
Abstract
Porcine reproductive and respiratory syndrome (PRRS) is a serious disease burdening global swine industry. Infection by its etiological agent, PRRS virus (PRRSV), shows a highly restricted tropism of host cells and has been demonstrated to be mediated by an essential scavenger receptor (SR) CD163. CD163 fifth SR cysteine-rich domain (SRCR5) is further proven to play a crucial role during viral infection. Despite intense research, the involvement of CD163 SRCR5 in PRRSV infection remains to be elucidated. In the current study, we prepared recombinant monkey CD163 (moCD163) SRCR5 and human CD163-like homolog (hCD163L1) SRCR8, and determined their crystal structures. After comparison with the previously reported crystal structure of porcine CD163 (pCD163) SRCR5, these structures showed almost identical structural folds but significantly different surface electrostatic potentials. Based on these differences, we carried out mutational research to identify that the charged residue at position 534 in association with the one at position 561 were important for PRRSV-2 infection in vitro. Altogether the current work sheds some light on CD163-mediated PRRSV-2 infection and deepens our understanding of the viral pathogenesis, which will provide clues for prevention and control of PRRS.
Similar content being viewed by others

Introduction
PRRS is characterized by reproductive failures in sows of late-term gestation and respiratory signs in pigs of all ages. It causes significant economic losses to global swine industry [1, 2]. PRRS has an estimated annual cost of $664 million in the USA alone [3]. PRRSV, as the causative agent, is an enveloped single-stranded positive-sense RNA virus [4, 5] and belongs to the genus Betaarterivirus, family Arteriviridae, order Nidovirales [6, 7]. All PRRSV isolates are classified into PRRSV-1 and PRRSV-2 [8, 9]. In China, PRRSV-2 strains are predominantly prevalent [10, 11], and highly pathogenic variants of PRRSV-2 (HP-PRRSVs) result in serious declines in pig production throughout the country [12, 13].
PRRSV infection shows a strongly restricted tropism for host species and target cells. Swine, including domestic pigs and wild boar, are the only known hosts for PRRSV [14,15,16,17,18,19]. CD163-positive macrophages, particularly porcine alveolar macrophages (PAMs), are PRRSV primary target cells in vivo [20]. In addition, African green monkey kidney epithelial cell MA-104 and its derivatives, MARC-145 and CL2621 cells, support viral infection in vitro [16, 21]. The specific tropism of PRRSV is mediated by host cell receptor(s) [22,23,24]. Stable expression of SR CD163 from different species (pig, human and monkey) was found to render various non-permissive cells susceptible to PRRSV infection, including porcine kidney (PK 032495), Norden Laboratories feline kidney (NLFK) and baby hamster kidney (BHK-21) cells [25]. Other cell lines, such as SV40-transformed PAM 3D4/21 (CRL-2843) [26], PK-15 [27, 28] and murine macrophage-derived cell lines [29], were also susceptible to PRRSV with expression of pCD163. Recent studies have demonstrated that gene-edited pigs lacking functional pCD163 are completely resistant to PRRSV, which confirms that it is an indispensable receptor for the viral infection [30,31,32,33].
CD163 contains nine class B SRCR domains (SRCR1-9) in its large ectodomain [34, 35]. SRCR5 has been shown to play a crucial role during PRRSV infection in vitro [36,37,38]. Additionally, a significantly reduced permissiveness to PRRSV-2, particularly HP-PRRSV, was shown in the PAMs with pCD163 SRCR5 substitution by homologous hCD163L1 SRCR8 [31, 39]. Despite these studies, the mechanisms by which CD163 SRCR5 takes effect in PRRSV infection are not fully elucidated.
In this work, we prepared recombinant moCD163 SRCR5 in Pichia pastoris X-33 and hCD163L1 SRCR8 in Drosophila melanogaster Schneider 2 (S2) cells, respectively. After purification and crystallization, we determined their crystal structures and aligned them with that of pCD163 SRCR5 we previously reported [40]. Based on the structural comparison, we carried out mutational assays to explore which residues are important for PRRSV-2 infection and how they influence the viral infection in vitro.
Materials and methods
Materials, cell lines and viruses
All chemicals were purchased from Sigma-Aldrich Co., Ltd. (St. Louis, USA) or Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) unless otherwise stated.
The Drosophila S2 cells were kept in Schneider’s insect medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Grand Island, USA) and antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin; Gibco) at 28 °C in a humidified incubator. The PK-15 cells and MARC-145 cells were maintained routinely in Gibco Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated FBS and antibiotics at 37 °C in a humidified incubator with a 5% CO2 atmosphere.
The PRRSV-2 strain BJ-4 (Genbank ID: AF331831) was isolated on MARC-145 cells in China and kindly provided by Professor Hanchun Yang of China Agricultural University [41]. The HP-PRRSV strain HN07-1 (Genbank ID: KX766378.1) was isolated on PAMs in Henan Province of China by our laboratory [42].
Preparation of moCD163 SRCR5 in Pichia pastoris X-33 cells
MoCD163 SRCR5 was prepared in Pichia pastoris X-33 cells according to our previous studies [43, 44]. Briefly, the cDNA encoding moCD163 SRCR5 (residues 478-578, the numbering is according to UniProt entry Q2VLG4) was synthesized by Shanghai Sangon Biotech Co., Ltd. (Shanghai, China) and inserted into the expression vector pPICZαA (Invitrogen, Carlsbad, USA) between the Xho I and Sal I sites. The recombinant moCD163 SRCR5 expression vector was verified by Shanghai Sangon Biotech Co., Ltd., linearized using Pme I (New England Biolabs, Ipswich, USA) and transformed into competent X-33 cells by electroporation (1500 V, 25 μF, 200 Ω for 6 ms). For a large scale expression, recombinant X-33 cells were first grown in buffered minimal glycerol-complex medium (BMGY) (1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% yeast nitrogen base (YNB), 4 × 10–5% biotin, 1% v/v glycerol) to an OD600 reading of 2–6 and then in buffered minimal methanol-complex medium (BMMY) (1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% YNB, 4 × 10–5% biotin, 1% v/v methanol). The protein expression was induced every 24 h with 1% (v/v) methanol for 4 days.
