
Abstract
Halomonas smyrnensis AAD6T is a gram negative, aerobic, and moderately halophilic bacterium, and is known to produce high levels of levan with many potential uses in foods, feeds, cosmetics, pharmaceutical and chemical industries due to its outstanding properties. Here, the whole-genome analysis was performed to gain more insight about the biological mechanisms, and the whole-genome organization of the bacterium. Industrially crucial genes, including the levansucrase, were detected and the genome-scale metabolic model of H. smyrnensis AAD6T was reconstructed. The bacterium was found to have many potential applications in biotechnology not only being a levan producer, but also because of its capacity to produce Pel exopolysaccharide, polyhydroxyalkanoates, and osmoprotectants. The genomic information presented here will not only provide additional information to enhance our understanding of the genetic and metabolic network of halophilic bacteria, but also accelerate the research on systematical design of engineering strategies for biotechnology applications.
Similar content being viewed by others

Background
Halomonas smyrnensis AAD6T is a rod-shaped, Gram-negative, aerobic, exopolysaccharide (EPS) producing and moderately halophilic bacterium, growing at salt concentration in the range of 3–25% (w/v) NaCl (optimum, 10%), at temperatures between 5 and 40°C (optimum, 37°C), and pH values between 5.5 and 8.5 (optimum, 7.0) (Poli et al. 2012). The strain utilizes glucose, sucrose, maltose, arabinose, raffinose, fructose, galactose, and mannose as a sole carbon and energy source, and alanine and serine as a sole carbon, nitrogen and energy source (Poli et al. 2009, 2012). The EPS production by H. smyrnensis AAD6T is growth-associated, and when sucrose was used as carbon source the bacteria excrete high levels of levan, which is a long linear homopolymeric EPS of ß(2-6)-linked fructose residues (Poli et al. 2009). Levan has been credited as one of the most promising polysaccharides for foods, feeds, cosmetics, pharmaceutical and chemical industries (Donot et al. 2012) and, potential uses of levan produced by AAD6T, as a bioactive polymer, were reported (Costa et al. 2013; Kucukasik et al. 2011; Sam et al. 2011; Sezer et al. 2011; Sima et al. 2011, 2012).
When compared to the other groups of extremophilic microorganisms such as the thermophiles, the halophiles have been relatively little exploited in biotechnological processes with a few exceptions (Pastor et al. 2010; Philip et al. 2007; Rodriguez-Valera and Lillo 1992; Sam et al. 2011). On the other hand, due to their osmoadaptation abilities and metabolic capabilities for the production of compatible solutes, bioplastics, nutritional products, biopolymers, and halophilic enzymes, they represent considerable potential in various industries including chemical, environmental, biofuel, medical, pharmaceutical and health care.
In systems biology research, the microbial genome sequence is the starting point for detailed analysis of identifying gene-protein associations and metabolic reconstruction. In this context, next generation sequencing (NGS) technologies play an important role since they provide high-throughput genomic data, including whole genome sequences (WGS), at unprecedented speed with relatively low cost (Gov and Arga 2014; Zhang et al. 2011). WGS data provide many advantages in comparative genomics and metabolic engineering. In addition to the nucleotide sequence and protein sequences of the encoded proteins, the gene content, the order of genes and the predicted enzymes on the genome may be informative for phylogenetic analyses and metabolic reconstructions. Moreover, WGS information facilitates the acquirement of functional genomics data.
To design economical production schemes, fermentation conditions and effective stimulatory factors for levan production by H. smyrnensis AAD6T was analyzed systematically (Ateş et al. 2013; Ates et al. 2011; Sarilmiser Kazak et al. 2014). However, towards the design of efficient microbial cell factories for levan overproduction, the main bottleneck was the lack of information on the genome of the microorganism. Considering this fact, we employed two different NGS technologies, namely, Roche 454 GS FLX+ System and Ion Torrent Sequencer, and a hybrid NGS approach to develop a high-quality WGS data for H. smyrnensis AAD6T (Sogutcu et al. 2012). Within the context of this study, the genome of AAD6T was further analyzed to reveal the essential biological mechanisms and the whole genomic organization of levan producing H. smyrnensis AAD6T, and the genome-scale metabolic model of H. smyrnensis AAD6T was reconstructed. This understanding is crucial for rational design and optimization of engineering strategies for levan overproduction. Furthermore, the information provided here will facilitate future studies on the genetic and metabolic diversity of halophilic bacteria and contribute to the employment of H. smyrnensis AAD6 in biotechnological processes.
Methods
Bacterial strain
Halophilic bacterial strain H. smyrnensis AAD6T (JCM 15723, DSM 21644) used in this study was isolated from Çamaltı Saltern Area in Turkey (Poli et al. 2012).
Sequencing and assembly
The genomic DNA was isolated from exponential-phase cultures using the quick protocol of Wizard® Genomic DNA Purification Kit (Promega, Madison, WI). The optimum semi-chemical medium (Poli et al. 2009) was used in growth cultures. To obtain the highest growth rate without EPS production, glucose was added as sole carbon source to chemical medium at concentration of 10 g/L.
