
Abstract
Hypertension (HTN) is a primary risk factor for cardiovascular (CV) events, target organ damage (TOD), premature death and disability worldwide. The pathophysiology of HTN is complex and influenced by many factors including biological sex. Studies show that the prevalence of HTN is higher among adults aged 60 and over, highlighting the increase of HTN after menopause in women. Estrogen (E2) plays an important role in the development of systemic HTN and TOD, exerting several modulatory effects. The influence of E2 leads to alterations in mechanisms regulating the sympathetic nervous system, renin-angiotensin-aldosterone system, body mass, oxidative stress, endothelial function and salt sensitivity; all associated with a crucial inflammatory state and influenced by genetic factors, ultimately resulting in cardiac, vascular and renal damage in HTN. In the present article, we discuss the role of E2 in mechanisms accounting for the development of HTN and TOD in a sex-specific manner. The identification of targets with therapeutic potential would contribute to the development of more efficient treatments according to individual needs.
Similar content being viewed by others

Background
Hypertension (HTN) is a multifactorial condition affecting around 1.13 billion people worldwide, with an estimated increase of 15–20% by 2025, reaching close to 1.5 billion [1, 2]. HTN is a primary risk factor for cardiovascular (CV) events, target organ damage (TOD), and premature death and disability worldwide [3,4,5,6]. Individual characteristics, such as age, race, body mass and genetic factors, as well as environmental factors, lifestyle and dietary habits, such as salt intake, may contribute to the development of HTN and TOD, i.e., cardiac, vascular and renal damage.
Notably, biological sex has been revealed as a key factor in understanding variation in the development of HTN and related CV implications. Recent data show that men have a higher prevalence of HTN than women among adults aged 18–39 years (9.2% men vs. 5.6% women) and 40–59 years (37.2% men vs. 29.4% women), but men have a lower prevalence of HTN than women in adults older than 60 years (58.5% men vs. 66.8% women) [7]. Therefore, aging is characterized by increases in blood pressure (BP) in both men and women, reaching 63.1% among adults aged 60 years and over, and it is well known that the incidence of HTN increases after menopause in women [8]. Actually, women experience steeper increases in BP than men as they age [9].
Along this line, 41% of postmenopausal women become hypertensive, while more than 75% of women older than 60 years are hypertensive in the USA [10]. The majority of women older than 60 years has stage 2 HTN (BP ≥ 160/100 mmHg) or receives antihypertensive therapy [11,12,13]. Notably, it is more difficult to achieve BP control in elderly women, and women are at a greater risk of developing resistance to antihypertensive treatment than men [3, 14]. Due to the dramatic increase of HTN in postmenopausal women, it is expected that the steroid hormone estrogen plays an important role in this process.
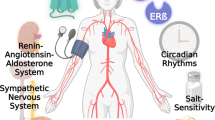
In fact, several studies have investigated the influence of 17β-estradiol (E2) in the development of systemic HTN and TOD. The influence of E2 leads to alterations in mechanisms regulating the sympathetic nervous system (SNS), renin-angiotensin-aldosterone system (RAAS), body mass, oxidative stress, endothelial function and salt sensitivity; all associated with a crucial inflammatory state and influenced by genetic factors, ultimately resulting in cardiac, vascular and renal damage in HTN. In the present article, we discuss the role of E2 in mechanisms accounting for the development of HTN and TOD in a sex-specific manner (Fig. 1). Accordingly, the goal of this article is not to provide an exhaustive review of the literature, but rather to focus on pertinent studies (Table 1).

Role of estrogen in sex differences in hypertension and related target organ damage. The influence of estrogen leads to alterations in mechanisms regulating the sympathetic nervous system (SNS), renin-angiotensin-aldosterone system (RAAS), body mass, endothelial (dys) function, oxidative stress and salt sensitivity; all associated with a crucial inflammatory state and influenced by genetic factors, ultimately resulting in cardiac, vascular and renal damage in hypertension in a sex-specific manner
Regulatory effects of estrogen in hypertension
Sympathetic nervous system
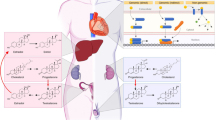
The role of the SNS in the development of HTN is well established, mediated by renal sympathetic nerves, increased renin release, alteration of glomerular filtration rate, and increased tubular sodium reabsorption [26]. Sympathetic nerve activity decreases with age, but it increases in the presence of weight gain and metabolic syndrome [27, 28], common to postmenopausal women. In addition, sympathetic nerve activity differs significantly between men and women [29] and E2 is expected to mediate sex differences by exerting several regulatory effects (Fig. 2a).

Regulatory effects of estrogen leading to sex differences in hypertension: a sympathetic nervous system, b renin-angiotensin-aldosterone system, c body mass, d oxidative stress, e inflammation, f endothelial (dys) function, g salt sensitivity, h genetic factors, i cardiac hypertrophy, j arterial stiffness, and k renal dysfunction. AT1R, angiotensin type 1 receptor; eNOs, endothelial nitric oxide synthase; ER, estrogen receptor; PI3K, phosphoinositide 3-kinase (PI3K); PKB, protein kinase B (also known as AKT); RAAS, renin-angiotensin-aldosterone system
In this context, E2 has an important role in the brain. Peripheral efferent nerves and signaling pathways that respond to neurotransmitters and neurons containing nuclear estrogen receptors (ER) have been identified in brain centers involved in the regulation of CV function [30,31,32]. Interestingly, lower baroreflex sensitivity has been described in postmenopausal women compared with age-matched men, while postmenopausal women with hormone therapy (HT) had higher baroreflex sensitivity than those not on HT [15]. Along this line, it was suggested that E2 exerts direct effects on central nervous autonomic centers, thereby leading to sympathoinhibitory effects, which could be important for conferring CV protection [33].
Several studies with experimental animals support this notion. For example, ovariectomized (OVX) rats showed enhanced sympathetic activation and attenuated baroreflex sensitivity or vagal tone, while these effects were attenuated with E2 treatment [34]. It was further shown that the activation of ERβ with a selective agonist in the paraventricular nucleus and rostroventrolateral medulla of OVX rats attenuates the sympathetic nerve activity reducing BP in aldosterone-induced HTN [35], suggesting that a potential decrease in ERβ levels or function with aging could contribute to SNS-mediated HTN in women. Furthermore, E2 treatment inhibited the development of left ventricular (LV) hypertrophy (LVH) in baroreceptor-denervated rats [36]. Together, these data highlight the regulatory role of E2 in the SNS and its influence in the development of CV pathology related to HTN.
The renal sympathetic nerves have also been shown to play a role in regulating HTN in young and old female spontaneously hypertensive rats (SHR), with a greater decrease in BP with adrenergic blockade occurring in old compared with young animals [37], suggesting an important contribution of the SNS to HTN in old animals. In addition, renal denervation was associated with reduced BP in both young and old females, with a more pronounced response in old females. However, after renal denervation the BP remained above 140 mmHg [37,38,39], indicating that mechanisms other than the renal nerves also contribute to HTN. In humans, clinical trials of renal denervation for resistant HTN showed inconsistent BP results [40,41,42]. However, a revised procedural method in the absence of antihypertensive treatment led to significant BP reductions [43]. Nevertheless, the efficacy of renal denervation in the setting of concurrent antihypertensive treatment was challenged [44, 45], but the latest findings showed a significant reduction in BP [46].