Preparation of hCD163L1 SRCR8 in Drosophila S2 cells
The cDNA encoding hCD163L1 SRCR8 (residues 795-895, the numbering is according to UniProt entry Q9NR16) was synthesized by Shanghai Sangon Biotech Co., Ltd. and inserted into the expression vector pMT/BiP/V5-HisA (Invitrogen) between the Bgl II and Mlu I sites. The recombinant expression vector was verified by Shanghai Sangon Biotech Co., Ltd. and transfected into Drosophila S2 cells with the pCoBlast vector by Cellfectin II reagent according to Invitrogen’s instructions. The selected stably transfected Drosophila S2 cells were grown in Sf-900 II serum-free medium (Invitrogen) and induced by 0.75 mM CuSO4 for five days according to our methods [45, 46].
Purification of recombinant target proteins
After centrifugation and clarification by filtration, the supernatant containing each target protein was applied to GE Ni Sepharose excel column (Boston, USA) pre-equilibrated with 20 mM Tris–HCl pH 8.0, 150 mM NaCl. Each target protein was then eluted with 20 mM Tris–HCl pH 8.0, 150 mM NaCl and 200 mM imidazole. The eluent containing each target protein was further purified by GE Superdex 200 10/300 GL prepacked column on the GE AKTA Pure system (Uppsala, Sweden) with 20 mM Tris–HCl pH 8.0, 150 mM NaCl as elution buffer. The fraction containing moCD163 SRCR5 or hCD163L1 SRCR8 was collected, dialyzed, and concentrated to 7.75 and 10 mg/mL in 20 mM Tris–HCl pH 8.0, 20 mM NaCl, respectively.
Crystallization, data collection, and structural determination of target proteins
Crystallization of each target protein was carried out at room temperature (RT; 25 °C) by the sitting-drop vapor diffusion method with an equal volume of each target protein and various crystallization reagents from the Hampton crystallization screening kits (Aliso Viejo, USA). Single crystals of moCD163 SRCR5 were acquired under 0.1 M Bis–Tris pH 5.5, 25% PEG 3350, 0.2 M NaCl, and those of hCD163L1 SRCR8 were acquired under 0.1 M citric acid pH 3.5, 15% PEG 3350. The crystals were flash-frozen in liquid nitrogen using a cryoprotection solution with 20% glycerol in the crystallization solution. X-ray data sets of the crystals were collected at a wavelength of 0.979 Å on the beamlines BL18U and BL19U1 at the Shanghai Synchrotron Radiation Facility (SSRF) [47]. Diffraction data sets were processed using the HKL-3000 package [48]. The crystal structures were solved by molecular replacement using pCD163 SRCR5 (PDB code 5JFB) as the search model [40] with the program Molrep in CCP4 suites [49]. The structures were refined by CCP4 program package and manually adjusted by the molecular graphics program COOT [50]. Solvent molecules were added using a Fo-Fc Fourier difference map at 2.5 σ in the final refinement step. Statistics of data collection and final model refinement were summarized in Table 1. The final structures were analyzed by the software PyMOL [51].
Site-directed mutagenesis of SRCR5 in pCD163
We utilized a construct with complete wild-type (WT) pCD163 cDNA integrated into the PiggyBac transposon system (kindly provided by Professor Enmin Zhou, Northwest Agriculture and Forestry University, China) as a template to generate each single-site mutant encoding pCD163 G499R, E509H, S512N, T522D, E534K, E543K, H549S, P560Q, R561H and G564D (the numbering is according to UniProt entry Q2VL90). The primers designed for mutation were listed in Table 2. All mutation constructs were verified by Shanghai Sangon Biotech Co. Ltd.
Cell transfection with WT or mutant pCD163
PK-15 cells were seeded at a density of 4.0 × 105 cells/mL and incubated overnight. The PK-15 cells were transfected with the same amount (1 μg/well, 24-well plate or 3 μg/well, 6-well plate) construct of WT or mutant pCD163 using Lipofectamine LTX reagent with Plus reagent according to the manufacturer’s instructions (Invitrogen). The expression levels of WT and each mutant pCD163 were measured by immunofluorescence assay (IFA) or immunoblotting (IB).
Immunofluorescence assay (IFA)
Cells were grown in 24-well plates, fixed with 4% paraformaldehyde (PFA) for 15 min and permeabilized with 0.1% Triton X-100 in PBS at RT for 5 min. Anti-CD163 antibody (MCA2311GA; AbD Serotec, Hercules, USA), anti-PRRSV nucleocapsid (N) protein antibody (kept in our laboratory) and DAPI were used to stain CD163, PRRSV N protein and nuclei, respectively. Then the cells were stained with the appropriate secondary antibodies. Images were representative as a single slice of a stack from three independent experiments. Quantitative analyses of single channel fluorescence were performed using ImageJ software [52, 53].
Immunoblotting (IB)
The cells were harvested and lysed in radio immunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China). The lysates were normalized to equal amounts of tubulin, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and electro-transferred onto Immobilon-P membranes (Merck Millipore, Darmstadt, Germany). The membranes were blocked in 5% skimmed milk for 1 h, and probed with the mouse anti-human CD163 (MCA1853, AbD serotec), mouse anti-PRRSV N protein or mouse anti-β-tubulin monoclonal antibody (3G6, Abbkine, Wuhan, China). After incubation with horseradish peroxidase (HRP)-labeled goat anti-mouse IgG antibody as secondary antibody, the indicated proteins were visualized by enhanced chemiluminescence (ECL) reagent (Solarbio, Beijing, China).
Quantitative real-time PCR (RT-qPCR)
Total RNAs from PRRSV-inoculated PK-15 cells were extracted with TRIzol reagent (Invitrogen). The reversely transcribed cDNAs were prepared using the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa, Dalian, China) and amplified by RT-qPCR to measure RNA abundance on a 7500 Fast RT-PCR system (Applied Biosystems, Foster City, USA). PRRSV open reading frame (ORF) 7 gene was normalized with housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and relatively quantified by the 2−ΔΔCT method [54], or quantitated using a plasmid containing PRRSV ORF7 as the template to generate a standard curve to calculate the actual RNA copies [40]. Three replicates were run, and each experiment was independently repeated for three times.