The genome of H. smyrnensis AAD6T was sequenced by the whole genome shotgun approach using a combination of 454 GS FLX+ (454 Life Sciences, Branford, CT) and Ion Torrent (Ion Torrent Systems, Inc., Guilford, CT) sequencers and the generated shotgun sequence data was assembled as described in (Sogutcu et al. 2012).
Genome annotation
Gene prediction and genome annotation were carried out using the RAST auto-annotation server (Aziz et al. 2008). Protein-encoding and transfer RNA genes were predicted by RAST server, while the ribosomal RNA genes were identified with RNAmmer (v1.2) (Lagesen et al. 2007). The gene predictions of essential biosystems were manually verified using BLAST searches against protein databases, the Universal Protein Resource (UniProt) (http://www.uniprot.org/) and National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/). The gene functions and classifications were based on the subsystem annotation of the RAST server. Information on enzyme-encoding genes was taken from Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2012) and ExPaSy databases (Artimo et al. 2012). Transport protein coding genes were annotated using the similarity searches against the Transfer Classification Database (TCDB) (Saier et al. 2009).
Phylogenetic and phylogenomic analyses
The phylogenetic analyses were performed using PhyML (v3.0) and phylogenetic trees were reconstructed using maximum likelihood method (Guindon et al. 2010). The phylogenetic classification of bacterial strains was based on complete 16S rRNA sequences. The phylogenetic classification of a set of bacterial levansucrase enzymes was based on nucleotide sequences taken from NCBI database. The phylogenomic comparison within the Halomonadaceae family was based on the encoded proteins with putative functions assigned by the RAST server. Encoded proteins were sorted out for all genomes and comparatively analyzed based on the presence/absence of proteins.
Reconstruction of the genome-scale metabolic model
The entire reconstruction procedure consists of an automated procedure followed by manual curation at the gap filing step. The preliminary model was built automatically via the Model SEED (Henry et al. 2010). The refinement of the model (for stand-alone reactions or metabolites, and missing connections in essential metabolic processes) was achieved via a non-automated but iterative decision-making process, as described in (Ates et al. 2011).
Results and discussion
Sequencing and developing the H. smyrnensis AAD6T genome
The WGS project of H. smyrnensis AAD6T (Sogutcu et al. 2012) was comprised of data from Roche 454 GS FLX+ platform and Ion Torrent sequencer (Table 1). GS FLX+ platform provided longer reads (with mean length of 499.46 bp) and higher coverage (76-fold in average), when compared to Ion Torrent System (with mean length of 113.01 bp and 11-fold coverage). On the other hand, Ion Torrent System represented a duplication rate of 0.09%, which is significantly lower than that of GS FLX+ platform, i.e. 9.47% in average. Consequently, the de novo assembly procedure resulted in a total assembly size of 3.56 Mbp and is presumed to be a single circular chromosome (Fig. 1).

Circular representation of draft genome sequence of H. smyrnensis AAD6T. The inner and outer scales designate the coordinates in two hundreds kilo base pair increments. The red and blue circles show the predicted coding regions of AAD6 on the forward and reverse strands, respectively. The clustering of putative genes on the forward and reverse strands to Clusters of Orthologous Group (COG) functional categories was represented by differential colors in the outer and inner circles, respectively.
General features of the H. smyrnensis AAD6T genome
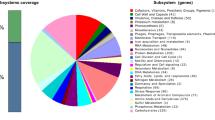
The gene prediction and genome annotation of draft genome of H. smyrnensis AAD6T resulted in a total of 3,237 coding sequences and 64 RNA genes (Table 2, Additional files 1, 2). Putative functions could be assigned to 2,436 protein-coding genes, whereas 801 hypothetical proteins had no match to any known proteins. The major part (55%) of the coding sequences was clustered in 449 subsystems (Additional file 3); however 1,481 coding sequences were not included in any subsystem. With regard to the subsystem distribution of coding sequences (Fig. 2), the largest number of these sequences were found in “Amino acids and Derivatives” and “Carbohydrates” categories with 391 and 328 coding sequences, respectively. H. smyrnensis AAD6T has significantly more coding sequences related to “osmotic stress” subsystem under “Stress Response” category (56 coding sequences) when compared to Escherichia coli K12 and Bacillus cereus AH187 (22 and 13 coding sequences, respectively). Most of the osmotic stress genes of H. smyrnensis AAD6T (47 coding sequences) encode proteins associated with choline and glycine betaine uptake, and glycine betaine biosynthesis pathways, which are involved in osmoprotection.

Subsystem distribution of coding sequences in the draft genome of H. smyrnensis AAD6T. The bar on the left indicates the clustered (green) and unclustered (blue) percentages of coding sequences. Distribution of coding sequences into subsystems is shown in pie graph. The legend on the right designates the color associated with each subsystem.
At a large-scale comparison, the size of the genome, the number of coding regions and the coding capacity of AAD6T were slightly smaller than those of other halophilic bacteria whose genome has already been sequenced (Table 3). On the other hand, the GC content of H. smyrnensis AAD6T seems to be slightly higher than that of H. elongata DSM 2581 (Schwibbert et al. 2011) and C. salexigens DSM 3043 (Copeland et al. 2011), but the GC contents of genomes of halophilic bacteria vary in a large range from 35.7% (Oceanobacillus iheyensis HTE831) (Takami et al. 2002) to 74.3% (Halomonas ventosae sp. nov.) (Martínez-Cánovas et al. 2004).
