
Abstract
Interleukin-33 (IL-33) is a member of the interleukin-1 (IL-1) cytokine family. It is preferentially and constitutively expressed in different structural cells such as epithelial cells, endothelial cells, and smooth muscle cells. During necrosis of these cells (after tissue injury or cell damage), the IL-33 that is released may be recognized by different types of immune cells, such as eosinophils, basophils and, especially, mast cells. IL-33 needs the specific receptor ST2 (membrane-bound receptor) and Interleukin-1 receptor accessory protein heterodimer for its binding, which instigates the production of different types of cytokines and chemokines that have crucial roles in the exacerbation of allergic diseases and inflammation. IL-33 and mast cells have been influentially associated to the pathophysiology of allergic diseases and inflammation. IL-33 is a crucial regulator of mast cell functions and might be an attractive therapeutic target for the treatment of allergic and inflammatory diseases. In this review, we summarize the current knowledge regarding the roles of IL-33 and mast cells in the pathogenesis of allergies and inflammation.
Similar content being viewed by others


Background
Interleukin-33 is a recently discovered cytokine that belongs to the IL-1 super-family and is mainly expressed by different types of structural cells [1, 2]. IL-33 binds to a specific receptor named T1/ST2 (also known as ST2) that belongs to the toll like receptor (TLR)/IL1R super family [3]. The T1/ST2 receptor forms heterodimer with the IL-1 receptor accessory protein (IL-1RAcP). Both receptors are required for the binding and action of IL-33. ST2 receptor has two major isoforms: a transmembrane form (ST2 or ST2L) and a soluble form (sST2) [2]. The ST2L isoform is mainly expressed on mast cells, basophils, dendritic cells, natural killer cells and Th2 lymphocytes [1–3]. IL-33 is considered an alarm in molecule due to its release after necrosis or tissue damage. In contrast, apoptosis leads to the inactivation of IL-33 because it is cleaved by caspases. Different stimuli such as bacterial, viral, fungal infections and allergen challenges can trigger the release of IL-33. Recent research suggests that IL-33 plays an important role in allergy and inflammation. In this review, we will focus on the recent advancements in the field of IL-33 and its association with mast cells in the context of allergy and inflammation.
Source of IL-33: expression, release and processing
Expression of IL-33 in physiological and pathological conditions
Interleukin-33 transcript and protein is widely expressed in different cell types including in cells of both hematopoietic as well as non-hematopoietic origin such as macrophages, dendritic cells, fibroblasts, adipocytes, smooth muscle cells, endothelial cells, bronchial, osteoblast and intestinal epithelial cells [4, 5]. Schmitz et al. [5] demonstrated that IL-33 mRNA is expressed in purified dendritic cells, epithelial cells, activated macrophages and it was also confirmed that its expression was much higher in stomach, lung, brain and skin tissues. A more detailed summary of IL-33 distribution is shown in Table 1.
Like IL-1α/β and IL-18, IL-33 functions as a transcriptional regulator [6] in high endothelial venules (HEVs), and has been reported to be also expressed within the nuclei of epithelial cells [7]. Wood et al. [8] studied the expression of IL-33, IL-1RL1 and IL-1RAcP gene in human pre-adipocytes and in adipocytes (SGBS cells). Expression of IL-33 has also been detected in epithelial cells, skin, lungs, and gastrointestinal tract [7, 9]. Andronicos et al. [10] established that damage caused by motile gastrointestinal nematode larvae in parasitic infection significantly induced IL-33 mRNA expression in epithelial cells. Later, it was reported that IL-33 also plays an important role in innate immune responses, for example during influenza virus infections in the lungs [11] and in helminth infections in the intestine [12, 13].