Renin-angiotensin-aldosterone system
The RAAS is a complex system involved in the regulation of BP through water and electrolyte balance, and the preservation of vascular tone. The vasoconstrictive properties of RAAS include the activation of angiotensin (ANG) II, a potent vasoconstrictor also involved in cell proliferation, hypertrophy, generation of oxidative radicals and inflammation. ANG II also stimulates the secretion of aldosterone increasing the reabsorption of sodium into the blood, contributing to increases in BP.
Data from clinical investigations, epidemiological surveys and experimental studies suggest that the components of the circulating, as well as tissue-based RAAS, are markedly affected by sex. In particular, men were reported to have higher renin activity than women regardless of age and ethnicity [47]. More recently, further important sex differences in elements of RAAS were reported showing that older men have lower aminopeptidase A and angiotensin-converting enzyme (ACE) serum activity compared with older women, while older women have higher ACE2 serum activity than younger women [48]. During sodium intake, compared with women, men were also found to have significantly higher plasma aldosterone levels, extracellular volume and systolic BP, as well as lower adrenal response to exogenous ANG II [49].
These sex differences could be attributed to differences in the balance of the RAAS between the sexes, with the so-called depressor arm of the RAAS, i.e., ACE2/ANG 1-7/Mas receptor and ANG type 2 receptor (AT2R) counter-regulating the classical pressor ACE/ANG II/AT1R pathway [50]. In this context, it was suggested that men have the pressor pathway enhanced, while premenopausal women have mainly the depressor pathway activated, whose activity is decreased in older women [51].
Experimental animals also demonstrate significant sex differences. In particular, young male SHR have higher mean BP than young female SHR [52,53,54], while this sex difference is eliminated by RAAS inhibition [55]. This also occurs after cessation of estrous cycling, which is due to an increase in BP in females rather than any change in BP in males [56, 57]. The sex-specific regulation of the RAAS includes higher renal angiotensinogen mRNA and protein levels in old males than females, as well as higher renal ANG II in old females than males, suggesting sex differences in the depressor response to AT1R even when BP is similar [58]. In addition, endothelin (ET) was shown to mediate increases in BP in old female SHR [59]. Taken together, these data highlight the important role of the RAAS and ET mediating the increase in BP subsequent to cessation of estrous cycling in aging female SHR.
In line with these findings, it has been widely hypothesized that E2 is a protective CV factor due to its capacity to control various components of the RAAS (Fig. 2b). For example, HT was associated with reduced renin levels and increased angiotensinogen levels [16], as well as reduced circulating levels of ACE activity [17], which may be mediated in part by a direct down-regulating effect of E2 on ACE mRNA [60]. Treatment of OVX rats with E2 led to reduced tissue responsiveness to ANG II and attenuated ANG-induced aldosterone secretion [61, 62], as well as reduced AT1R levels in ANG II-target tissues [62,63,64]. In addition, the administration of E2 significantly increased the production of ANG 1-7 [65, 66], an angiotensin with cardioprotective effects. Overall, E2 appears to attenuate the production of ANG II and the levels of AT1R, thereby leading to a decreased RAAS activity.
Body mass
Higher body mass is associated with increased risk for the development of HTN [67, 68], underlain by increased sympathetic activity, increased ANG II formation and renin release, leading to adrenal aldosterone production, thereby resulting in sodium retention. Increased visceral fat is associated with increased inflammatory mediators, increased oxidative stress and decreased endothelial vasodilation [8, 69].
Not surprisingly, one sex may be more vulnerable than the other. In particular, women may have higher levels of body fat (adipose tissue) compared with men and a greater risk of developing metabolic syndrome [70]. Notably, overweight and obese women have a higher risk of developing HTN compared with overweight and obese men [71]. Decreased E2 levels after menopause are markedly associated with lipid profile variations and abdominal fat accumulation. Therefore, alterations in E2 levels may lead to metabolic and adipocyte physiology disturbances contributing to obesity (Fig. 2c).
Subcutaneous and visceral adipose tissues express both ER, ERα, and ERβ, with ERα playing a major role in the activity of adipocytes and sex-specific fat distribution [72, 73]. It has been shown that non-classical ERα signaling mediates major effects of E2 on energy balance, suggesting that selective ERα agonists could reduce the risk of obesity and metabolic alterations in postmenopausal women [74]. In addition, E2 directly increased the number of anti-lipolytic α2A-adrenergic receptors in subcutaneous adipocytes in vivo and in vitro [75], but not in intra-abdominal adipose tissue, and it increased the lipolytic β-adrenergic receptor expression through ERα in vitro [76]. Further complex regulatory effects of E2 include the down-regulation of peroxisome proliferator-activated receptor-γ in the adipose tissue and concomitant adipogenesis-related genes [77], as well as the up-regulation of peroxisome proliferator-activated receptor-α in the liver [78]. These results provide a mechanistic insight for the effects of E2 on the maintenance of fat distribution with an increased use of lipids as an energy source, which may partially promote the fat reduction in abdominal fat [79].
The regulation of adipokines and cytokines released by the adipose tissue can be impaired in obesity and metabolic syndrome, thereby contributing to CV complications, including arterial stiffness, vascular and renal damage, ultimately contributing to the development of HTN [6, 80]. Adipokines, such as leptin (a metabolic regulator and feedback signal of body fat to regulate appetite also with a lipolytic effect) and adiponectin (an anti-inflammatory hormone) are cytokines released by the adipose tissue. These hormones have gained attention due to their capacity to influence the inflammatory system with pro- and anti-inflammatory actions. Increased levels of leptin and resistin—another adipokine that contributes to obesity—and lower levels of adiponectin were previously associated with uncontrolled BP [80]. In obese postmenopausal women, increased leptin and resistin levels and decreased adiponectin levels were reported, while HT was shown to be beneficial in reducing many of the parameters of metabolic syndrome [81]. In addition, E2 may increase leptin sensitivity by controlling the expression of leptin-specific receptors [24]. Furthermore, the administration of estradiol benzoate reduced resistin levels in adipocytes [82]. Together, these data suggest that E2 is a pivotal regulator of adipokines.
Oxidative stress
Oxidative stress is a condition that occurs when the rate of reactive oxygen species (ROS) formation exceeds the rate of the antioxidant defense system. ROS has an important role in cell signaling and tissue homeostasis. In pathological conditions and environmental stress, ROS levels can increase dramatically and may result in significant damage to cellular structures. Oxidative stress has been linked to the development of HTN.
Women prior to menopause appear to have lower levels of oxidative stress compared with age-matched men, mediated by anti-oxidant activities of E2 scavenging-free radicals [83,84,85]. In contrast, postmenopausal women have higher oxidative stress levels than age-matched men [86]. Experimental data suggest a greater antioxidant potential in females over males [87], with higher levels of superoxide generation and lower levels of nitric oxide (NO) in males compared with females [88, 89]. Furthermore, female SHR showed increased renal NADPH oxidase activity and urinary F2-isoprostanes compared with male SHR [88, 90], corroborating the idea that E2 has a prominent role in sex differences in oxidative stress (Fig. 2d).