Phylogenetic and phylogenomic relatedness to other microorganisms
Phylogenetic classification based on 16S rRNA sequences places H. smyrnensis AAD6T in the Halomonas genus of the Halomonadaceae family (Poli et al. 2012). H. salina F8-11 (99.5%) and H. halophila CCM 3662 (99.5%) were found to be the closest species based on 16S rRNA gene sequence comparison (Additional file 4: Figure S1a). On the other hand, based on the phylogenetic classification of bacterial strains whose WGS data were available (comprising of 2 extreme halophilic, 10 moderate halophilic, 15 mesophilic, and 29 non-halophilic bacterial strains), two halophilic bacteria, H. elongata DSM 2581 and C. salexigens DSM 3043, were the most closely related organisms to H. smyrnensis AAD6T (Additional file 4: Figure S1b).
Rapid Annotation using Subsystem Technology (RAST) server establishes the phylogenetic context with using representative sequences such as tRNA synthetases, which are admitted as universal or nearly universal in prokaryotes (Aziz et al. 2008). The phylogenetic comparison of H. smyrnensis AAD6T genome based on these representative sequences with genomes available pointed out the closest similarity with C. salexigens DSM 3043 (with a score of 524), followed by H. elongata DSM 2581 (with the score of 346). Transfer RNAs (tRNAs) are thought to be among the oldest and highly conserved sequences on earth, and are often involved in horizontal gene transfer (Widmann et al. 2010). The partnership of horizontal genes between H. smyrnensis AAD6T and C. salexigens DSM 3043 may cause such a different phylogenetic relatedness compared to 16S rRNA based analysis.
The close relationships between these three moderately halophilic strains from Halomonadaceae family were also confirmed with the phylogenomic analysis within three genomes, which was based on the encoded proteins with putative function assignment (Fig. 3). Accordingly, 92.3% of the total predicted proteins in H. smyrnensis AAD6T were encoded by H. elongata DSM 2581; while 1,811 proteins that were 83.3% of the total predicted proteins in H. smyrnensis AAD6T were also encoded by C. salexigens DSM 3043. In addition, 80.4% of the total proteins encoded by the H. smyrnensis AAD6T (1,748 proteins) were also encoded by the genomes of all three strains. This finding indicates that at least 80% of the H. smyrnensis AAD6T genome has been vertically transmitted from a common ancestor of the Halomonadaceae family, comprising the backbone of the H. smyrnensis AAD6T genome. The rest of the genome encodes proteins similar to those produced by bacterial species other than those in the Halomonadaceae family. A large fraction of the 1,748 core proteins (Fig. 3) was assigned to categories with general cellular processes such as energy production and conversion, amino acid transport and metabolism, central carbon metabolism, translation, and transcription. On the other hand, 104 proteins from diverse categories were unique to H. smyrnensis AAD6T. Among these categories, Arginine biosynthesis with ten enzymes (EC 1.2.1.38, EC 2.1.3.3, EC 2.3.1.1, EC 2.3.1.35, EC 2.6.1.11, EC 2.7.2.8, EC 3.5.1.16, EC 3.5.1.18, EC 4.3.2.1, EC 6.3.4.5) and a regulatory protein (ArgR), and auxin biosynthesis with 4 proteins (EC 2.4.2.18, EC 5.3.1.24, alpha and beta chains of EC 4.2.1.20) drew the attention.

Venn diagram comparing the encoded proteins of three halophilic bacteria. Total numbers of proteins encoded in the genomes are given in parenthesis. The numbers of shared and unique proteins are shown inside the diagram.
Metabolism
To facilitate reconstruction of genome scale metabolic network of AAD6T, metabolic proteins and enzymes were identified with the open frame shifts that are encoded these proteins and enzymes via whole genome annotation (Additional file 5). The gene annotations revealed that the complete pathways of glycolysis (Embden–Meyerhof–Parnas, EMP), Entner–Doudoroff (ED), gluconeogenesis, pentose-phosphate and all de novo amino acid biosynthesis were represented in the H. smyrnensis AAD6T genome. The TCA cycle and glyoxylate shunt were also complete and genes corresponding to aerobic respiration pathways were detected. The biosynthetic pathways for most vitamins were found; however, pathways for riboflavin (vitamin B2) and pyrroloquinoline quinone syntheses were notably absent.