Interleukin-33 expression was found to be increased in several pathological conditions such as airway smooth muscle cell [14] and lung epithelial cells of asthmatic patients [9] as well as in airway epithelial cells in COPD patients [15]. Expression of IL-33 was also reported in liver cells (hepatocytes) and in inflamed colon in a mouse model of acute hepatitis [16] and colitis [4], respectively. IL-33 also plays a crucial roles in the initiation as well as amplification of a type 2 response in group 2 innate lymphoid cells (ILC2s) [17]. These results were further confirmed in IL-33-deficient mice. Moreover, Th2 cell differentiation and eosinophilic lung inflammation were found to be impaired in intranasally challenged IL-33-deficient mice [18, 19]. IL-33 is the essential factor for severe allergic lung inflammation [20]. IL-33 signaling is required for causing airway eosinophilia and production of IL-5 as well as IL-13 from lung ILC2s following fungal allergen challenge Alternaria alternate [21]. IL-33 also seems to be essential for development of allergic rhinitis induced by ragweed pollen challenge as IL-33 knockout mice failed to induce ragweed pollen induce allergic rhinitis [22]. IL-33 is also an effective stimulator for skin ILC2s, and it is directly associated with skin inflammation and mouse model of atopic dermatitis [23].
The type 2 (ILC2) innate lymphoid cells localized in mucosal tissues (lung and intestine), adipose tissue and lymphoid organs (spleen, lymph node) are the major target cells of IL-33 [12, 24, 25]. These cells express high expression levels of ST2 and secretion of significant quantities of the type 2 cytokines IL-5 and IL-13, and pro-inflammatory IL-6 from ILC2. Further studies have shown that IL-33 not only activates mast cells, but also other immune cells, such as granulocytes, macrophages, NK and Th2 cells [26].
Release of IL-33
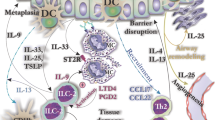
Interleukin-33 is stored in the nucleus and secreted upon necrosis or damage and released in response to cell injury, infection or mechanical damage [27, 28]. The high levels of constitutive IL-33 may act as a novel alarmin (intracellular alarm signal released after cell injury) to alert the immune system after endothelial or epithelial cell damage during trauma or infection (Fig. 1) [7].

The IL-33/ST2 signaling pathway in mast cells. IL-33 is primarily expressed by different types of structural cells. Damage to these cells by external stimuli results in cell injury followed by release of IL-33. IL-33 can be neutralized by binding to soluble ST2 (sST2) or recognized by membrane bound ST2 receptor, which subsequently leads to activation of MyD88 dependent signaling pathways and the release of mast cell mediators that play important roles in the progression of allergic diseases
The IL-1 family members do not possess signal peptide to release the proteins via endoplasmic reticulum and Golgi pathway [29]. Indeed, these interleukins are already translated and stored in the nucleus of the cells and released immediately during emergency conditions such as infection, injury or inflammation due to other stressors [30]. Extracellular IL-33 has also been detected in human blood and synovial fluids in pathological conditions [31], where cells have been damaged (during rheumatoid arthritis or infection with influenza virus, respectively) and in mouse peritoneal and bronchoalveolar lavage fluid [32]. However, in recent studies, it has been proposed that IL-33 may be released without cell damage and necrosis. Kouzaki et al. [33] found that exposing the human airway epithelial cells to A. alternata induces an acute extracellular danger signal, ATP, which releases IL-33 through activation of P2 purinergic receptors.
Processing of IL-33
Interleukin-33 has been proposed to be a cytokine with dual function; it acts as a traditional cytokine through activation of the ST2L receptor complex as well as an intracellular nuclear factor with transcriptional regulatory properties. IL-33 shows the closest homology to IL-18 among the members of the IL-1 family of cytokines which are synthesized as full length pro-peptides. It was reported that IL-33 is synthesized as a 30 kDa peptide. It consists of N-terminal helix-turn-helix (HTH) motif responsible for nuclear translocation and chromatin binding [6] and an IL-1-like C-terminal domain, which is cleaved by caspase 1 to form an active 18 kDa mature peptide [28, 34, 35]. Later, it was confirmed that IL-33 protein contains the cleavage sites for caspase-3 and caspase-7 and it can be expressed and released by activated macrophages [36]. The processing of IL-33 by caspases results in its inactivation [28, 35]. IL-33 inactivation by caspases during apoptosis could be to prevent IL-33 release during programmed cell death, which does not require an inflammatory response.