PRRSV titration assay
The transfected cells were inoculated with PRRSV at a multiplicity of infection (MOI) of 1 and incubated at 37 °C for 3 h. The viruses not entering into the cells were then washed away. At 48 h post-infection (hpi), the progeny virus titers were measured by the 50% tissue culture infected dose (TCID50) assay in MARC-145 cells according to Reed and Muench [55].
PRRSV binding, entry and infection assays
For PRRSV binding assay [56,57,58], PRRSV strain BJ-4 or HN07-1 at a MOI of 1 was inoculated in the transfected cells at 4 °C for 1 h. After the unbound viruses was washed away, the level of cell-bound viral RNA (PRRSV ORF7) was measured by RT-qPCR. For PRRSV entry assay [59, 60], the unbound viruses were washed away and the inoculated cells were cultured at 37 °C for 3 h to allow viral entry. The viruses not entering into the cells were washed, and the entering viral RNA was analyzed by RT-qPCR. For PRRSV infection assay, the infected cells were further cultured for 9 h, and at 12 hpi, viral RNA abundance was analyzed by RT-qPCR. The PK-15 cells transfected with empty vector were inoculated with PRRSV and assayed in parallel as negative control. Three replicates were run, and each experiment was independently repeated for three times.
Statistical analysis
All experimental data were presented as group means and standard errors of the means (SEM). The experimental data were analyzed using the unpaired, 2-tailed Student t test with Origin software. Differences at the 95% confidence level (p < 0.05) were considered statistically significant.
Results
Crystal structures of moCD163 SRCR5 and hCD163L1 SRCR8
The recombinant moCD163 SRCR5 and hCD163L1 SRCR8 were eluted as monomer during purification. Their crystal structures were determined and refined to 1.58 Å in P1 space group and 2.0 Å in C2 space group, respectively (Table 1). Both these two target proteins were crystallized with two molecules in each asymmetric unit (data not shown). We analyzed only one representative molecule for each protein since the root mean square deviation (RMSD) differences of the two molecules were slight (0.603 Å for 86 matching Cα atoms of moCD163 SRCR5, 0.068 for 92 matching Cα atoms of hCD163L1 SRCR8).
In Figures 1A and 2A, both moCD163 SRCR5 and hCD163L1 SRCR8 adopted a compact heart shape and were characterized by the presence of a long flexible loop region. MoCD163 SRCR5 was comprised of two antiparallel β-sheets (β1-4, β7 and β5-6) and three helices (a single α-helix α2 and two 310-helices α1, α3), whereas hCD163L1 SRCR8 contained two antiparallel β-sheets (β1-4, β7 and β5-6) and two helices (a single α-helix α1 and a 310-helix α2). Four disulfide bonds were bridged in moCD163 SRCR5 (Cys487-Cys521, Cys503-Cys567, Cys516-Cys577 and Cys547-Cys557, the numbering is according to UniProt entry Q2VLG4, Figure 1B) and hCD163L1 SRCR8 (Cys804-Cys838, Cys820-Cys884, Cys833-Cys894 and Cys864-Cys874, the numbering is according to UniProt entry Q9NR16, Figure 2B), consistent with the typical disulfide linkage pattern of class B SRCR [34].

Crystal structure of moCD163 SRCR5 and comparison with pCD163 SRCR5. A The cartoon diagrams of moCD163 SRCR5 represented in the 180° rotation. Seven β-strands β1-7, three helices α1-3 and the loop regions are colored in blue, magenta and cyan, respectively. The N- and C-termini are labeled. B The ribbon diagrams of moCD163 SRCR5 showing the disulfide bonds represented in the 180° rotation. The disulfide bonds are colored in yellow and the bridged cysteines are labeled. C Structural comparison of moCD163 SRCR5 with pCD163 SRCR5. The crystal structures of moCD163 SRCR5 was aligned with pCD163 SRCR5 in cartoon diagrams. The pCD163 SRCR5 and moCD163 SRCR5 are in green and cyan, respectively. Their N- and C-termini are labeled, and their minor differences are circled in dashed lines.

Crystal structure of hCD163L1 SRCR8 and comparison with pCD163 SRCR5. A The cartoon diagrams of hCD163L1 SRCR8 represented in the 180° rotation. Seven β-strands β1-7, two helices α1-2 and the loop regions are colored as in Figure 1A. The N- and C-termini are labeled. B The ribbon diagrams of hCD163L1 SRCR8 showing the disulfide bonds represented in the 180° rotation as in Figure 1B. C Structural comparison of hCD163L1 SRCR8 with pCD163 SRCR5.The crystal structures of hCD163L1 SRCR8 was aligned with pCD163 SRCR5 in cartoon diagrams. The pCD163 SRCR5 and hCD163L1 SRCR are in green and magenta, respectively. Their N- and C-termini are labeled, and their differences are circled in dashed lines.
Structural comparison of moCD163 SRCR5 or hCD163L1 SRCR8 with pCD163 SRCR5
As shown in Figures 1C and 2C, a close comparison of moCD163 SRCR5 or hCD163L1 SRCR8 with pCD163 SRCR5 revealed that they shared almost identical structural folds (RMSD = 0.355 Å for 79 matching Cα atoms between moCD163 SRCR5 and pCD163 SRCR5, RMSD = 0.249 Å for 95 matching Cα atoms between hCD163L1 SRCR8 and pCD163 SRCR5). The minor differences between moCD163 SRCR5 and pCD163 SRCR5 were only observed in the long loop regions (Figure 1C). In addition to minor differences in the long loop region, hCD163L1 SRCR8 contained a shorter β4 (Ile842-Ser845) than that of pCD163 SRCR5 (Thr522-Leu527) (Figure 2C).
As shown in Figures 3A and B, moCD163 SRCR5 displayed a similar surface electrostatic potential as pCD163 SRCR5 did, so-called “D/E-R-rich” charge distribution [40]. In contrast, hCD163L1 SRCR8 showed a significantly different surface electrostatic potential from the other two proteins (Figure 3). There were more positively and negatively charged regions in hCD163L1 SRCR8 due to different amino acid contents (Figures 3 and 4A). Especially, certain regions with the opposed charge were clearly observed (Figure 3), where the acidic residues were replaced by the basic ones (Figure 4A). Based on the comparison, we hypothesize that the difference in the surface electrostatic potentials between hCD163L1 SRCR8 and pCD163 SRCR5 may result in reduced cell permissiveness to PRRSV-2 infection, rather than their structural folds.

