecitabine in advanced colorectal cancer. Int J Colorectal Dis 17:46–49
Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci USA 83:8440–8441
Peters GJ, Backus HHJ, Freemantle S, Van Triest B, Codacci-Pisanelli G, Van der Wilt CL et al (2002) Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta Mol Basis Dis 1587:194–205
Phan J, Koli S, Minor W, Dunlap RB, Berger SH, Lebioda L (2001) Human thymidylate synthase is in the closed conformation when complexed with dUMP and raltitrexed, an antifolate drug. Biochemistry 40:1897–1902
Politzer P, Lane P, Concha MC, Ma Y, Murray JS (2007) An overview of halogen bonding. J Mol Model 13:305–311
Saif MW (2009) Targeting cancers in the gastrointestinal tract: role of capecitabine. Onco Targets Ther 2:29–41
Salo-Ahen OMH, Wade RC (2011) The active-inactive transition of human thymidylate synthase: targeted molecular dynamics simulations. Proteins Struct Funct Bioinform 79:2886–2899
Santi DV, McHenry CS, Sommer H (1974) Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 13:471–481
Scheithauer W, McKendrick J, Begbie S, Borner M, Burns WI, Burris HA et al (2003) Oral capecitabine as an alternative to i.v. 5-fluorouracil-based adjuvant therapy for colon cancer: safety results of a randomized, phase III trial. Ann Oncol 14:1735–1743
Schmoll HJ, Arnold D (2006) Update on capecitabine in colorectal cancer. Oncologist 11:1003–1009
Scott WRP, Hünenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J et al (1999) The GROMOS biomolecular simulation program package. J Phys Chem A 103:3596–3607
Shen J, Cheng F, Xu Y, Li W, Tang Y (2010) Estimation of ADME properties with substructure pattern recognition. J Chem Inf Model 50:1034–1041
Sirimulla S, Bailey JB, Vegesna R, Narayan M (2013) Halogen interactions in protein-ligand complexes: implications of halogen bonding for rational drug design. J Chem Inf Model 53:2781–2791
Smart BE (2001) Fluorine substituent effects (on bioactivity). J Fluor Chem 109:3–11
Snyder SH, Merril CR (1965) A relationship between the hallucinogenic activity of drugs and their electronic configuration. Proc Natl Acad Sci USA 54:258–266
Song CM, Lim SJ, Tong JC (2009) Recent advances in computer-aided drug design. Brief Bioinform 10:579–591
Stella VJ, Charman WN, Naringrekar VH (1985) Prodrugs: do they have advantages in clinical practice? Drugs. 5:455–473
Świniarska M, Leś A, Rode W, Cieśla J, Millán-Pacheco C, Blake IO et al (2010) Segmental motions of rat thymidylate synthase leading to half-the-sites behavior. Biopolymers 93:549–559
Tang Y, Zhu W, Chen K, Jiang H (2006) New technologies in computer-aided drug design: toward target identification and new chemical entity discovery. Drug Discov Today Technol 3:307–313
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321
Trott O, Olson A (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Varma AK, Patil R, Das S, Stanley A, Yadav L, Sudhakar A (2010) Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS One 5:e12029
Wang S, Li Y, Wang J, Chen L, Zhang L et al (2012) ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of HERG potassium channel blockage. Mol Pharm 9:996–1010
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH (2009) PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res 37:W623–W633
Wheeler SE (2011) Local nature of substituent effects in stacking interactions. J Am Chem Soc 133:10262–10274
Wheeler SE, Bloom JWG (2014) Towards a more complete understanding of non- covalent interactions involving aromatic rings. J Phys Chem 118:6133–6147
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P et al (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672
Yale HL (1959) The trifluoromethyl group in medical chemistry. J Med Pharm Chem 2:121–133
Zhou Z, Felts AK, Friesner RA, Levy RM (2007) Comparative performance of several flexible docking programs and scoring functions: enrichment studies for a diverse set of pharmaceutically relevant targets. J Chem Inf Model 47:1599–1608
Zhou P, Zou J, Tian F, Shang Z (2009) Fluorine bonding—how does it work in protein–ligand interactions? J Chem Inf Model 49:2344–2355
Authors’ contributions
MAH, MGS and MAKK developed the idea. MMH, MGS and MAKK designed the drugs. MAH performed the quantum calculation and trained AR for this work. AR performed the molecular docking, data collection and draft writing. All authors read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional file
40064_2016_1844_MOESM1_ESM.docx
Additional file: 1. Figure S1 to Figure S5 depict binding pockets, partial charge maps and non-covalent interactions. Table S1 shows the Frontier orbitals.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rahman, A., Hoque, M.M., Khan, M.A.K. et al. Non-covalent interactions involving halogenated derivatives of capecitabine and thymidylate synthase: a computational approach. SpringerPlus 5, 146 (2016). https://doi.org/10.1186/s40064-016-1844-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-1844-y