Lingel et al. [37] studied the structure and interaction of IL-33 with its receptor ST2 and IL-1RAcP by X-ray crystallography as well as by NMR spectroscopy. Lefrancais et al. [38] demonstrated that mature form of IL-33 (after cleavage by mast cell proteases) are more potent than full length IL-33. During Inflammation, neutrophil-released proteases may regulate IL-33 activity. Mast cell serine proteases cleave the full-length IL-33 (IL-331–270) and liberate active forms: IL-3395–270, IL-3399–270, and IL-33109–270. These cleaved IL-33 forms have 10 times greater potency than the full-length protein [38]. Roy et al. [39] reported that mast cell chymase but not tryptase cleaves IL-33 and results in increased bioactivity. This finding suggests that IL-33 activity could be exacerbated by the inflammatory environment. It has also been shown that serine proteases released by inflammatory cells play a critical role in the generation of super active forms of IL-33 and enhance immune response in asthma, rheumatoid arthritis, intestinal inflammation and other diseases [40, 41].
Interleukin-33 signaling: involvement of the MyD88 and activation of MAP Kinases
The IL-33 mediated downstream signaling pathway is governed through ST2 and IL-1RAcP receptors. In an in vivo model, mice deficient either in IL-1RAcP or ST2 did not show an inflammatory reaction in response to IL-33 administration [42]. IL-33 binding by the ST2 receptor leads to the activation and recruitment of MyD88 adapter protein along with IL-1R-associated kinase1 (IRAK1), IRAK4 and TNFR-associated factor 6 (TRAF6) [5, 43]. This signaling cascade further leads to the activation of transcription factors such as NF-ĸB and MAP kinases and the production of inflammatory mediators (Fig. 1) [3]. MyD88 is crucial for several functional responses to IL-33 such as survival cytokine production and MCs proliferation [44, 45]. IL-33 treatment further leads to the activation of different kinases such as ERK1/2, p38MAPK, and JNK (Fig. 1) [5].
Interleukin-33 mediated signaling pathways further modulate MC functions. It has also been reported that IL-33 can activate MCs [46], and act directly on Th2 cells to increase secretion of Th2 cytokines such as IL-5 and IL-13 [5, 47]. Furthermore, IL-33 functions as a chemo-attractant for Th2 cells [48]. Research from several studies has indicated that IL-33 also acts as a potent activator of mast cells and basophils and reported to induce migration, maturation, adhesion, promote survival and the production of several pro-inflammatory cytokines in these cells [44, 49–51]. The IL-33 mediated downstream signaling pathway including functional aspects is shown in Table 2.
Role of IL-33 in allergic disease
Interleukin-33 is considered to be linked to the development of several allergic diseases such as asthma and atopic dermatitis. IL-33 is also thought to accelerate Th17 cell-mediated airway inflammation via MCs [52]. Thus, it is evident from studies that IL-33 acts not only as a Th2-inducing cytokine, but also as a proinflammatory cytokine in various immune responses as do IL-1 and IL-18.
Asthma is characterized by chronic inflammation of the airways which is associated with variable airflow obstruction arising from various genetic and environmental factors. It involves the activation of MCs, Th2 cells, IgE producing B cells, basophils, eosinophils and lungs epithelial, smooth muscle cells and macrophages. Oshikawa et al. [53] and Hayakawa et al. [54] observed elevated levels of soluble ST2 as well as IL-33 mRNA in the serum and lung tissues, respectively in an ovalbumin (OVA)-induced murine asthma model of airway inflammation. Different advanced approaches, such as the use of anti-ST2 antibody (clone E310) [55, 56], anti-IL-33 antibody [57], or soluble ST2-Fc fusion protein [53] have been used to investigate the role of the ST2/IL-33 pathway in asthma models. Pre-treatment with these antibodies significantly inhibits airway inflammation and the Th2-associated responses. These antibodies also reduced IgE level in serum and the numbers of eosinophils in bronchoalveolar lavage in a murine model of allergic asthma. Kurowska et al. [47] detected IL-33 protein in the lungs of mice with OVA/alum-induced airway inflammation. IL-33 has been reported to also induce allergic bronchoconstriction through mast cell activation in mice [58]. IL-33 increases the expression of tryptophan hydroxylase 1, serotonin synthesis, and storage and thus results in airway obstruction in asthma [58].