Surface electrostatic potentials of pCD163 SRCR5 (A), moCD163 SRCR5 (B) and hCD163L1 SRCR8 (C). The figures are represented in the 180° rotation to depict basic residues in the positively-charged clusters (blue) and acidic residues in the negatively-charged areas (red). The differences of hCD163L1 SRCR8 from the other two proteins are indicated with dashed circles. The electrostatic potentials are colored from − 62 to + 62 kiloteslas/charge.

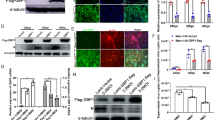
IFA analyses of WT and mutant pCD163 expression. A Sequence alignment of pCD163 SRCR5, moCD163 SRCR5 and hCD163L1 SRCR8. The amino acid sequences of pCD163 SRCR5 (UniProt entry Q2VL90), moCD163 SRCR5 (UniProt entry Q2VLG4) and hCD163L1 SRCR8 (UniProt entry Q9NR16) are aligned. The chosen mutated residues are marked by asterisks (*). B IFA analyses of WT and mutant pCD163 expression. The PK-15 cells were transfected with WT or pCD163 mutated constructs (1 μg/well, 24-well plate). Twenty four hours post-transfection, the cells were fixed and stained with a commercial mouse anti-pCD163 monoclonal antibody (MCA2311GA), and then examined by IFA. The total fluorescence intensity of pCD163 was calculated using ImageJ software. Data represent means ± SEM of three independent experiments. ns, not significant.
Site-directed mutagenesis of SRCR5 in pCD163
Based on the analyses above, we chose the residues Gly499, Glu509, Ser512, Thr522, Glu534, Glu543, His549, Pro560, Arg561 and Gly564 in pCD163 SRCR5, whose surface electrostatic potential was significantly different from that in hCD163L1 SRCR8. We mutated each residue to the corresponding one in hCD163L1 SRCR8, namely G499R, E509H, S512N, T522D, E534K, E543K, H549S, P560Q, R561H and G564D (Figure 4A). WT or each mutated CD163 construct was transfected into PK-15 cells. Native PK-15 cells express no pCD163 and are refractory to PRRSV infection, while they are permissive to the viral infection after transfection with pCD163 [40]. After 24 h, the transfected PK-15 cells were monitored by IFA with a commercial anti-CD163 antibody. As shown in Figure 4B, all mutated and WT pCD163 receptors were expressed at almost the same level at 24 h post-transfection in PK-15 cells. In addition, IB detected their identical expression patterns at 36 h and 48 h post-transfection in PK-15 cells in Figures 5A and B, respectively. These results ruled out the influence of their expression levels in our subsequent experiments.

Identification of residues in pCD163 SRCR5 important for PRRSV-2 infection. A RT-qPCR analyses of the effect of mutated pCD163 on PRRSV-2 replication. WT or mutant pCD163 constructs (3 μg/well, 6-well plate) were transfected into PK-15 cells. After 24 h, the transfected PK-15 cells were inoculated with PRRSV-2 strain BJ-4 at a MOI of 1. At 12 hpi, total RNAs of infected PK-15 cells were extracted and then the viral RNA was measured by RT-qPCR. In parallel, expression levels of WT and mutant pCD163 were tested by IB using a commercial mouse anti-human CD163 antibody (MCA1853). Data represent means ± SEM from three independent experiments. *p < 0.05. B Analyses of PRRSV N protein expression in WT or mutant pCD163-expressed cells. The infected WT or mutant pCD163-expressed cells were harvested and lysed at 24 hpi. PRRSV N protein in WT or mutant pCD163-expressed cells was measured by IB. Significant reduction in PRRSV N protein expression is marked by an arrow. In parallel, expression levels of WT and mutant pCD163 were tested by IB using a commercial mouse anti-human CD163 antibody (MCA1853). C PRRSV infectivity in WT or mutant pCD163-expressed cells. The infected cells (24 hpi) were fixed and stained with PRRSV N protein (red) antibody. Nuclei were stained with DAPI and examined by IFA. The total fluorescence intensity of PRRSV N protein was calculated using ImageJ software. Data represent means ± SEM of three independent experiments. *p < 0.05.
Identification of residues in pCD163 SRCR5 important for PRRSV-2 infection
In order to detect the effect of each mutated residue on PRRSV-2 infection, we first utilized a typical PRRSV-2 VR2332-like strain BJ-4 to measure viral infections. As shown in Figure 5A, RT-qPCR analysis demonstrated that the transfected cells with the mutated construct G499R, E509H, S512N, T522D, E543K, H549S, P560Q or G564D showed comparable PRRSV RNA abundance as the WT construct-transfected ones did (p > 0.05). In contrast, the transfected cells showed 0.72 × 106 copies/μg in viral RNA abundance with site-directed mutagenesis of pCD163 at position 534 and 0.65 × 106 copies/μg at position 561, respectively, corresponding to > 50% reduction compared to the WT ones (1.45 × 106 copies/μg). These results of RT-qPCR showed statistically significant in reduction of viral RNA abundance (p < 0.05). In addition, in Figures 5B and C, compared to the WT one, the mutated pCD163 at position 534 or 561 showed a strong inhibitory effect on PRRSV N protein expression and infectivity (> 50%; p < 0.05). The TCID50 results further corroborated the importance of the residue 534 or 561 for PRRSV-2 infection, where the progeny virus titers were decreased by more than tenfold (~5.1 log10TCID50/mL for the mutated pCD163 at position 534 or 561, compared to 6.3 log10TCID50/mL for the WT one; namely > 1 log10TCID50/mL, p < 0.01; Figure 6A).

Analyses of simultaneous mutagenesis of the identified two residues for PRRSV-2 infection. A TCID50 analyses of the effect of mutated CD163 on PRRSV-2 viral titers. PK-15 cells were transfected with WT or mutated pCD163 constructs. After 24 h, the transfected PK-15 cells were inoculated with PRRSV strain BJ-4 or HN07-1 at a MOI of 1 for 48 h. The viral yields were measured by TCID50 assay in MARC-145 cells. Data represent means ± SEM of three independent experiments. **p < 0.01, ***p < 0.001 for mutant CD163 compared to the WT one. #p < 0.05 for simultaneous mutagenesis at position 534 and 561 of CD163 compared to the single-site mutagenesis. B Analyses of PRRSV N protein expression in WT or mutant pCD163-expressed cells. PRRSV N protein expression was tested by IB as described in Figure 5B. C PRRSV infectivity in WT or mutant pCD163-expressed cells. The infectivity was measured by IFA as described in Figure 5C. Data represent means ± SEM of three independent experiments. *p < 0.05.