In OVX rats, E2 deficiency was associated with increased H2O2 production, as well as increased AT1R levels, leading to increased oxidative stress and endothelial dysfunction [64, 91]. In contrast, the administration of E2 in OVX rats led to the down-regulation of AT1R and E2 treatment of vascular smooth muscle cells resulted in a time-dependent downregulation of AT1R mRNA [64], indicating that the antioxidant properties of E2 may be mediated through the downregulation of the AT1R gene expression. In addition, E2 stimulates NO level increases in endothelial cells via endothelial NO synthase activation and ER-mediated mechanisms [18, 92, 93]. Ovariectomy-related loss of E2 leads to reduced endothelial NO synthase levels and activity [94], which appears to exert broad cardioprotective actions, including the attenuation of chemotherapeutic drug-induced cardiomyopathy [95]. E2 was further shown to reduce NADPH oxidase levels in endothelial cells [96], as well as to increase the levels of the antioxidant enzyme heme oxygenase 1 in in vivo and in vitro models [97,98,99,100,101,102]. Also, E2 administration in OVX rats led to an increase in catalase activity [103,104,105], and acute E2 treatment substantially enhanced myocardial catalase activity and restored LV oxidative stress and dysfunction caused by ethanol in OVX rats [106]. Overall, E2 modulates several factors, including pro- and antioxidant enzymes, thereby attenuating the production of ROS.
Inflammation
Inflammation plays a central role in the CV system, underlying several CV pathologies. CV alterations trigger activation of inflammatory responses due to systemic damage, releasing pro- and anti-inflammatory factors, and activating cellular stress pathways.
Adipose tissue dysregulation is one of the main sources of inflammatory signaling in obesity-associated metabolic alterations, and E2 plays an important role. Menopause is associated with increases in fat mass, as well as elevated circulating inflammatory markers, such as tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6) and plasminogen activator inhibitor-1 [107, 108]. Similarly, the loss of E2 in OVX rodents, as well as the deficiency of ERα, has been linked with increased adiposity in part mediated by increased food intake and decreased energy expenditure, accompanied by increased inflammation, while E2 treatment attenuated these effects [109,110,111]. In fact, ERα protects against obesity-related diseases and inflammation [112, 113]. In this context, ERα polymorphisms result in insulin resistance, body fat accumulation and inflammation [114, 115].
Many of the alterations occurring in comorbidities associated with decreased E2 levels and decreased ER expression, including the metabolic syndrome and obesity accompanied by inflammation, are attenuated by E2 (Fig. 2e). In particular, the administration of E2 has been shown to be important in the regulation of metabolic and inflammatory processes, leading to decreased expression of genes involved in lipogenesis [116], increased glucose clearance [117], lowered levels of inflammation soluble intracellular adhesion molecule 1, vascular cell adhesion molecule 1, E-selectin and ET in postmenopausal women [19, 118], as well as lowered circulatory cytokine levels, including TNFα, IL-1β and IL-10, in OVX rats [119]. In addition, E2 induces the transcriptional activation of Fas ligand via an ER-mediated, NO-dependent mechanism, thereby resulting in the inhibition of leukocyte traffic across the endothelium [120].
E2 also has a key role in the regulation of NFκB, which is a central modulator of a variety of inflammatory pathways and cellular responses. In particular, E2 repressed the activity of NFκB by inhibiting its DNA-binding ability, as well as reducing the NFκB-p65 subunit expression, thereby down-regulating NFκB-dependent activation of genes, such as TNFα and IL-6 [121, 122]. In addition, E2 appears to suppress inflammatory cell adhesion to endothelial cells via an ERα-dependent mechanism, which may involve inhibition of NFκB-mediated up-regulation of vascular cell adhesion molecule 1 [123]. Overall, E2 is important in the regulation of inflammatory pathways and signaling.
Endothelial dysfunction
Endothelial dysfunction is associated with increased systemic oxidative stress and vascular inflammation. It is characterized by reduced levels of vasodilators, such as NO, and increased ET levels, thereby modulating vascular tone.
Clinical data have indicated an important association between endothelial dysfunction and reduced E2 levels in postmenopausal women. In particular, vascular and hemodynamic parameters and arterial stiffness were elevated, while the endothelial function was reduced across different stages of the menopausal transition [124]. In line, oophorectomy associated with acute E2 deprivation resulted in impaired endothelium-dependent vasodilation, due to reduced NO availability [125], while HT improved endothelium-dependent vasodilation after oophorectomy, as well as after menopause [126]. Interestingly, this beneficial effect of HT was reported to be greater in hypertensive postmenopausal women [127]. Furthermore, the levels of plasma ET were higher in postmenopausal women than in premenopausal women, while HT attenuated this increase [128].
E2 may act on the CV system directly in the vessels or indirectly by regulating the lipid profile (Fig. 2f). These E2 actions are associated with lower coronary vascular resistance, enhanced coronary blood flow and improved coronary vasodilatory responses [129, 130]. The effects of E2 in the vascular system comprise increases in the synthesis of NO [92], modulation of serum- and ANG II-stimulated synthesis of ET [131, 132], and long-term modulation of vascular tonus, inducing the production of prostaglandins [133]. Along this line, E2 administration in OVX rats restored flow-induced dilation mediated by epoxyeicosatrienoic acids, which are generated following metabolism of arachidonic acid by cytochrome P450 epoxygenases [134,135,136]. However, it is not clear whether E2 regulates the synthesis of epoxyeicosatrienoic acids. E2 also has an anti-proliferative role in vascular remodeling [137], inhibiting the proliferation of the inner layer after injury [138]. Studies have also demonstrated antioxidant effects of E2. In particular, E2 appears to be involved in the regulation of the uptake of oxidized low-density lipoprotein, which was found to be dependent upon ER activation [139]. In addition, E2 reduced cholesteryl ester accumulation in human monocyte-derived macrophages [139]. Collectively, these data indicate that E2 regulates endothelial function through multiple mechanisms.
Salt sensitivity
Salt sensitivity refers to BP responses to changes in dietary salt intake. It has been described that salt intake has pathological effects on the vasculature and sodium homeostasis, and salt sensitivity appears to be related not only to kidney malfunction but also to endothelial dysfunction [140]. Interventional studies with essential hypertensive patients receiving diets with varying salt levels demonstrated that patients who were salt-sensitive more often had LVH, CV events, and/or endothelial dysfunction than non-salt sensitive hypertensive patients [141,142,143].