An important point to be discussed is the pathway utilized in glucose degradation in halophilic bacteria. Although complete EMP pathway was annotated in the genome of C. salexigens, glucose was degraded via the ED pathway (Pastor et al. 2013). Performing a bioinformatics analysis on phosphofructokinases (PFKs), it was shown that the PFKs from C. salexigens (Csal_1534) and H. elongata (Helo_2186) are only remotely similar to functional characterized PFKs. As a result, they hypothesized that the Csal_1534 protein functions as fructose-bisphosphatase, rather than as PFK. The genome annotation of H. smyrnensis AAD6T presented Peg.1123 as PFK and also failed to identify the presence of any fructose-bisphosphatase. However, PFK of H. smyrnensis AAD6T displays significant sequence similarity with the PFKs from C. salexigens (Csal_1534) and H. elongata (Helo_2186), raising the possibility that Peg.1123 may also be a fructose-biphosphatase. It is therefore doubtful whether H. smyrnensis AAD6T has a functional EMP pathway. On the other hand, on the basis of the genome annotation of H. smyrnensis AAD6T, one of the three variants of the ED pathway, the phosphorylative ED pathway from glucose or d-gluconate to 6-phosphogluconate, could be reconstructed, as in the case of C. salexigens (Pastor et al. 2013).
The genome annotation results for H. smyrnensis AAD6T include information required to reconstruct the stoichiometric metabolic model of H. smyrnensis AAD6T, which reflects the metabolic capabilities of the bacterium. The metabolic model was reconstructed systematically via automatic building followed by manual curation at the gap filling step (Additional file 6). The resulting iKYA1142 metabolic model comprises 980 metabolites and 1,142 metabolic reactions. A total of 819 protein-encoding genes were included in the metabolic model, which represents approximately 33.5% of the genes with assigned function in the H. smyrnensis AAD6T genome.
As a consequence of validation and constraints-based flux analyses, the genome-scale reconstructed metabolic model of H. smyrnensis AAD6T, iKYA1142, is expected to be useful for medium design and the development of metabolic engineering strategies for increased levan production, and systematical design of strain development strategies for biotechnology applications. Consequently, the reconstructed model together with the whole genome information presented here will be a crucial pathfinder towards future in silico and in vivo studies of this organism.
Transport
Transport systems are vitally important for all organisms and also play a key role in halophiles for adaptation to high salinity environments. In the H. smyrnensis AAD6T genome, 307 open reading frames (ORFs) (9.5% of total) were found to be involved in various transport systems which enable the bacteria to accumulate needed nutrients, extrude unwanted by-products and maintain cytoplasmic content of protons and salts conducive to growth and development. This proportion does not show a major difference in other halophilic and non-halophilic microorganisms taken into comparison (Blattner et al. 1997; Copeland et al. 2011; Kunst et al. 1997; Pastor et al. 2013; Schwibbert et al. 2011).
Major part of transporter genes in H. smyrnensis AAD6T genome belongs to Tripartite ATP-independent periplasmic (TRAP) transporters that are a large family of solute transporters ubiquitous in bacteria and archaea, but absent in eukaryotes, however the number of TRAP transporter genes in E. coli (Blattner et al. 1997) genome is much lower compared to H. smyrnensis AAD6T genome.
At the same time, halophilic genomes exhibit significant differences in ion transport systems. For instance, H. smyrnensis AAD6T was shown specific requirement to presence of Na+ and Cl− ions, in absence of these ions the strain does not grow (Poli et al. 2009). Accordingly, its genome has significantly more (i.e., ten genes) Na+ and Cl− transporter genes like other halophilic bacteria [Eight genes in C. salexigens (Copeland et al. 2011) and nine genes in H. elongata DSM 2581 (Schwibbert et al. 2011)] than non-halophilic bacteria [two genes in E. coli K12 (Blattner et al. 1997) and three genes in B. subtilis (Kunst et al. 1997)]. This larger Na+ transporter portion may provide more Na+ gradients to drive transport processes in the cell membrane (Ventosa et al. 1998). Among the ORFs, 183 were categorized under subsystems whereas 124 were identified as putative transporters. They include transporters for sugars and carbohydrates (maltose/maltodextrin, inositol, d-xylose, succinoglycan, mannitol), ions (Na+, K+, Mg2+, Zn2+, Mn2+, Fe2+/3+, Co2+, Ni2+ and Mo2+), anions and cations (sulphate, ammonium, nitrate, nitrite, phosphate, phosphonates, sulfonates, and malonate), amino acids (arginine, ornithine, histidine, methionine, glutamic acid, glutamine, alanine, glycine, aspartate and threonine), and other molecules (cobalamin, thiamine, urea, C4 dicarboxylates, uracil, xanthine, auxin, spermidine/putrescine, glycine betaine, l-proline, benzoate, N-acetylneuraminate, glycerol-3-phosphate, l-lactate, cyclase, oligoketide and various polyols). In addition, utilization systems were present for glucose, fructose, galactose, sucrose, arabinose, mannose and rhamnose.
Osmoregulation
Osmolarity resistance (or tolerance) is one of the most important characteristics of H. smyrnensis AAD6T, since the microorganism can grow in media containing up to 3.4 mol/L sodium chloride (Poli et al. 2012). Accumulating intracellular organic solutes, such as ectoine and betaine, and rapidly releasing those solutes when extracellular osmolarity declines, is a frequently observed osmolarity resistance strategy when extracellular osmolarity rises (Oren 2002). Here, using genome annotation predictions, pathways associated with uptake of choline-betaine, biosynthesis of glycine betaine (GB), ectoine and hydroxyectoine were reported for H. smyrnensis AAD6T.