Analyses of simultaneous mutagenesis of the identified two residues for PRRSV-2 infection
Next, we examined the effect of simultaneous mutagenesis of the identified two residues on PRRSV-2 infection. As shown in Figure 6A, TCID50 analysis demonstrated that the transfected cells with simultaneous mutagenesis at position 534 and 561 showed an additive decrease in the viral titer compared with the single-site mutagenesis of pCD163 at position 534 or 561 (p < 0.05). Importantly, compared to the WT one (6.3 log10TCID50/mL), simultaneous mutagenesis (4.4 log10TCID50/mL) showed an almost 100-fold decreased viral titer (~2 log10TCID50/mL; p < 0.001). We also utilized a HP-PRRSV strain, HN07-1, to carry out the viral titration assay and observed similar results (Figure 6A). Furthermore, we measured PRRSV N protein expression and infectivity to test the effect of simultaneous mutagenesis on viral infection. As shown in Figures 6B and C, IB and IFA analyses demonstrated that simultaneous mutagenesis showed a significant reduction in PRRSV infection compared with the WT one (p < 0.05).
Finally, we explored how these residues influenced PRRSV-2 infection through viral binding and entry assays. The results showed that these two residues actually took effect during the viral binding stage (Figure 7).

Analyses of the effect of the mutated pCD163 on PRRSV-2 invasion. A, B The binding, entry and infection assays with mutated pCD163 for PRRSV-2 strain BJ-4 and HN07-1, respectively. WT or mutated pCD163 constructs were transfected into PK-15 cells. After 24 h, the transfected PK-15 cells were inoculated with PRRSV strain BJ-4 or HN07-1 at a MOI of 1 at 4 °C for 1 h, and then relative quantitation of viral RNA abundance was carried out. For PRRSV entry assay, the unbound viruses were washed away and the inoculated cells were then cultured at 37 °C for 3 h to allow viral entry. The entering viral RNA was analyzed by RT-qPCR. For PRRSV infection, PRRSV strain BJ-4 or HN07-1 was inoculated in the transfected PK-15 cells as described above. PRRSV RNA abundance was tested by RT-qPCR. For the RT-qPCR, PRRSV ORF7 gene was normalized with GAPDH mRNA and relatively quantified by the 2−ΔΔCT method. In parallel, we have transfected empty vector and inoculated PRRSV as negative control. However, the C(t) value of the negative control was comparable to that of water, which was not included in the manuscript. Data represent means ± SEM of three independent experiments. *p < 0.05, **p < 0.01.
All these results demonstrated that residues 534 and 561 were important for PRRSV-2 infection in vitro.
Discussion
Heparin sulfate [61, 62], sialoadhesin (Sn/CD169) [63, 64], CD163 [25], CD151 [65], vimentin [66], dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin [67] and non-muscle myosin heavy chain 9 [60] were previously proposed to be associated with PRRSV infection. Among them, Sn and CD163 have been intensively studied; the former was reported to be responsible for viral attachment and internalization, and the latter contributed to viral membrane fusion and uncoating [27]. However, Sn knockout pigs were still susceptible to PRRSV infection [68]. Increasing evidence supports the view that CD163 is an indispensable receptor and its SRCR5 domain is crucial for PRRSV infection both in vitro and in vivo [25, 30,31,32,33, 36, 37, 39, 69,70,71]. However, the detailed mechanisms of CD163, especially the SRCR5, involved in PRRSV infection are not fully understood.
In this study, we determined the crystal structures of moCD163 SRCR5 and hCD163L1 SRCR8 (Figures 1 and 2). Compared to the crystal structure of pCD163 SRCR5, these three SRCR domains shared almost identical structural folds (Figures 1C and 2C). Intriguingly, hCD163L1 SRCR8 showed a significantly different surface electrostatic potential compared to the other two proteins (Figure 3). The comparison provided a structural basis to explain why stable expression of moCD163 rendered comparable infectivity to PRRSV as pCD163 did, since their crucial SRCR5 shared the overall conformations and surface electrostatic potentials [25]. Furthermore, this comparison suggested that the surface electrostatic potential might lead to different cell susceptibility to PRRSV infection between pCD163 and hCD163L1 [31, 39], where pCD163 SRCR5 substitution by homologous hCD163L1 SRCR8 showed a significantly reduced permissiveness to PRRSV-2. Therefore, we focused on the surface electrostatic potential differences between pCD163 SRCR5 and hCD163L1 SRCR8.
We carried out mutational studies to identify that the residue at position 561 was important for PRRSV-2 infection in vitro (Figures 5 and 6). The arginine residue at position 561 (Arg561) was reported to participate during viral infection in our previous study [40]. In addition, we identified a novel residue at position 534 important for PRRSV-2 infection (Figures 5 and 6). Moreover, both these two residues influenced the viral invasion process (Figure 7). Importantly, simultaneous mutagenesis of these two residues conferred additive resistance to PRRSV-2 infection in vitro as shown in Figure 6. These results demonstrate that the two residues showed a biological significance regarding PRRSV-2 actual infection and the charge may contribute to their involvement. Of course, whether the in vitro results will be applicable to the in vivo viral infection needs further demonstration. For example, although PAMs from the genetically modified pigs with hCD163L1 SRCR8 substitution showed a significantly reduced permissiveness, the corresponding pigs were resistant to PRRSV-1 and HP-PRRSV, but not to typical PRRSV-2 [31, 39]. The underlying mechanisms are interesting to be explored. Additionally, whether these two residues are important for PRRSV-1 infection should be addressed. It is worth mentioning that in addition to SRCR5, other SRCR domains of CD163 may be involved in PRRSV infection, which will be addressed in the future.