Stolarski et al. [59] reported that IL-33 induces eosinophil mediated massive airway inflammation of the lung tissue and markedly elevated local concentrations of IL-5 and IL-13 and induced goblet cell hyperplasia in ova induced asthma model in mice. Lee et al. [60] investigated the role of anti-IL-33 antibodies and sST2 in the blockade of airway inflammation in a murine model of asthma and confirmed that both treatments were successful in reducing the total cell count and may serve as therapeutic agents for allergic asthma. Several studies have shown that IL-33 is expressed more abundantly in asthma patients than healthy individuals [47, 61]. Additionally, these results were also confirmed by elevated IL-33 expression in bronchial epithelial cells of asthma patients compared to healthy individuals by immuno-histochemical studies [59]. Role of IL-33 in mast cell activation and airway smooth muscle wound repair has also been reported [62] which suggests that IL-33 presents important target to modulate mast cell-airway smooth muscle (ASM) crosstalk in asthma.
Atopic dermatitis (AD) is a chronic inflammatory skin disease. Shimizu et al. [63] explored the association of AD with a polymorphism of the ST2 gene and suggested that the IL-33-ST2 axis plays a pivotal role in AD. Recently, Savinko et al. [64] and Meephansan et al. [65] reported the up-regulation of IL-33 in the epidermis and the infiltration of ST2-positive cells in the dermis of the skin lesion of AD patients. Imai et al. [66] reported that IL-33 from epidermal keratinocytes activates ILC2s in the skin and lymph nodes and stimulates the production of IL-5 from those cells to induce AD-like dermatitis with eosinophil infiltrates. On the basis of these observations IL-33 is considered as a unique danger alarmin and pathogenic driver in AD [67].
Conclusions
Interleukin-33 is a unique cytokine that plays an essential role in regulating MC associated immune responses in allergic diseases. In the present scenario, the IL-33/ST2 pathway is being used as a novel therapeutic target for understanding the role of IL-33 in diseases associated with MCs. However, the elementary mechanisms of the release, expression, processing and regulation of IL‑33 in allergic diseases are not yet defined properly and may be crucial for the development of future therapeutic targets. Future studies are essential to recognize the biological and clinical significance of IL-33 in allergic diseases.
Abbreviations
- BMMCs:
-
bone marrow-derived murine mast cell
- ERK:
-
extracellular signal-regulated kinases
- HUCBMCs:
-
human umbilical cord blood mast cells
- IL-1RAcP:
-
IL-1 receptor accessory protein
- IL-1RL1:
-
IL-1 receptor-like 1
- ILCs:
-
innate lymphoid cells
- JNK:
-
c-Jun N-terminal kinases
- MyD88:
-
myeloid differentiation primary response gene (88)
- NF- κB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- p38 MAPK:
-
p38 mitogen-activated protein kinases
- SGBS:
-
Simpson-Golabi-Behmel syndrome
References
Yasuoka S, Kawanokuchi J, Parajuli B, Jin S, Doi Y, Noda M, et al. Production and functions of IL-33 in the central nervous system. Brain Res. 2011;1385:8–17. doi:10.1016/j.brainres.2011.02.045.
Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10(2):103–10. doi:10.1038/nri2692.
Miller AM. Role of IL-33 in inflammation and disease. J Inflamm (Lond). 2011;8(1):22. doi:10.1186/1476-9255-8-22.
Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. 2012;188(7):3488–95. doi:10.4049/jimmunol.1101977.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90. doi:10.1016/j.immuni.2005.09.015.
Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104(1):282–7. doi:10.1073/pnas.0606854104.
Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One. 2008;3(10):e3331. doi:10.1371/journal.pone.0003331.
Wood IS, Wang B, Trayhurn P. IL-33, a recently identified interleukin-1 gene family member, is expressed in human adipocytes. Biochem Biophys Res Commun. 2009;384(1):105–9. doi:10.1016/j.bbrc.2009.04.081.
Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125(3):752–4. doi:10.1016/j.jaci.2009.12.935.