Halomonas smyrnensis AAD6T possesses two different GB/proline betaine (PB) high-affinity uptake systems, OpuA, known from B. subtilis (Kempf et al. 1997) and ProU, known from E. coli (Schiefner et al. 2004). The OpuA system is a member of ABC transporter superfamily and consists of a membrane-associated ATPase (OpuAA), the integral membrane protein (OpuAB), and the extracellular ligand-binding protein (OpuAC). The extracellular OpuAC protein binds GB or PB with high affinity, and delivers it to the OpuAA/OpuAB protein complex for the release of substrate into the cytosol with an ATP-dependent substrate translocation mechanism. H. smyrnensis AAD6T also has a second ABC transporter system ProU for GB/PB import into the cytoplasm. ProU transport system is composed of a GB/PB binding protein ProX and permease protein ProW and ProV. The ProX protein is binding to GB/PB for the substrate translocation via ProWV permease protein complex into the cytosol. GB can also be synthesized from the precursor choline. In this sense, annotations also included a high-affinity choline uptake protein BetT and two enzymes: choline dehydrogenase (EC 1.1.99.1) that catalyzes the conversion of choline into betaine aldehyde as the intermediate and betaine aldehyde dehyrogenase (EC 1.2.1.8) that catalyzes the conversion of betaine aldehyde to GB (Additional file 4: Figure S2). Choline and GB may function as stress protectants, not only against osmotic stress but also against high-temperature and low-temperature challenges (Hoffmann and Bremer 2011).
Ectoine and hydroxyectoine are widely distributed compatible solutes accumulated by halophilic and halotolerant microorganisms to prevent osmotic stress in highly saline environments. Ectoine presents industrial importance with current and potential applications as protecting agents for macromolecules, cells and tissues, together with its potential as therapeutic agents for certain diseases (Pastor et al. 2010). Genome analysis revealed three genes (Peg.2770, Peg.2771, and Peg.2772), which were located together on the chromosome of H. smyrnensis AAD6T, encoding enzymes of ectoine biosynthesis pathway, i.e. EctA (l-2,4-diaminobutyric acid acetyltransferase, EC 2.3.1.-), EctB (diaminobutyrate-pyruvate aminotransferase, EC 2.6.1.46), and EctC (l-ectoine synthase, EC 4.2.1.-,), respectively. In addition, the ectD gene (peg.3147) encoding ectoine hydroxylase (EC 1.17.-.-), which is involved in the conversion of ectoine to hydroxyectoine, is also present. On the other hand, any gene encoding neither a transcriptional regulator nor an aspartokinase specific for ectoine biosynthesis could not found adjacent to ectABC genes. Unlike C. salexigens and H. elongata genomes, the genome of H. smyrnensis AAD6T does not possess any genes for the degradation of ectoine.
Levan exopolysaccharide biosynthesis
Genome analysis revealed Hs_SacB gene encoding the extracellular levansucrase enzyme (EC 2.4.1.10) which catalyzes levan synthesis from sucrose-based substrates by transfructosylation (Donot et al. 2012). The levansucrase belongs to the family of glycosyltransferases, specifically the hexosyltransferases. The length of the Hs_SacB gene was 1,251 bp and it is predicted to encode a protein of 416 amino acids. The amino acid distribution in Hs_SacB indicates the dominance of hydrophobic amino acids (with 45.2%), whereas polar amino acids and charged amino acids constitute 32.2 and 22.5%, respectively (Additional file 4: Figure S3). The high frequency of glycine (G) and proline (P) (10.3 and 6.3%, respectively) were not surprising since these two amino acids are often highly conserved in protein families due to their essential role in preserving the protein tertiary structure.
The phylogenetic classification based on nucleotide sequence for a set of bacterial levansucrase enzymes indicated that the most closely related levansucrases to Hs_SacB were those from Pseudomonas strains (Fig. 4). Similar results were obtained in classification based on the amino acid sequences, where Hs_SacB enzyme exhibited high similarities with levansucrases from Pseudomonas strains with 96–99% coverage.

Phylogenetic classification of levansucrase enzymes. The phylogenetic tree was constructed using maximum likelihood method and nucleotide sequences of enzymes taken from NCBI database.
Levansucrases follow different secretion routes, and are generally secreted by a signal-peptide-independent pathway in the gram-negative bacteria (Arrieta et al. 2004). Multiple sequence alignment analysis indicated the absence of such a signal-peptide, and pointed out a signal-peptide-independent secretion pathway in H. smyrnensis AAD6T like other gram-negative bacteria.
Biosynthesis of Pel exopolysaccharide
One strategy for bacterial adaptation to environmental stresses is to self-encapsulate with a matrix material, primarily composed of secreted extracellular polysaccharides (Sutherland 1998). H. smyrnensis AAD6T is known to produce high levels of levan EPS (Poli et al. 2009), but there is no experimental study reporting biosynthesis of any other EPSs. The genome analysis revealed a putative Pel polysaccharide gene cluster (PelA, PelB, PelC, PelD, PelE, PelF, and PelG) in H. smyrnensis AAD6T (Additional file 4: Figure S4). PelB gene, annotated as a conserved hypothetical protein via RAST server, was presumed to encode a polysaccharide biosynthesis protein due to both its position in the genome and amino acid sequence similarity with “biofilm formation protein PelB” of Pseudomonas mendocina (99% coverage) and Pseudomonas aeruginosa (96% coverage).