A recent study with human subjects and experimental animals indicated that females have 30% higher salt sensitivity of BP than males, regardless of menopausal status or HTN and altered aldosterone production, and differences in the kidney seem to be responsible for this sex-specific effect [144]. Another previous study examined BP responses to dietary sodium and potassium interventions by sex, age and baseline BP subgroups among men and women aged 16 years or older. Also, this study showed that the female sex, as well as older age and elevated baseline BP levels, increases BP responses to dietary sodium intake [145]. Salt sensitivity increases with age and is likely mediated by impaired vasodilation of the renal circulation, possibly due to reduced NO availability, increased vasoconstriction response to ANG II, leading to a disturbed renal sodium handling, oxidative stress, and HTN [146, 147]. As postmenopausal women appear to be more salt-sensitive than pre-menopausal women [148], decreases in ovarian hormone levels and increased sensitivity to dietary sodium may be important factors in the development of HTN at menopause. Furthermore, the surgical removal of the ovaries is associated with the development of salt sensitivity [149], while the administration of E2 reduced salt sensitivity of BP in postmenopausal women [150]. These data support further the view that salt sensitivity may be associated at least in part with changes of the hormonal profile, particularly E2 (Fig. 2g), that occur in women after menopause.
HTN due to salt sensitivity has been linked to decreased renal NO production and inappropriate activation of the RAAS [151]. As previously discussed, E2 has an important role in both systems, NO and RAAS, and through its antioxidant properties, it has the ability to increase the bioavailability of endothelium-derived NO. Subsequently, in the presence of salt sensitivity, decreases in E2 levels may impair the bioavailability of NO. In addition, in OVX salt-sensitive rats, HTN was correlated with increased renal AT1R protein levels, while treatment with E2 or an AT1R antagonist prevented the development of HTN [151]. Therefore, menopause-related E2 deficiency leads to the over-expression of renal AT1R, thereby resulting in oxidative stress and disturbed renal sodium handling, ultimately contributing to the development of HTN.
Genetic factors
It is well known that genetic factors play a major role in CV pathology [152, 153] and that they influence the development of HTN [154]. Interestingly, through modeling gene-environment interactions, several genetic variants associated with HTN-related phenotypes have been discovered [155]. Thus, mechanisms related to individual genetic variation may lead to specific responses in HTN, which may differ significantly between the sexes [156].
In this context, various studies have reported sex-specific associations between HTN and polymorphisms of components of the RAAS [157], endothelial NO synthase [158] and aldosterone synthase [159]. In a large-scale study of the general population, double homozygosity for Thr235 and Thr174 in the angiotensinogen gene was associated with a 10% increase in angiotensinogen levels and was considered a risk factor for elevated BP in women but not in men [157]. Gene variants of the endothelial NO synthase were reported to influence the long-term burden and trend of BP since childhood in females contributing to their predisposition to HTN [158]. Polymorphisms in the β1-adrenergic receptor and α2A adrenergic receptor were also associated with BP in women [160]. Investigation of the association between the insertion/deletion (I/D) polymorphism of the gene that codes for ACE and HTN in black and white men and women revealed a significant association between the D variant and HTN only in black women, highlighting the importance of sex-specific ethnic differences in the association between genetic variation and expression of a hypertensive phenotype [161]. Furthermore, this ACE polymorphism was associated with BP in a sex- and age-dependent manner [162].
As previously mentioned, the effects of E2 in the CV system are mainly mediated by the two ERs, i.e., ERα encoded by the ESR1 gene and ERβ encoded by the ESR2 gene. Polymorphisms in ESR1 have been associated with diastolic BP in women [163]. Moreover, ESR1 genotypes and alterations in its expression have been linked with increased body mass and body fat distribution [115, 164, 165]. The binding of E2 to ERβ has been reported to lead to vasodilation [166]. In this context, it has been shown that women that are heterozygous for certain genotypic polymorphisms of ESR2 present increased risk of HTN, especially those who use oral contraceptives [167], suggesting that specific single-nucleotide polymorphisms in ESR2 may transform the interaction of E2 with ERβ to a harmful axis regarding BP instead of a protective one.
In addition, variation at rs10144225 in ESR2 was associated with salt sensitivity of BP in premenopausal women but not in men or postmenopausal women [168]. In premenopausal women with the major allele, E2 is expected to bind to ERβ leading to vasodilation, thereby acting to protect against salt sensitivity of BP. However, in women with the risk allele, the binding affinity between E2 and ERβ may decrease, thereby attenuating the vasodilatory effects of E2, ultimately leading to salt sensitivity of BP. Together, polymorphic variants within ESR2 may inhibit its binding to E2, thereby hindering the vasodilatory effects of E2, ultimately leading to a loss of its protective actions against HTN [168]. Interestingly, polymorphisms in the human follicle-stimulating hormone receptor gene, which may cause hereditary hypergonadotropic ovarian failure, have also been linked to HTN in women [169]. Therefore, genetic variation influences the regulatory effects of E2, thereby impacting sex-specific phenotypes in the development of HTN (Fig. 2h).
Regulatory effects of estrogen in hypertension-induced target organ damage
Patients with HTN and lack of BP control have a higher probability to develop TOD, such as cardiac hypertrophy, vascular alterations—including arterial stiffness—and renal damage. Individuals can respond differently to the development and manifestation of the disease, response to treatment, outcome and recovery process. The development of these CV complications also differs significantly between men and women [156, 170, 171], and E2 appears to be crucial in sex-specific pathophysiology [172].
Cardiac hypertrophy
The heart responds to pathological stimuli, such as HTN, aortic stenosis, or cardiac injury, with hypertrophy of the cardiac muscle, accompanied by several tissue and cellular alterations, including increases in cardiomyocyte size and changes in the extracellular matrix. Although initially this is an adaptive and compensatory response, upon the persistence of the stress factor, there is maladaptive remodeling leading to pathological hypertrophy. Consequently, HTN-induced LVH is a major risk factor for heart failure and sudden death [173]. In the hypertrophic process, distinct molecular mechanisms may occur between men and women, many of which are expected to be mediated by E2 (Fig. 2i).
Despite antihypertensive therapy, hypertensive women have a greater risk to develop LVH than hypertensive men [174], and the presence of LVH in HTN offsets the protection in a cardiovascular risk linked with the female sex [175]. As in patients with aortic stenosis [176,177,178,179,180,181,182], another major precursor inducing LVH, the hypertrophic response of the heart in hypertensive patients exhibits significant sex differences in its structural and functional adaptation [183, 184]. These differences include greater indexed LV mass, better systolic function and increased risk of incident heart failure with preserved ejection fraction in hypertensive women compared with hypertensive men [79, 185, 186]. Studies with experimental animals also demonstrate major sex differences in the development of HTN-induced LVH, where males develop greater hypertrophy and dysfunction than females [172].
Notably, HT in hypertensive postmenopausal women contributed to a reduction in LV mass [20, 187, 188], thereby indicating a modulatory role of E2 in HTN-induced LVH. Similarly, in OVX spontaneously hypertensive heart failure rats treated with E2, the development of HTN and related LVH were attenuated [189]. Direct effects of E2 and its receptors in the myocardium have been previously shown [190,191,192,193,194,195,196,197,198], which might affect several processes in a sex-specific manner [199,200,201,202,203,204,205,206]. The loss of E2 by ovariectomy suggests that E2 influences cardiac hypertrophy in part via the phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB, also known as AKT) signaling pathway [94]. In this context, the hearts of premenopausal women exhibit greater PKB activity than those of men or of postmenopausal women, and treatment of rat cardiomyocytes with E2 led to higher levels of phosphorylated PKB [22]. Therefore, E2 influences signal transduction in the myocardium that might exert regulatory actions on the hypertrophic response in a sex-specific manner.