In fact, we have tried to determine the crystal structure of human CD163 (hCD163) SRCR5. Unexpectedly, recombinant hCD163 SRCR5, moCD163 SRCR5 and hCD163L1 SRCR8 showed a different expression pattern in the same expression system despite their high sequence identities (data not shown). Furthermore, crystals of hCD163 SRCR5 diffracted to a low quality and was unable to be processed although it was successfully prepared in Drosophila S2 cells and crystallized under the same condition as those of hCD163L1 SRCR8 (data not shown). All these phenomena are attractive to clarify.
In conclusion, we have compared the crystal structures among pCD163 SRCR5, moCD163 SRCR5 and hCD163L1 SRCR8. Based on the structural comparison, we identified that the charged residue at position 534 in association with the one at position 561 in the long loop region were important for PRRSV-2 infection in vitro. The results provide clues for CD163-mediated PRRSV infection and deepen our understanding of the viral pathogenesis, which will support the genome-edited implications to select pigs resistant to PRRSV.
Availability of data and materials
The coordinates of moCD163 SRCR5 and hCD163L1 SRCR8 were deposited in the Protein Data Bank (PDB code 6K0L and 6K0O).
Abbreviations
- PRRSV:
-
Porcine reproductive and respiratory syndrome virus
- SRCR:
-
Scavenger receptor cysteine-rich domain
- HP-PRRSVs:
-
Highly pathogenic variants of PRRSV-2
- moCD163:
-
Monkey CD163
- hCD163L1:
-
Human CD163-like homolog
- pCD163:
-
Porcine CD163
- PAMs:
-
Porcine alveolar macrophages
- NLFK:
-
Norden Laboratories feline kidney
- BHK-21:
-
Baby hamster kidney-21
- TCID50 :
-
50% Tissue culture infective dose
- hCD163:
-
Human CD163
- RIPA:
-
Radio immunoprecipitation assay
- PDB:
-
Protein Data Bank
- SEM:
-
Standard errors of the means
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- PFA:
-
Paraformaldehyde
- PK:
-
Porcine kidney
- SSRF:
-
Shanghai Synchrotron Radiation Facility
References
Done SH, Paton DJ (1995) Porcine reproductive and respiratory syndrome: clinical disease, pathology and immunosuppression. Vet Rec 136:32–35
Rossow KD (1998) Porcine reproductive and respiratory syndrome. Vet Pathol 35:1–20
Holtkamp DJ, Kliebenstein JB, Neumann EJ (2013) Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. JSHAP 21:72–84
Zimmerman JJ, Yoon KJ, Wills RW, Swenson SL (1997) General overview of PRRSV: a perspective from the United States. Vet Microbiol 55:187–196
Snijder EJ, Meulenberg JJ (1998) The molecular biology of arteriviruses. J Gen Virol 79:961–979
Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfacon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ (2017) Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses. Arch Virol 162:2505–2538
International Committee on Taxonomy of Viruses (ICTV) Release EC 50, Washington, DC, July 2018; Email Ratification October 2018 (MSL #33). https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=201851832
Meng XJ, Paul PS, Halbur PG, Lum MA (1995) Phylogenetic analyses of the putative M (ORF 6) and N (ORF 7) genes of porcine reproductive and respiratory syndrome virus (PRRSV): implication for the existence of two genotypes of PRRSV in the U.S.A. and Europe. Arch Virol 140:745–755
Nelsen CJ, Murtaugh MP, Faaberg KS (1999) Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol 73:270–280
Han J, Zhou L, Ge X, Guo X, Yang H (2017) Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Microbiol 209:30–47
Guo Z, Chen X, Li R, Qiao S, Zhang G (2018) The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: a molecular epidemiological perspective. Virol J 15:2
Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF (2007) Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One 2:e526
Tong G, Zhou Y, Hao X, Tian Z, An T, Qiu H (2007) Highly pathogenic porcine reproductive and respiratory syndrome, China. Emerg Infect Dis 13:1434–1436
Keffaber K (1989) Reproductive failure of unknown etiology. Am Assoc Swine Pract Newsl 1:1–9
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F (1991) Mystery swine disease in the Netherlands: the isolation of Lelystad virus. Vet Q 13:121–130
Collins JE, Benfield DA, Christianson WT, Harris L, Hennings JC, Shaw DP, Goyal SM, McCullough S, Morrison RB, Joo HS (1992) Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J Vet Diagn Invest 4:117–126
Reiner G, Fresen C, Bronnert S, Willems H (2009) Porcine reproductive and respiratory syndrome virus (PRRSV) infection in wild boars. Vet Microbiol 136:250–258
Wu J, Liu S, Zhou S, Wang Z, Li K, Zhang Y, Yu J, Cong X, Chi X, Li J, Xu S, Du Y, Ren S, Wang J (2011) Porcine reproductive and respiratory syndrome in hybrid wild boars, china. Emerg Infect Dis 17:1071–1073
Molina-Barrios R, Luevano-Adame J, Henao-Diaz YA, Gimenez-Lirola L, Pineyro P, Magtoto R, Cedillo-Cobian J, Diaz-Rayo C, Hernandez J, Zimmerman J (2018) Collared peccary (Pecari tajacu) are susceptible to porcine reproductive and respiratory syndrome virus (PRRSV). Transbound Emerg Dis 65:1712–1719
Duan X, Nauwynck HJ, Pensaert MB (1997) Virus quantification and identification of cellular targets in the lungs and lymphoid tissues of pigs at different time intervals after inoculation with porcine reproductive and respiratory syndrome virus (PRRSV). Vet Microbiol 56:9–19
Kim HS, Kwang J, Yoon IJ, Joo HS, Frey ML (1993) Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol 133:477–483
Nauwynck HJ, Duan X, Favoreel HW, Van Oostveldt P, Pensaert MB (1999) Entry of porcine reproductive and respiratory syndrome virus into porcine alveolar macrophages via receptor-mediated endocytosis. J Gen Virol 80:297–305