Pel polysaccharide is a glucose-rich, cellulose-like polymer, which is essential for the formation of a pellicle at the air–liquid interface. P. aeruginosa is the most-well known Pel polysaccharide producer, and this bacterium also produces Alginate and Psl polysaccharides as well as Pel polysaccharide, all of which has been shown to be important for biofilm formation (Colvin et al. 2012). In AAD6T genome, Psl related genes were not detected, however, “Alginate lyase precursor (EC 4.2.2.3)” and “Alginate biosynthesis protein Alg8” genes were predicted.
Biosynthesis of polyhydroxyalkanoates (PHAs)
In addition to genes related to EPS biosynthesis, genes that are involved in intracellular polyhydroxyalkanoate (PHA) biosynthesis were also detected. Fifteen genes related to “Acetyl-CoA fermentation to Butyrate-Polyhydroxybutyrate” metabolism along with “Polyhydroxyalkanoate synthesis repressor PhaR” and “3-ketoacyl-CoA reductase PhaB” genes were present in the draft genome. PHAs are natural polyesters produced as carbon and energy reserve materials by many microorganisms under stressful growth conditions in the presence of excess carbon sources. The stored PHA can be degraded by intracellular depolymerases and metabolized as carbon and energy source as soon as the supply of the limiting nutrient is restored (Philip et al. 2007).
In addition to H. smyrnensis AAD6T, ten Halomonas strains (H. eurihalina, H. sinaiensis, H. almeriensis, H. salina, H. maura, H. ventosae, H. anticariensis, H. alkaliphila, H. almeriensis, H. rifensis) were also reported to secrete EPSs along with PHAs (Amjres et al. 2011; Llamas et al. 2012; Rodriguez-Valera and Lillo 1992). This potential of the bacteria should be considered in the design of bioreactor experiments, since the co-production of EPS and PHA may cause difficulties in bioreactors as they tend to increase the viscosity of the medium rapidly and may also interfere with purification of the polyester (Rodriguez-Valera and Lillo 1992).
Other features of H. smyrnensis AAD6T genome
The genome annotation results pointed out several genes and gene clusters associated with other biological processes such as cell division, transcription process, lipopolysaccharide (LPS) biosynthesis, quorum sensing and arsenic resistance (Additional file 7). Briefly, fifteen genes were identified, which encode proteins associated with cell division. A significant portion of these proteins belonged to the Fts (filamentation thermosensitive) family. H. smyrnensis AAD6T genome presents a Min system, consisting of three proteins MinCDE, which controls the division site selection. The rod-shape of the microorganism is provided by functioning of four rod-shape determining proteins: RodA, MreB, MreC and MreD, which in turn serves as a genetic support to the observations. The genome annotation identified several transcription process associated genes, which show high similarities to the corresponding orthologous of C. salexigens. 16S, 23S, 5S rRNA and 61 tRNA regions (for all amino acids) were also annotated.
As a gram-negative bacterium, H. smyrnensis AAD6T possesses LPSs in the external leaflet of its outer membrane. Several genes associated with lipopolysaccharide assembly were identified in H. smyrnensis AAD6T. Several genes such as LuxS, LuxP, RpoS, RpoN that play roles in cell-density-dependent gene-expression mechanism (quorum sensing) and bacterial cell-to-cell communication were identified.
The H. smyrnensis AAD6T genome also carries multiple genes that are potentially involved in arsenic resistance, as has also been reported for other Halomonas species (Lin et al. 2012; Wolfe-Simon et al. 2011). Presence of these genes suggests that H. smyrnensis AAD6T is an arsenite-specific expulsion prokaryote rather than a dissimilatory arsenic-reducing prokaryote. The predicted arsenic resistance genes in the genome, as well as scientific reports for arsenic resistance of several Halomonas species support the genomic potential of arsenic resistance of H. smyrnensis AAD6T and put it as a candidate organism for bioremediation studies.
Conclusions
The microbial genome data form the basis of systems biology research including functional genomics and metabolic engineering studies. Therefore, whole genome analysis of H. smyrnensis AAD6T was performed. The genome analysis reveals the biotechnological and industrial potential of this levan producing halophilic extremophile. The bacterium was found to have many potential applications in biotechnology not only being a levan producer, but also because of its capacity to produce Pel exopolysaccharide, PHAs, and osmoprotectants as well as its resistance to arsenic.
The genomic information of H. smyrnensis AAD6T together with the reconstructed genome-scale metabolic model presented here will not only provide additional information to enhance our understanding of the genetic and metabolic network of halophilic bacteria, but also accelerate research on medium design and development of metabolic engineering strategies for enhanced levan biosynthesis, systematical design of strain development strategies for biotechnology applications, and purification and characterization of levansucrase enzyme. Consequently, the information presented here will be a crucial pathfinder towards future in silico and in vivo studies of this organism.