In addition, E2 inhibits the cardiomyocyte response to hypertrophic stimuli by preventing new protein synthesis and skeletal muscle actin expression [21]. ET stimulates the tyrosine phosphatase calcineurin resulting in new protein synthesis. Both ET and calcineurin were inhibited by E2, which also induced the gene encoding MCIP1, an anti-hypertrophic protein that prevents calcineurin activity [21]. E2 also stimulates the production and release of natriuretic peptides [21, 207], thereby inhibiting the hypertrophic response [208, 209]. Several studies revealed that E2 acts through ERβ to mitigate the deleterious signaling by ANG II that produces cardiac hypertrophy [208], as well as to protect against LVH in rodents with transverse aortic constriction [190, 210] in a sex-specific manner [201, 204, 211]. Interestingly, polymorphisms in the ESR2 gene are associated with LV mass and wall thickness in women with HTN but not in men [212], thereby indicating an important role of ERβ in the development of cardiac hypertrophy and sex-specific responses.
Arterial stiffness
Arterial stiffness is characterized by the reduced capability of an artery to expand and contract in response to pressure changes. This process is tightly associated with HTN, and arterial stiffness has emerged as an important predictor of CV events and mortality [213]. Sex differences in arterial stiffness have been reported, and E2 has been implicated in vascular and endothelial protection (Fig. 2j).
The structure of the arterial wall is maintained by the balance between collagen and elastin—extracellular matrix components responsible for the compliance and stability of the arterial wall. On the one hand, increased collagen content and density have been associated with increased vascular stiffness [214,215,216,217]. On the other hand, elastin is an essential determinant of arterial morphogenesis and vascular disease [218, 219]. In fact, mutations in the gene coding for the most abundant elastic fiber proteins result in a broad spectrum of elastic tissue disorders, ranging from skeletal abnormalities to ocular and vascular defects [220,221,222,223,224,225,226]. Along this line, elastin haploinsufficiency in mice leads to altered mechanical properties of large arteries, thereby contributing to increases in BP [227, 228]. Similarly, age-related proteolytic degradation and chemical alterations of elastic fibers result in changes in their mechanical properties [229], thereby conferring the arterial wall a more rigid structure, ultimately contributing to arterial stiffness.
Major determinants of arterial stiffness include age, sex and BP [230,231,232,233]. Accordingly, markers of arterial stiffness differ significantly between men and women. In particular, compared with elderly hypertensive men, elderly hypertensive women have a longer ejection time, earlier arterial wave reflection and smaller vessel size, independent of body size and heart rate [234]. Also in end-stage renal disease, arterial wave reflection is greater in women compared with men [235]. Furthermore, aortic stiffness is greater in women than men and is associated with diastolic dysfunction, impaired ventricular coupling and LV remodeling, potentially contributing to the greater risk of heart failure with preserved ejection fraction in women [236, 237]. Notably, compared with premenopausal women, postmenopausal women have greater pulse wave velocity, indicating that the deficiency of E2 associated with menopause may account for the augmented increase in arterial stiffness with aging in women [238,239,240,241]. Along this line, investigations of postmenopausal women indicate that HT ameliorates arterial stiffness [242, 243].
The molecular processes that contribute to the changes in vascular properties accounting for these differences are incompletely understood, as well as the role of biological sex influencing genes and proteins of the extracellular matrix in older males and females. Indeed, potential mechanisms include sex differences in collagen isoforms, elastin levels and abundance of other extracellular matrix proteins [244,245,246]. Along this line, the decrease of E2 at menopause may lead to arterial stiffness through mechanisms related to changes in the components of the arterial wall, such as collagen and elastin deposition, leading to alterations in the arterial biomechanical properties. In this context, E2 has been shown to decrease collagen deposition and increase elastin production in human aortic smooth muscle cells [23]. Notably, acute endogenous E2 deprivation leads to impaired NO release [125], thereby resulting in loss of vasodilation, ultimately contributing to arterial stiffness. However, E2 administration promotes vasodilation in part by stimulating endothelial NO synthase and NO release, thereby promoting vasodilation [247, 248], as well as by up-regulating the endothelial NO synthase messenger RNA and protein levels [18, 93]. E2 also inhibits vascular smooth muscle cell proliferation [249] mediated by ERα [138, 250]. Therefore, E2 exerting direct effects in the vessel wall contributes to sex differences in arterial stiffening.
Renal dysfunction
Chronic kidney disease (CKD), defined by albuminuria and/or reduced estimated glomerular filtration rate, is a common TOD in HTN, associated with high rates of morbidity and mortality [251]. The prevalence of CKD increases with aging, underlain by changes in kidney morphology, hemodynamics and function, which increase the incidence of CV events. Sex differences in CKD have been reported, and E2 has been shown to influence renal disease development (Fig. 2k).
Considering chronic renal disease of various etiologies reveals a complicated role for biological sex. For example, the prevalence of CKD appears to be higher in older women than older men [252, 253]. However, the male sex is an independent risk factor for end-stage renal disease [254]. Further studies have reported different clinical features and prognosis of renal diseases between men and women. In particular, men present with a more rapid rate of progression of renal diseases, such as polycystic kidney disease, IgA nephropathy, membranous nephropathy, chronic renal failure, than women [255, 256]. Similarly, there is a faster decline in renal function and worse prognosis of CKD in men than in women [257, 258]. Collectively, these data indicate that the male sex is a major determinant of the progression of renal dysfunction, while younger, premenopausal women may be protected from the development of CKD.
Experimental studies of CKD with rats also showed that males present with faster progression and worse outcome of renal disease than females [259]. In particular, male rats exhibit marked albuminuria, augmented cortical histological damage, interstitial inflammation and fibrosis, while these are all significantly less pronounced in female rats [260]. Similarly, renal function is worse in male than in female rats following ischemia/reperfusion injury [261]. However, a more recent study reported no sex differences in acute renal injury due to ischemia alone, but only male rats developed CKD [262]. Notably, OVX rats also developed CKD and ovariectomy was associated with increased proteinuria, oxidative stress, increased glomerular and tubular damage, whereas E2 is thought to protect against renal disease [25, 262, 263]. Along this line, HT has been suggested for the management of CKD in postmenopausal women [264].
Although the molecular processes regulated by E2 that might affect renal function are incompletely understood, E2 exerts modulatory actions on renal morphology, such as anti-growth effects on glomerular mesangial cells and inhibition of mesangial extracellular matrix accumulation, common to the development of glomerular sclerosis [265,266,267,268]. In particular, E2 inhibits collagen synthesis induced by transforming growth factor β in glomerular mesangial cells, suggesting that E2 may limit the progression of glomerulosclerosis, thereby attenuating deleterious effects in the kidney [266, 268, 269]. In addition, the inhibitory effects of E2 on various components of the RAAS may protect the kidney against glomerular remodeling, damage and glomerulosclerosis. On the other hand, the stimulatory effects of E2 on NO may attenuate mesangial cell growth, matrix production, vasoconstriction and renal sodium reabsorption, which contribute to the progression of CKD [270]. It has been suggested that the relative renal protection observed in females may be mediated by ERα [271]. Further research is warranted.