Xiao S, Wang X, Ni H, Li N, Zhang A, Liu H, Pu F, Xu L, Gao J, Zhao Q, Mu Y, Wang C, Sun Y, Du T, Xu X, Zhang G, Hiscox JA, Goodfellow IG, Zhou E (2015) MicroRNA miR-24-3p promotes porcine reproductive and respiratory syndrome virus replication through suppression of heme oxygenase-1 expression. J Virol 89:4494–4503
Shi C, Liu Y, Ding Y, Zhang Y, Zhang J (2015) PRRSV receptors and their roles in virus infection. Arch Microbiol 197:503–512
Calvert JG, Slade DE, Shields SL, Jolie R, Mannan RM, Ankenbauer RG, Welch SK (2007) CD163 expression confers susceptibility to porcine reproductive and respiratory syndrome viruses. J Virol 81:7371–7379
Lee YJ, Park CK, Nam E, Kim SH, Lee OS, du Lee S, Lee C (2010) Generation of a porcine alveolar macrophage cell line for the growth of porcine reproductive and respiratory syndrome virus. J Virol Methods 163:410–415
Van Gorp H, Van Breedam W, Delputte PL, Nauwynck HJ (2008) Sialoadhesin and CD163 join forces during entry of the porcine reproductive and respiratory syndrome virus. J Gen Virol 89:2943–2953
Wang X, Wei R, Li Q, Liu H, Huang B, Gao J, Mu Y, Wang C, Hsu WH, Hiscox JA, Zhou E (2013) PK-15 cells transfected with porcine CD163 by PiggyBac transposon system are susceptible to porcine reproductive and respiratory syndrome virus. J Virol Methods 193:383–390
Li L, Wu C, Hou G, Xue B, Xie S, Zhao Q, Nan Y, Zhang G, Zhou E (2017) Generation of murine macrophage-derived cell lines expressing porcine CD163 that support porcine reproductive and respiratory syndrome virus infection. BMC Biotechnol 17:77
Whitworth KM, Rowland RR, Ewen CL, Trible BR, Kerrigan MA, Cino-Ozuna AG, Samuel MS, Lightner JE, McLaren DG, Mileham AJ, Wells KD, Prather RS (2016) Gene-edited pigs are protected from porcine reproductive and respiratory syndrome virus. Nat Biotechnol 34:20–22
Wells KD, Bardot R, Whitworth KM, Trible BR, Fang Y, Mileham A, Kerrigan MA, Samuel MS, Prather RS, Rowland RR (2017) Replacement of porcine CD163 scavenger receptor cysteine-rich domain 5 with a CD163-like homolog confers resistance of pigs to genotype 1 but not genotype 2 porcine reproductive and respiratory syndrome virus. J Virol 91:e01521-e1616
Prather RS, Wells KD, Whitworth KM, Kerrigan MA, Samuel MS, Mileham A, Popescu LN, Rowland RRR (2017) Knockout of maternal CD163 protects fetuses from infection with porcine reproductive and respiratory syndrome virus (PRRSV). Sci Rep 7:13371
Yang H, Zhang J, Zhang X, Shi J, Pan Y, Zhou R, Li G, Li Z, Cai G, Wu Z (2018) CD163 knockout pigs are fully resistant to highly pathogenic porcine reproductive and respiratory syndrome virus. Antiviral Res 151:63–70
Sarrias MR, Gronlund J, Padilla O, Madsen J, Holmskov U, Lozano F (2004) The scavenger receptor cysteine-rich (SRCR) domain: an ancient and highly conserved protein module of the innate immune system. Crit Rev Immunol 24:1–37
Van Gorp H, Delputte PL, Nauwynck HJ (2010) Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol 47:1650–1660
Van Gorp H, Van Breedam W, Van Doorsselaere J, Delputte PL, Nauwynck HJ (2010) Identification of the CD163 protein domains involved in infection of the porcine reproductive and respiratory syndrome virus. J Virol 84:3101–3105
Burkard C, Lillico SG, Reid E, Jackson B, Mileham AJ, Ait-Ali T, Whitelaw CB, Archibald AL (2017) Precision engineering for PRRSV resistance in pigs: macrophages from genome edited pigs lacking CD163 SRCR5 domain are fully resistant to both PRRSV genotypes while maintaining biological function. PLoS Pathog 13:e1006206
Yu P, Wei R, Dong W, Zhu Z, Zhang X, Chen Y, Liu X, Guo C (2019) CD163(ΔSRCR5) MARC-145 cells resist PRRSV-2 infection via inhibiting virus uncoating, which requires the interaction of CD163 with calpain 1. Front Microbiol 10:3115
Chen J, Wang H, Bai J, Liu W, Liu X, Yu D, Feng T, Sun Z, Zhang L, Ma L, Hu Y, Zou Y, Tan T, Zhong J, Hu M, Bai X, Pan D, Xing Y, Zhao Y, Tian K, Hu X, Li N (2019) Generation of pigs resistant to highly pathogenic-porcine reproductive and respiratory syndrome virus through gene editing of CD163. Int J Biol Sci 15:481–492
Ma H, Jiang L, Qiao S, Zhi Y, Chen X, Yang Y, Huang X, Huang M, Li R, Zhang G (2017) The crystal structure of the fifth scavenger receptor cysteine-rich domain of porcine CD163 reveals an important residue involved in porcine reproductive and respiratory syndrome virus infection. J Virol 91:e01897-e1916
Yang H, Guan S, Yin X (1997) Isolation and identification of porcine reproductive and respiratory syndrome virus. Chin Vet Med 23:9–10 (in Chinese)
Qiao S, Feng L, Bao D, Guo J, Wan B, Xiao Z, Yang S, Zhang G (2011) Porcine reproductive and respiratory syndrome virus and bacterial endotoxin act in synergy to amplify the inflammatory response of infected macrophages. Vet Microbiol 149:213–220
Li R, Zheng K, Hu P, Chen Z, Zhou S, Chen J, Yuan C, Chen S, Zheng W, Ma E, Zhang F, Xue J, Chen X, Huang M (2014) A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics 4:642–659
Ma H, Qiao S, Huang M, Jiang L, Li R, Zhang G (2017) Crystal structure of a CD163 scavenger receptor cysteine-rich domain produced in Pichia pastoris. Chinese J Struct Chem 36:1409–1417
Hou J, Li R, Ma H, Qiao S, Zhang G (2016) Structural prediction of porcine sialoadhesin V-set Ig-like domain sheds some light on its role in porcine reproductive and respiratory syndrome virus (PRRSV) infection. Front Agr Sci Eng 3:65–71
Li R, Ma H, Jiang L, Qiao S, Zhi Y, Huang M, Deng R, Zhang G (2018) The CD163 long-range scavenger receptor cysteine-rich repeat: expression, purification and X-ray crystallographic characterization. Acta Crystallogr F Struct Biol Commun 74:322–326