Andronicos NM, McNally J, Kotze AC, Hunt PW, Ingham A. Trichostrongylus colubriformis larvae induce necrosis and release of IL33 from intestinal epithelial cells in vitro: implications for gastrointestinal nematode vaccine design. Int J Parasitol. 2012;42(3):295–304. doi:10.1016/j.ijpara.2012.01.007.
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8. doi:10.1038/ni.2045.
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70. doi:10.1038/nature08900.
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107(25):11489–94. doi:10.1073/pnas.1003988107.
Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183(8):5094–103. doi:10.4049/jimmunol.0802387.
Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Investig. 2013;123(9):3967–82. doi:10.1172/JCI65570.
Arshad MI, Piquet-Pellorce C, L’Helgoualc’h A, Rauch M, Patrat-Delon S, Ezan F, et al. TRAIL but not FasL and TNFalpha, regulates IL-33 expression in murine hepatocytes during acute hepatitis. Hepatology. 2012;56(6):2353–62. doi:10.1002/hep.25893.
Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–7. doi:10.1016/j.coi.2014.09.004.
Kamijo S, Takeda H, Tokura T, Suzuki M, Inui K, Hara M, et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J Immunol. 2013;190(9):4489–99. doi:10.4049/jimmunol.1201212.
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–35. doi:10.1016/j.immuni.2014.01.011.
Louten J, Rankin AL, Li Y, Murphy EE, Beaumont M, Moon C, et al. Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int Immunol. 2011;23(5):307–15. doi:10.1093/intimm/dxr006.
Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage—CD25 + CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188(3):1503–13. doi:10.4049/jimmunol.1102832.
Haenuki Y, Matsushita K, Futatsugi-Yumikura S, Ishii KJ, Kawagoe T, Imoto Y, et al. A critical role of IL-33 in experimental allergic rhinitis. J Allergy Clin Immunol. 2012;130(1):184 e11–194 e11. doi:10.1016/j.jaci.2012.02.013.
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–50. doi:10.1084/jem.20130351.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4. doi:10.1038/nature08636.
Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62. doi:10.1038/ni.2104.
Oboki K, Ohno T, Kajiwara N, Saito H, Nakae S. IL-33 and IL-33 receptors in host defense and diseases. Allergol Int. 2010;59(2):143–60. doi:10.2332/allergolint.10-RAI-0186.
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13(7):851–6. doi:10.1038/nm1603.
Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. 2009;106(22):9021–6. doi:10.1073/pnas.0812690106.
Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol. 2015;33:49–77. doi:10.1146/annurev-immunol-032414-112306.
Martin MU. Special aspects of interleukin-33 and the IL-33 receptor complex. Semin Immunol. 2013;25(6):449–57. doi:10.1016/j.smim.2013.10.006.
Matsuyama Y, Okazaki H, Tamemoto H, Kimura H, Kamata Y, Nagatani K, et al. Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J Rheumatol. 2010;37(1):18–25. doi:10.3899/jrheum.090492.
Le Goffic R, Arshad MI, Rauch M, L’Helgoualc’h A, Delmas B, Piquet-Pellorce C, et al. Infection with influenza virus induces IL-33 in murine lungs. Am J Respir Cell Mol Biol. 2011;45(6):1125–32. doi:10.1165/rcmb.2010-0516OC.
Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186(7):4375–87. doi:10.4049/jimmunol.1003020.
Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31(1):84–98. doi:10.1016/j.immuni.2009.05.007.
Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 2009;284(29):19420–6. doi:10.1074/jbc.M901744200.
Ohno T, Oboki K, Kajiwara N, Morii E, Aozasa K, Flavell RA, et al. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J Immunol. 2009;183(12):7890–7. doi:10.4049/jimmunol.0802449.
Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, et al. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors—insight into heterotrimeric IL-1 signaling complexes. Structure. 2009;17(10):1398–410. doi:10.1016/j.str.2009.08.009.
Lefrancais E, Cayrol C. Mechanisms of IL-33 processing and secretion: differences and similarities between IL-1 family members. Eur Cytokine Netw. 2012;23(4):120–7. doi:10.1684/ecn.2012.0320.
Roy A, Ganesh G, Sippola H, Bolin S, Sawesi O, Dagalv A, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem. 2014;289(1):237–50. doi:10.1074/jbc.M112.435156.