Nucleotide sequence accession numbers
The genome project for H. smyrnensis AAD6T has been deposited at DDBJ/EMBL/GenBank under the accession AJKS00000000. This is the second version, with accession numbers AJKS02000001 to AJKS02000034. The 16S rRNA gene and levansucrase (Hs_SacB) gene sequences are available in DDBJ/EMBL/GenBank under the accession numbers DQ131909.2 and KC480580.1, respectively.
References
Amjres H, Béjar V, Quesada E, Abrini J, Llamas I (2011) Halomonas rifensis sp. nov., an exopolysaccharide-producing, halophilic bacterium isolated from a solar saltern. Int J Syst Evol Microbiol 61:2600–2605
Arrieta JG, Sotolongo M, Menéndez C, Alfonso D, Trujillo LE, Soto M et al (2004) A type II protein secretory pathway required for levansucrase secretion by Gluconacetobacter diazotrophicus. J Bacteriol 186:5031–5039
Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E et al (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40:597–603
Ates O, Toksoy Oner E, Arga KY (2011) Genome scale reconstruction of metabolic network for a halophilic extremophile, Chromohalobacter salexigens DSM 3043. BMC Syst Biol 5:12
Ateş Ö, Arga KY, Toksoy Öner E (2013) The stimulatory effect of mannitol on levan biosynthesis: lessons from metabolic systems analysis of Halomonas smyrnensis AAD6T. Biotechnol Prog 29:1386–1397
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA et al (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genom 9:75
Blattner FR, Plunkett G, Bloch CA, Perna MT, Burland V, Riley M et al (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474
Colvin KM, Irie Y, Cs Tart, Urbano R, Whitney JC, Ryder C et al (2012) The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ Microbiol 14:1913–1928
Copeland A, O’Connor K, Lucas S, Lapidus A, Berry KW, Detter JC et al (2011) Complete genome sequence of the halophilic and highly halotolerant Chromohalobacter salexigens type strain (1H11T). Stand Genomic Sci 5:379–388
Costa RR, Neto AI, Calgeris I, Correia CR, Pinho ACM, Fonseca J (2013) Adhesive nanostructured multilayer films using a bacterial exopolysaccharide for biomedical applications. J Mater Chem B 1:2367–2374
Donot F, Fontana A, Baccou JC, Schorr-Galindo S (2012) Microbial exopolysaccharides: main examples of synthesis, excretion, genetics and extraction. Carbohydr Polym 87:951–962
Gov E, Arga KY (2014) Systems biology solutions to challenges in marine biotechnology. Front Mar Sci 1:14
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Henry CS, DeJongh M, Best AB, Frybarger PM, Linsay B, Stevens RL (2010) High-throughput generation and optimization of genome-scale metabolic models. Nat Biotechnol 28:977–982
Hoffmann T, Bremer E (2011) Protection of Bacillus subtilis against cold stress via compatible solute acquisition. J Bacteriol 193:1552–1562
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res 40:109–114
Kempf B, Gade J, Bremer E (1997) Lipoprotein from the osmoregulated ABC transport system OpuA of Bacillus subtilis: purification of the glycine betaine binding protein and characterization of a functional lipidless mutant. J Bacteriol 179:6213–6220
Kucukasik F, Kazak H, Güney D, Finore I, Poli A, Yenigün O (2011) Molasses as fermentation substrate for levan production by Halomonas sp. Appl Microbiol Biotechnol 89:1729–1740
Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V et al (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108
Lin Y, Fan H, Hao X, Johnstone L, Hu Y, Wei G et al (2012) Draft genome sequence of Halomonas sp. strain HAL1, a moderately halophilic arsenite-oxidizing bacterium isolated from goldmine soil. J Bacteriol 194:199–200
Llamas I, Amjres H, Mata JA, Quesada E, Béjar V (2012) The potential biotechnological applications of the exopolysaccharide produced by the halophilic bacterium Halomonas almeriensis. Molecules 17:7103–7120
Martínez-Cánovas MJ, Quesada E, Llamas I, Béjar V (2004) Halomonas ventosae sp. nov., a moderately halophilic, denitrifying, exopolysaccharide-producing bacterium. Int J Syst Evol Microbiol 54:733–737
Oren A (2002) Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J Ind Microbiol Biotechnol 28:56–63
Pastor JM, Salvador M, Argandoña M, Bernal V, Reina-Bueno M, Csonka LN et al (2010) Ectoines in cell stress protection: uses and biotechnological production. Biotechnol Adv 28:782–801
Pastor JM, Bernal V, Salvador M, Argandoña M, Vargas C, Csonka L et al (2013) Role of central metabolism in the osmoadaptation of the halophilic bacterium Chromohalobacter salexigens. J Biol Chem 288:17769–17781
Philip S, Keshavarz T, Roy I (2007) Polyhydroxyalkanoates: biodegradable polymers with a range of application. J Chem Technol Biotechnol 82:233–247
Poli A, Kazak H, Gürleyendağ B, Tommonaro G, Pieretti G, Toksoy Öner E et al (2009) High level synthesis of levan by a novel Halomonas species growing on defined media. Carbohydr Polym 78:651–657