Perspectives and significance
Although the role of biological sex has yet underestimated consequences for physiology and pathology [272], several experimental and clinical studies have demonstrated the importance of understanding its effects and the underlying mechanisms in many diseases, highlighting that sex differences represent important biological phenomena that need further investigation. At least in part, E2 accounts for these sex differences and has a key role in the development of HTN and associated TOD. However, there are several pitfalls of HT and risks that depend on the type, dose, duration of use, route of administration and timing of initiation [273]. In this context, the elucidation of E2-related mechanisms and the identification of targets with therapeutic potential will contribute to the development of more efficient therapies for men and women, improving the treatment and care of patients with HTN and CV diseases according to individual needs.
Availability of data and materials
Not applicable.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ANG:
-
Angiotensin
- AT1R:
-
Angiotensin type 1 receptor
- BP:
-
Blood pressure
- CV:
-
Cardiovascular
- E2:
-
17β-Estradiol
- ER:
-
Estrogen receptor
- ET:
-
Endothelin
- HT:
-
Hormone therapy
- HTN:
-
Hypertension
- IL-6:
-
Interleukin-6
- LV:
-
Left ventricular
- LVH:
-
LV hypertrophy
- NO:
-
Nitric oxide
- OVX:
-
Ovariectomized
- RAAS:
-
Renin-angiotensin-aldosterone system
- ROS:
-
Reactive oxygen species
- SHR:
-
Spontaneously hypertensive rats
- SNS:
-
Sympathetic nervous system
- TOD:
-
Target organ damage
References
Collaboration NCDRF. Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet. 2017;389(10064):37–55.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005 Jan 15-21;365(9455):217-223.
Daugherty SL, Powers JD, Magid DJ, Tavel HM, Masoudi FA, Margolis KL, et al. Incidence and prognosis of resistant hypertension in hypertensive patients. Circulation. 2012;125(13):1635–42.
Fatemi O, Goa C, Faselis C, Kokkinos P, Papademetriou V. Improvement in all-cause mortality with blood pressure control in a group of US veterans with drug-resistant hypertension. J Clin Hypertens (Greenwich). 2016;18(1):33–9.
Forouzanfar MH, Alexander L, Anderson HR, Bachman VF, Biryukov S, Brauer M, et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. [Comparative Study Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t]. 2015 Dec 05;386(10010):2287-323.
Sabbatini AR, Fontana V, Laurent S, Moreno H. An update on the role of adipokines in arterial stiffness and hypertension. J Hypertens. [Research Support, Non-U.S. Gov’t Review]. 2015 Mar;33(3):435-444.
Fryar CD, Ostchega Y, Hales CM, Zhang G, Kruszon-Moran D. Hypertension prevalence and control among adults: United States, 2015–2016. NCHS Data Brief. 2017;289:1–8.
Coylewright M, Reckelhoff JF, Ouyang P. Menopause and hypertension: an age-old debate. Hypertension. [Research Support, N.I.H., Extramural Review]. 2008 Apr;51(4):952-959.
Ji H, Kim A, Ebinger JE, Niiranen TJ, Claggett BL, Bairey Merz CN, et al. Sex differences in blood pressure trajectories over the life course. JAMA Cardiol. 2020 Jan 15.
Lima R, Wofford M, Reckelhoff JF. Hypertension in postmenopausal women. Current hypertension reports. [Research Support, N.I.H., Extramural Review]. 2012 Jun;14(3):254-60.
Wassertheil-Smoller S, Anderson G, Psaty BM, Black HR, Manson J, Wong N, et al. Hypertension and its treatment in postmenopausal women: baseline data from the Women’s health initiative. Hypertension. 2000 Nov;36(5):780–9.
Lloyd-Jones DM, Evans JC, Levy D. Hypertension in adults across the age spectrum: current outcomes and control in the community. JAMA. 2005 Jul 27;294(4):466–72.
Ong KL, Tso AW, Lam KS, Cheung BM. Gender difference in blood pressure control and cardiovascular risk factors in Americans with diagnosed hypertension. Hypertension. 2008 Apr;51(4):1142–8.
Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. [Practice Guideline]. 2008 Jun;51(6):1403-19.
Huikuri HV, Pikkujamsa SM, Airaksinen KE, Ikaheimo MJ, Rantala AO, Kauma H, et al. Sex-related differences in autonomic modulation of heart rate in middle-aged subjects. Circulation. [Comparative Study Research Support, Non-U.S. Gov’t]. 1996 Jul 15;94(2):122-5.
Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S, Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation. 1997 Jan 7;95(1):39–45.
Proudler AJ, Ahmed AI, Crook D, Fogelman I, Rymer JM, Stevenson JC. Hormone replacement therapy and serum angiotensin-converting-enzyme activity in postmenopausal women. Lancet. 1995 Jul 8;346(8967):89–90.
Hayashi T, Yamada K, Esaki T, Kuzuya M, Satake S, Ishikawa T, et al. Estrogen increases endothelial nitric oxide by a receptor-mediated system. Biochem Biophys Res Commun. 1995 Sep 25;214(3):847–55.
Silvestri A, Gebara O, Vitale C, Wajngarten M, Leonardo F, Ramires JA, et al. Increased levels of C-reactive protein after oral hormone replacement therapy may not be related to an increased inflammatory response. Circulation. 2003 Jul 1;107(25):3165–9.
Lim WK, Wren B, Jepson N, Roy S, Caplan G. Effect of hormone replacement therapy on left ventricular hypertrophy. Am J Cardiol. 1999 Apr 01;83(7):1132–4 A9.
Pedram A, Razandi M, Aitkenhead M, Levin ER. Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1. The Journal of biological chemistry. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.]. 2005 Jul 15;280(28):26339-48.
Camper-Kirby D, Welch S, Walker A, Shiraishi I, Setchell KD, Schaefer E, et al. Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ Res. 2001 May 25;88(10):1020–7.
Natoli AK, Medley TL, Ahimastos AA, Drew BG, Thearle DJ, Dilley RJ, et al. Sex steroids modulate human aortic smooth muscle cell matrix protein deposition and matrix metalloproteinase expression. Hypertension. 2005 Nov;46(5):1129–34.
Clegg DJ, Brown LM, Woods SC, Benoit SC. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes. 2006 Apr;55(4):978–87.
Antus B, Hamar P, Kokeny G, Szollosi Z, Mucsi I, Nemes Z, et al. Estradiol is nephroprotective in the rat remnant kidney. Nephrol Dial Transplant. 2003 Jan;18(1):54–61.
DiBona GF. Sympathetic nervous system and hypertension. Hypertension. [Review]. 2013 Mar;61(3):556-60.
Seals DR, Esler MD. Human ageing and the sympathoadrenal system. J Physiol. [Research Support, U.S. Gov't, P.H.S. Review]. 2000 Nov 01;528(Pt 3):407-17.
Esler M, Rumantir M, Wiesner G, Kaye D, Hastings J, Lambert G. Sympathetic nervous system and insulin resistance: from obesity to diabetes. American journal of hypertension. [Review]. 2001 Nov;14(11 Pt 2):304S-9S.