Cascao R, Rosario HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: more than simple final effectors. Autoimmun Rev. 2010;9(8):531–5. doi:10.1016/j.autrev.2009.12.013.
Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5(4):354–66. doi:10.1038/mi.2012.24.
Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179(4):2551–5.
Funakoshi-Tago M, Tago K, Hayakawa M, Tominaga S, Ohshio T, Sonoda Y, et al. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal. 2008;20(9):1679–86. doi:10.1016/j.cellsig.2008.05.013.
Saluja R, Ketelaar ME, Hawro T, Church MK, Maurer M, Nawijn MC. The role of the IL-33/IL-1RL1 axis in mast cell and basophil activation in allergic disorders. Mol Immunol. 2015;63(1):80–5. doi:10.1016/j.molimm.2014.06.018.
Saluja R, Hawro T, Eberle J, Church MK, Maurer M. Interleukin-33 promotes the proliferation of mouse mast cells through ST2/MyD88 and p38 MAPK-dependent and Kit-independent pathways. J Biol Regul Homeost Agents. 2014;28(4):575–85.
Enoksson M, Lyberg K, Moller-Westerberg C, Fallon PG, Nilsson G, Lunderius-Andersson C. Mast cells as sensors of cell injury through IL-33 recognition. J Immunol. 2011;186(4):2523–8. doi:10.4049/jimmunol.1003383.
Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5 + T cells and promotes allergic-induced airway inflammation independent of IL-4. Journal of immunology. 2008;181(7):4780–90.
Komai-Koma M, Brombacher F, Pushparaj PN, Arendse B, McSharry C, Alexander J, et al. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naive mice. Allergy. 2012;67(9):1118–26. doi:10.1111/j.1398-9995.2012.02859.x.
Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. 2007;87(10):971–8. doi:10.1038/labinvest.3700663.
Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA. 2008;105(31):10913–8. doi:10.1073/pnas.0801898105.
Schneider E, Petit-Bertron AF, Bricard R, Levasseur M, Ramadan A, Girard JP, et al. IL-33 activates unprimed murine basophils directly in vitro and induces their in vivo expansion indirectly by promoting hematopoietic growth factor production. J Immunol. 2009;183(6):3591–7. doi:10.4049/jimmunol.0900328.
Cho KA, Suh JW, Sohn JH, Park JW, Lee H, Kang JL, et al. IL-33 induces Th17-mediated airway inflammation via mast cells in ovalbumin-challenged mice. Am J Physiol Lung Cell Mol Physiol. 2012;302(4):L429–40. doi:10.1152/ajplung.00252.2011.
Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y. Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin Exp Allergy. 2002;32(10):1520–6.
Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282(36):26369–80. doi:10.1074/jbc.M704916200.
Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med. 1999;190(7):895–902.
Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am J Respir Crit Care Med. 2009;179(9):772–81. doi:10.1164/rccm.200805-666OC.
Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun. 2009;386(1):181–5. doi:10.1016/j.bbrc.2009.06.008.
Sjoberg LC, Gregory JA, Dahlen SE, Nilsson GP, Adner M. Interleukin-33 exacerbates allergic bronchoconstriction in the mice via activation of mast cells. Allergy. 2015;70(5):514–21. doi:10.1111/all.12590.
Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185(6):3472–80. doi:10.4049/jimmunol.1000730.
Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, et al. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res. 2014;40(2):66–76. doi:10.3109/01902148.2013.870261.
Raeiszadeh Jahromi S, Mahesh PA, Jayaraj BS, Madhunapantula SR, Holla AD, Vishweswaraiah S, et al. Serum levels of IL-10, IL-17F and IL-33 in patients with asthma: a case-control study. J Asthma. 2014;51(10):1004–13. doi:10.3109/02770903.2014.938353.
Kaur D, Gomez E, Doe C, Berair R, Woodman L, Saunders R, et al. IL-33 drives airway hyper-responsiveness through IL-13-mediated mast cell: airway smooth muscle crosstalk. Allergy. 2015;70(5):556–67. doi:10.1111/all.12593.