Zhang W, Tang J, Wang S, Wang Z, Qin W, He J (2019) The protein complex crystallography beamline (BL19U1) at the Shanghai Synchrotron Radiation Facility. Nucl Sci Tech 30:170
Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62:859–866
CCP4 (1994) The CCP4 suite; programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132
DeLano WL (2002) The PyMOL molecular graphics system. DeLano scientific, Palo Alto, CA, USA. http://www.pymol.org
Jensen EC (2013) Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec 296:378–381
Wiesmann V, Franz D, Held C, Münzenmayer C, Palmisano R, Wittenberg T (2015) Review of free software tools for image analysis of fluorescence cell micrographs. J Microsc 257:39–53
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108
Reed LJ, Muench H (1938) A simple method of estimating fifty percent endpoints12. Am J Epidemiol 27:493–497
Wang R, Wang X, Ni B, Huan C, Wu J, Wen L, Liao Y, Tong G, Ding C, Fan H, Mao X (2016) Syndecan-4, a PRRSV attachment factor, mediates PRRSV entry through its interaction with EGFR. Biochem Biophys Res Commun 475:230–237
Wang T, Liu Y, Li L, Wang G, Wang H, Zhang H, Zhao S, Gao J, An T, Tian Z, Tang Y, Cai X (2018) Porcine alveolar macrophage CD163 abundance is a pivotal switch for porcine reproductive and respiratory syndrome virus infection. Oncotarget 9:12174–12185
Huang C, Bernard D, Zhu J, Dash RC, Chu A, Knupp A, Hakey A, Hadden MK, Garmendia A, Tang Y (2020) Small molecules block the interaction between porcine reproductive and respiratory syndrome virus and CD163 receptor and the infection of pig cells. Virol J 17:116
Yang Q, Zhang Q, Tang J, Feng W (2015) Lipid rafts both in cellular membrane and viral envelope are critical for PRRSV efficient infection. Virology 484:170–180
Gao J, Xiao S, Xiao Y, Wang X, Zhang C, Zhao Q, Nan Y, Huang B, Liu H, Liu N, Lv J, Du T, Sun Y, Mu Y, Wang G, Syed SF, Zhang G, Hiscox JA, Goodfellow I, Zhou E (2016) MYH9 is an essential factor for porcine reproductive and respiratory syndrome virus infection. Sci Rep 6:25120
Jusa ER, Inaba Y, Kouno M, Hirose O (1997) Effect of heparin on infection of cells by porcine reproductive and respiratory syndrome virus. Am J Vet Res 58:488–491
Delputte PL, Costers S, Nauwynck HJ (2005) Analysis of porcine reproductive and respiratory syndrome virus attachment and internalization: distinctive roles for heparan sulphate and sialoadhesin. J Gen Virol 86:1441–1445
Duan X, Nauwynck HJ, Favoreel HW, Pensaert MB (1998) Identification of a putative receptor for porcine reproductive and respiratory syndrome virus on porcine alveolar macrophages. J Virol 72:4520–4523
Vanderheijden N, Delputte PL, Favoreel HW, Vandekerckhove J, Van Damme J, van Woensel PA, Nauwynck HJ (2003) Involvement of sialoadhesin in entry of porcine reproductive and respiratory syndrome virus into porcine alveolar macrophages. J Virol 77:8207–8215
Shanmukhappa K, Kim JK, Kapil S (2007) Role of CD151, A tetraspanin, in porcine reproductive and respiratory syndrome virus infection. Virol J 4:62
Kim JK, Fahad AM, Shanmukhappa K, Kapil S (2006) Defining the cellular target(s) of porcine reproductive and respiratory syndrome virus blocking monoclonal antibody 7G10. J Virol 80:689–696
Huang YW, Dryman BA, Li W, Meng XJ (2009) Porcine DC-SIGN: molecular cloning, gene structure, tissue distribution and binding characteristics. Dev Comp Immunol 33:464–480
Prather RS, Rowland RR, Ewen C, Trible B, Kerrigan M, Bawa B, Teson JM, Mao J, Lee K, Samuel MS, Whitworth KM, Murphy CN, Egen T, Green JA (2013) An intact sialoadhesin (Sn/SIGLEC1/CD169) is not required for attachment/internalization of the porcine reproductive and respiratory syndrome virus. J Virol 87:9538–9546
Welch SK, Calvert JG (2010) A brief review of CD163 and its role in PRRSV infection. Virus Res 154:98–103
Wang L, Zhang H, Suo X, Zheng S, Feng W (2011) Increase of CD163 but not sialoadhesin on cultured peripheral blood monocytes is coordinated with enhanced susceptibility to porcine reproductive and respiratory syndrome virus infection. Vet Immunol Immunopathol 141:209–220
Burkard C, Opriessnig T, Mileham AJ, Stadejek T, Ait-Ali T, Lillico SG, Whitelaw CBA, Archibald AL (2018) Pigs lacking the scavenger receptor cysteine-rich domain 5 of CD163 are resistant to porcine reproductive and respiratory syndrome virus 1 infection. J Virol 92:e00415-e418
Acknowledgements
We thank the staff of the BL18U and BL19U1 beamlines at SSRF, Shanghai, China, for assistance during data collection. We also thank Professor Hanchun Yang from China Agricultural University for providing PRRSV-2 strain BJ-4.
Funding
This work was supported by Grants from the National Natural Science Foundation of China (32002267 and 31972690), the Natural Science Foundation of Henan Province (202300410525), the Fund for Distinguished Young Scholars from Henan Academy of Agricultural Sciences (2021JQ01), China Agriculture Research System of MOF and MARA and the Special Fund for Henan Agriculture Research System (S2012-06). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
HM, RL, AW, and GZ designed the work. HM performed the experiments. RL and LJ performed the crystal data analyses. HM and RL wrote the manuscript. LJ, SQ, AW and XC revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, H., Li, R., Jiang, L. et al. Structural comparison of CD163 SRCR5 from different species sheds some light on its involvement in porcine reproductive and respiratory syndrome virus-2 infection in vitro. Vet Res 52, 97 (2021). https://doi.org/10.1186/s13567-021-00969-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-021-00969-z