Poli A, Nicolaus B, Denizci AA, Yavuzturk B, Kazan D (2012) Halomonas smyrnensis sp. nov., a moderately halophilic, exopolysaccharide-producing bacterium from Çamaltı Saltern Area. Int J Syst Evol Microbiol 63:10–18
Rodriguez-Valera F, Lillo JAG (1992) Halobacteria as producers of polyhydroxyalknoates. FEMS Microbiol Rev 103:181–186
Saier MH, Yen MR, Noto K, Tamang DG, Elkan C (2009) The transporter classification database (TCDB): recent advances. Nucleic Acids Res 37:274–278
Sam S, Kucukasik F, Yenigun O, Nicolaus B, Toksoy Oner E, Yukselen MA (2011) Flocculating performances of exopolysaccharides produced by a halophilic bacterial strain cultivated on agro-industrial waste. Bioresour Technol 102:1788–1794
Sarilmiser Kazak H, Ates O, Ozdemir G, Arga KY, Toksoy Oner E (2014) Effective stimulating factors for microbial levan production by Halomonas smyrnensis AAD6T. J Biosci Bioeng 92:28–34
Schiefner A, Breed J, Bösser L, Kneip S, Gade J, Holtmann G et al (2004) Cation–pi interactions as determinants for binding of the compatible solutes glycine betaine and proline betaine by the periplasmic ligand-binding protein ProX from Escherichia coli. J Biol Chem 279:5588–5596
Schwibbert K, Marin-Sanguino A, Bagyan I, Heidrich G, Lentzen G, Seitz H et al (2011) A blueprint of ectoine metabolism from the genome of the industrial producer Halomonas elongata DSM 2581 T. Environ Microbiol 13:1973–1994
Sezer AD, Kazak H, Toksoy Oner E, Akbuğa J (2011) Levan-based nanocarrier system for peptide and protein drug delivery: optimization and influence of experimental parameters on the nanoparticle characteristics. Carbohydr Polym 84:358–363
Sima F, Cansever Mutlu E, Eroglu MS, Sima LE, Serban N, Ristoscu C (2011) Levan nanostructured thin films by MAPLE assembling. Biomacromolecules 12:2251–2256
Sima F, Axente E, Sima LE, Tuyel U, Eroglu MS, Serban N (2012) Combinatorial matrix-assisted pulsed laser evaporation: single-step synthesis of biopolymer compositional gradient thin film assemblies. Appl Phys Lett 101:233705
Sogutcu E, Emrence Z, Arikan M, Cakiris A, Abaci N, Öner ET (2012) Draft genome sequence of Halomonas smyrnensis AAD6T. J Bacteriol 194:5690–5691
Sutherland IW (1998) Bacterial surface polysaccharide: structure and function. Int Rev Cytol 113:187–231
Takami H, Takaki Y, Uchiyama I (2002) Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res 15:3927–3935
Ventosa A, Nieto JJ, Oren A (1998) Biology of moderately halophilic aerobic bacteria. Microbiol Mol Biol Rev 62:504–544
Widmann J, Harris JK, Lozupone C, Wolfson A, Knight R (2010) Stable tRNA-based phylogenies using only 76 nucleotides. RNA 16:1469–1477
Wolfe-Simon F, Switzer Blum J, Kulp TR, Gordon GW, Hoeft SE, Pett-Ridge J et al (2011) A bacterium that can grow by using arsenic instead of phosphorus. Science 332:1163–1166
Zhang J, Chiodini R, Badr A, Zhang G (2011) The impact of next-generation sequencing on genomics. J Genet Genomics 38:95–109
Authors’ contributions
ED conceived and designed the sequencing experiments, carried out the bioinformatics analysis, analyzed the genome data and wrote the manuscript; TO participated in analysis of the genome data and reconstructed the metabolic model; ZE carried out the bioinformatics analysis, contributed to sequencing experiments and analysis of genome data; MA contributed to bioinformatics analysis and sequencing experiments; ETÖ participated in analysis of the genome data and helped to write the manuscript; DÜ designed and coordinated the sequencing experiments as well as the bioinformatics analysis and helped to write the manuscript; KYA coordinated, conceived and designed the whole project, analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This research has been financially supported by The Scientific and Technological Research Council of Turkey (TUBITAK) through Grant MAG/110M613 and by Marmara University Research Fund through Grant FEN-C-YLP-060911-0280.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Table S1. The coding sequences, their genomic positions, and function annotation results.
Additional file 2:
Table S2. Nucleotide and amino acid sequences of predicted coding sequences.
Additional file 3:
Table S3. Subsystem annotation results of the predicted coding sequences.
Additional file 5:
Table S4. The distribution of the proteins and associated genes of the global metabolism.
40064_2015_1184_MOESM7_ESM.docx
Additional file 7: Text document including the additional information on other cellular processes represented in H. smyrnensis AAD6T genome.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Diken, E., Ozer, T., Arikan, M. et al. Genomic analysis reveals the biotechnological and industrial potential of levan producing halophilic extremophile, Halomonas smyrnensis AAD6T. SpringerPlus 4, 393 (2015). https://doi.org/10.1186/s40064-015-1184-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-015-1184-3