Shimizu M, Matsuda A, Yanagisawa K, Hirota T, Akahoshi M, Inomata N, et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum Mol Genet. 2005;14(19):2919–27. doi:10.1093/hmg/ddi323.
Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimaki S, et al. IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol. 2012;132(5):1392–400. doi:10.1038/jid.2011.446.
Meephansan J, Tsuda H, Komine M, Tominaga S, Ohtsuki M. Regulation of IL-33 expression by IFN-gamma and tumor necrosis factor-alpha in normal human epidermal keratinocytes. J Invest Dermatol. 2012;132(11):2593–600. doi:10.1038/jid.2012.185.
Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci USA. 2013;110(34):13921–6. doi:10.1073/pnas.1307321110.
Cevikbas F, Steinhoff M. IL-33: a novel danger signal system in atopic dermatitis. J Invest Dermatol. 2012;132(5):1326–9. doi:10.1038/jid.2012.66.
Demyanets S, Konya V, Kastl SP, Kaun C, Rauscher S, Niessner A, et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011;31(9):2080–9. doi:10.1161/ATVBAHA.111.231431.
Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G821–32. doi:10.1152/ajpgi.00178.2010.
Velickovic M, Pejnovic N, Petrovic R, Mitrovic S, Jeftic I, Kanjevac T, et al. Expression of interleukin-33 and its receptor ST2 in periapical granulomas and radicular cysts. J Oral Pathol Med. 2015;. doi:10.1111/jop.12312.
Kay AB, Clark P, Maurer M, Ying S. Elevations in T-helper-2-initiating cytokines (interleukin-33, interleukin-25 and thymic stromal lymphopoietin) in lesional skin from chronic spontaneous (‘idiopathic’) urticaria. Br J Dermatol. 2014;. doi:10.1111/bjd.13621.
Tong X, Lu F. IL-33/ST2 involves the immunopathology of ocular toxoplasmosis in murine model. Parasitol Res. 2015;114(5):1897–905. doi:10.1007/s00436-015-4377-3.
Tjota MY, Hrusch CL, Blaine KM, Williams JW, Barrett NA, Sperling AI. Signaling through FcRgamma-associated receptors on dendritic cells drives IL-33-dependent TH2-type responses. J Allergy Clin Immunol. 2014;134(3):706 e8–713 e8. doi:10.1016/j.jaci.2014.06.013.
Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol. 2007;82(6):1481–90. doi:10.1189/jlb.0407200.
Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol. 2011;41(3):760–72. doi:10.1002/eji.201040718.
Yang Z, Sun R, Grinchuk V, Fernandez-Blanco JA, Notari L, Bohl JA, et al. IL-33-induced alterations in murine intestinal function and cytokine responses are MyD88, STAT6, and IL-13 dependent. Am J Physiol Gastrointest Liver Physiol. 2013;304(4):G381–9. doi:10.1152/ajpgi.00357.2012.
Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20(6):791–800. doi:10.1093/intimm/dxn037.
Tung HY, Plunkett B, Huang SK, Zhou Y. Murine mast cells secrete and respond to interleukin-33. J Interferon Cytokine Res. 2014;34(3):141–7. doi:10.1089/jir.2012.0066.
Matsuda A, Okayama Y, Terai N, Yokoi N, Ebihara N, Tanioka H, et al. The role of interleukin-33 in chronic allergic conjunctivitis. Invest Ophthalmol Vis Sci. 2009;50(10):4646–52. doi:10.1167/iovs.08-3365.
Authors’ contributions
RS, MK, MKC and MM designed the manuscript. RS and MK wrote the first draft and all authors were involved in drafting/revising the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge the BMBS COST ActionBM1007 (Mast Cells and Basophils—Targets for Innovative Therapies) of the European community. RS acknowledges support from the Department of Biotechnology, India for Ramalingaswami Re-entry Fellowship.
Compliance with ethical guidelines
Competing interests The authors declare no financial or commercial conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Saluja, R., Khan, M., Church, M.K. et al. The role of IL-33 and mast cells in allergy and inflammation. Clin Transl Allergy 5, 33 (2015). https://doi.org/10.1186/s13601-015-0076-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13601-015-0076-5