Hart EC, Charkoudian N, Wallin BG, Curry TB, Eisenach JH, Joyner MJ. Sex differences in sympathetic neural-hemodynamic balance: implications for human blood pressure regulation. Hypertension. [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. 2009 Mar;53(3):571-6.
Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997 Dec 1;388(4):507–25.
Shughrue PJ, Scrimo PJ, Merchenthaler I. Evidence for the colocalization of estrogen receptor-beta mRNA and estrogen receptor-alpha immunoreactivity in neurons of the rat forebrain. Endocrinology. 1998 Dec;139(12):5267–70.
Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. [Research Support, Non-U.S. Gov't]. 1990 Apr 01;294(1):76-95.
Weitz G, Elam M, Born J, Fehm HL, Dodt C. Postmenopausal estrogen administration suppresses muscle sympathetic nerve activity. The Journal of clinical endocrinology and metabolism. [Clinical Trial Randomized Controlled Trial Research Support, Non-U.S. Gov’t]. 2001 Jan;86(1):344-8.
Mohamed MK, El-Mas MM, Abdel-Rahman AA. Estrogen enhancement of baroreflex sensitivity is centrally mediated. The American journal of physiology. [Research Support, U.S. Gov’t, P.H.S.]. 1999 Apr;276(4 Pt 2):R1030-7.
Xue B, Zhang Z, Beltz TG, Johnson RF, Guo F, Hay M, et al. Estrogen receptor-beta in the paraventricular nucleus and rostroventrolateral medulla plays an essential protective role in aldosterone/salt-induced hypertension in female rats. Hypertension. [Comparative Study Research Support, N.I.H., Extramural]. 2013 Jun;61(6):1255-62.
Cabral AM, Vasquez EC, Moyses MR, Antonio A. Sex hormone modulation of ventricular hypertrophy in sinoaortic denervated rats. Hypertension. 1988 Feb;11(2 Pt 2):I93–7.
Maranon RO, Lima R, Mathbout M, do Carmo JM, Hall JE, Roman RJ, et al. Postmenopausal hypertension: role of the sympathetic nervous system in an animal model. Am J Physiol Regul Integr Comp Physiol. [Research Support, N.I.H., Extramural]. 2014 Feb 15;306(4):R248-56.
Iliescu R, Yanes LL, Bell W, Dwyer T, Baltatu OC, Reckelhoff JF. Role of the renal nerves in blood pressure in male and female SHR. Am J Physiol Regul Integr Comp Physiol. 2006 Feb;290(2):R341–4.
Maranon RO, Reckelhoff JF. Mechanisms responsible for postmenopausal hypertension in a rat model: roles of the renal sympathetic nervous system and the renin-angiotensin system. Physiol Rep. 2016 Feb;4(2).
Azizi M, Sapoval M, Gosse P, Monge M, Bobrie G, Delsart P, et al. Optimum and stepped care standardised antihypertensive treatment with or without renal denervation for resistant hypertension (DENERHTN): a multicentre, open-label, randomised controlled trial. Lancet. 2015 May 16;385(9981):1957–65.
Bhatt DL, Kandzari DE, O'Neill WW, D'Agostino R, Flack JM, Katzen BT, et al. A controlled trial of renal denervation for resistant hypertension. N Engl J Med. 2014 Apr 10;370(15):1393–401.
Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Bohm M. Renal sympathetic denervation in patients with treatment-resistant hypertension (the Symplicity HTN-2 trial): a randomised controlled trial. Lancet. 2010 Dec 4;376(9756):1903–9.
Townsend RR, Mahfoud F, Kandzari DE, Kario K, Pocock S, Weber MA, et al. Catheter-based renal denervation in patients with uncontrolled hypertension in the absence of antihypertensive medications (SPYRAL HTN-OFF MED): a randomised, sham-controlled, proof-of-concept trial. Lancet. 2017 Nov 11;390(10108):2160–70.
Azizi M, Pereira H, Hamdidouche I, Gosse P, Monge M, Bobrie G, et al. Adherence to antihypertensive treatment and the blood pressure-lowering effects of renal denervation in the renal denervation for hypertension (DENERHTN) trial. Circulation. 2016 Sep 20;134(12):847–57.
Kandzari DE, Bhatt DL, Brar S, Devireddy CM, Esler M, Fahy M, et al. Predictors of blood pressure response in the SYMPLICITY HTN-3 trial. Eur Heart J. 2015 Jan 21;36(4):219–27.
Kandzari DE, Bohm M, Mahfoud F, Townsend RR, Weber MA, Pocock S, et al. Effect of renal denervation on blood pressure in the presence of antihypertensive drugs: 6-month efficacy and safety results from the SPYRAL HTN-ON MED proof-of-concept randomised trial. Lancet. 2018 Jun 9;391(10137):2346–55.
Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov't, P.H.S. Review]. 2001 May;37(5):1199-208.
Fernandez-Atucha A, Izagirre A, Fraile-Bermudez AB, Kortajarena M, Larrinaga G, Martinez-Lage P, et al. Sex differences in the aging pattern of renin-angiotensin system serum peptidases. Biol Sex Differ. 2017;8:5.
Toering TJ, Gant CM, Visser FW, van der Graaf AM, Laverman GD, Danser AHJ, et al. Sex differences in renin-angiotensin-aldosterone system affect extracellular volume in healthy subjects. Am J Physiol Renal Physiol. 2018 May 1;314(5):F873–F8.
Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther. 2008 Dec;120(3):292–316.
Hilliard LM, Sampson AK, Brown RD, Denton KM. The “his and hers” of the renin-angiotensin system. Curr Hypertens Rep. 2013 Feb;15(1):71–9.
Ganten U, Schroder G, Witt M, Zimmermann F, Ganten D, Stock G. Sexual dimorphism of blood pressure in spontaneously hypertensive rats: effects of anti-androgen treatment. J Hypertens. 1989 Sep;7(9):721–6.
Masubuchi Y, Kumai T, Uematsu A, Komoriyama K, Hirai M. Gonadectomy-induced reduction of blood pressure in adult spontaneously hypertensive rats. Acta Endocrinol. 1982 Sep;101(1):154–60.
Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension. 1998 Jan;31(1 Pt 2):435–9.
Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension. 2000 Jan;35(1 Pt 2):480–3.
Fortepiani LA, Zhang H, Racusen L, Roberts LJ, 2nd, Reckelhoff JF. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension. 2003 Mar;41(3 Pt 2):640-645.
Reckelhoff JF, Fortepiani LA. Novel mechanisms responsible for postmenopausal hypertension. Hypertension. 2004 May;43(5):918–23.
Yanes LL, Romero DG, Iles JW, Iliescu R, Gomez-Sanchez C, Reckelhoff JF. Sexual dimorphism in the renin-angiotensin system in aging spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. 2006 Aug;291(2):R383-90.
Yanes LL, Romero DG, Cucchiarelli VE, Fortepiani LA, Gomez-Sanchez CE, Santacruz F, et al. Role of endothelin in mediating postmenopausal hypertension in a rat model. Am J Physiol Regul Integr Comp Physiol. [Research Support, U.S. Gov’t, P.H.S.]. 2005 Jan;288(1):R229-33.
Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. 1999 Jan;33(1 Pt 2):323–8.
Roesch DM, Tian Y, Zheng W, Shi M, Verbalis JG, Sandberg K. Estradiol attenuates angiotensin-induced aldosterone secretion in ovariectomized rats. Endocrinology. [Research Support, U.S. Gov't, P.H.S.]. 2000 Dec;141(12):4629-36.
Silva-Antonialli MM, Tostes RC, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, et al. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res. 2004 Jun 1;62(3):587–93.
Hinojosa-Laborde C, Craig T, Zheng W, Ji H, Haywood JR, Sandberg K. Ovariectomy augments hypertension in aging female Dahl salt-sensitive rats. Hypertension. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov't, P.H.S.]. 2004 Oct;44(4):405-9.
Nickenig G, Baumer AT, Grohe C, Kahlert S, Strehlow K, Rosenkranz S, et al. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation. [Research Support, Non-U.S. Gov’t]. 1998 Jun 09;97(22):2197-201.
Mompeon A, Lazaro-Franco M, Bueno-Beti C, Perez-Cremades D, Vidal-Gomez X, Monsalve E, et al. Estradiol, acting through ERalpha, induces endothelial non-classic renin-angiotensin system increasing angiotensin 1-7 production. Mol Cell Endocrinol. [Research Support, Non-U.S. Gov’t]. 2016 Feb 15;422:1-8.
Brosnihan KB, Li P, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Phys. 1997 Dec;273(6 Pt 2):R1908–15.
Zanchetti A, Facchetti R, Cesana GC, Modena MG, Pirrelli A, Sega R. Menopause-related blood pressure increase and its relationship to age and body mass index: the SIMONA epidemiological study. J Hypertens. 2005 Dec;23(12):2269–76.
Juntunen M, Niskanen L, Saarelainen J, Tuppurainen M, Saarikoski S, Honkanen R. Changes in body weight and onset of hypertension in perimenopausal women. J Hum Hypertens. 2003 Nov;17(11):775–9.
Reckelhoff JF, Romero DG, Yanes Cardozo LL. Sex, oxidative stress, and hypertension: insights from animal models. Physiology (Bethesda). 2019 May 1;34(3):178–88.
Karastergiou K, Smith SR, Greenberg AS, Fried SK. Sex differences in human adipose tissues - the biology of pear shape. Biol Sex Differ. 2012 May 31;3(1):13.
Wilson PW, D’Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Archives of internal medicine. [Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. 2002 Sep 09;162(16):1867-72.
Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. [Review]. 2011 Feb;32(1):81-151.
Weigt C, Hertrampf T, Zoth N, Fritzemeier KH, Diel P. Impact of estradiol, ER subtype specific agonists and genistein on energy homeostasis in a rat model of nutrition induced obesity. Mol Cell Endocrinol. 2012 Apr 04;351(2):227–38.
Park CJ, Zhao Z, Glidewell-Kenney C, Lazic M, Chambon P, Krust A, et al. Genetic rescue of nonclassical ERalpha signaling normalizes energy balance in obese Eralpha-null mutant mice. J Clin Invest. 2011 Feb;121(2):604–12.
Pedersen SB, Kristensen K, Hermann PA, Katzenellenbogen JA, Richelsen B. Estrogen controls lipolysis by up-regulating alpha2A-adrenergic receptors directly in human adipose tissue through the estrogen receptor alpha. Implications for the female fat distribution. The Journal of clinical endocrinology and metabolism. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. 2004 Apr;89(4):1869-78.
Monjo M, Pujol E, Roca P. alpha2- to beta3-Adrenoceptor switch in 3T3-L1 preadipocytes and adipocytes: modulation by testosterone, 17beta-estradiol, and progesterone. Am J Physiol Endocrinol Metab. [Research Support, Non-U.S. Gov’t]. 2005 Jul;289(1):E145-50.
Jeong S, Yoon M. 17beta-estradiol inhibition of PPARgamma-induced adipogenesis and adipocyte-specific gene expression. Acta Pharmacol Sin. 2011 Feb;32(2):230–8.
Abeles E, Cordeiro LM, Martins Ade S, Pesquero JL, Reis AM, Andrade SP, et al. Estrogen therapy attenuates adiposity markers in spontaneously hypertensive rats. Metabolism. 2012 Aug;61(8):1100–7.
Sabbatini AR, Kararigas G. Menopause-related estrogen decrease as a possible contributor to the pathogenesis of heart failure with preserved ejection fraction: potential mechanisms of action. J Am Coll Cardiol. 2020:In press.
Sabbatini AR, Faria AP, Barbaro NR, Gordo WM, Modolo RG, Pinho C, et al. Deregulation of adipokines related to target organ damage on resistant hypertension. Journal of human hypertension. [Comparative Study Research Support, Non-U.S. Gov’t]. 2014 Jun;28(6):388-92.
Chu MC, Cosper P, Orio F, Carmina E, Lobo RA. Insulin resistance in postmenopausal women with metabolic syndrome and the measurements of adiponectin, leptin, resistin, and ghrelin. Am J Obstet Gynecol. 2006 Jan;194(1):100–4.
Huang SW, Seow KM, Ho LT, Chien Y, Chung DY, Chang CL, et al. Resistin mRNA levels are downregulated by estrogen in vivo and in vitro. FEBS Lett. 2005 Jan 17;579(2):449–54.
Barp J, Araujo AS, Fernandes TR, Rigatto KV, Llesuy S, Bello-Klein A, et al. Myocardial antioxidant and oxidative stress changes due to sex hormones. Braz J Med Biol Res. [Research Support, Non-U.S. Gov’t]. 2002 Sep;35(9):1075-81.
Ruiz-Larrea MB, Martin C, Martinez R, Navarro R, Lacort M, Miller NJ. Antioxidant activities of estrogens against aqueous and lipophilic radicals; differences between phenol and catechol estrogens. Chem Phys Lipids. 2000 Apr;105(2):179–88.
Martin C, Barturen K, Martinez R, Lacort M, Ruiz-Larrea MB. In vitro inhibition by estrogens of the oxidative modifications of human lipoproteins. J Physiol Biochem. 1998 Dec;54(4):195–202.
Vassalle C, Sciarrino R, Bianchi S, Battaglia D, Mercuri A, Maffei S. Sex-related differences in association of oxidative stress status with coronary artery disease. Fertil Steril. [Comparative Study]. 2012 Feb;97(2):414-9.
Kander MC, Cui Y, Liu Z. Gender difference in oxidative stress: a new look at the mechanisms for cardiovascular diseases. J Cell Mol Med. [Review]. 2016 Dec 13.
Dantas AP, Franco Mdo C, Silva-Antonialli MM, Tostes RC, Fortes ZB, Nigro D, et al. Gender differences in superoxide generation in microvessels of hypertensive rats: role of NAD(P)H-oxidase. Cardiovascular research. [Comparative Study Research Support, Non-U.S. Gov’t]. 2004 Jan 01;61(1):22-9.
Brandes RP, Mugge A. Gender differences in the generation of superoxide anions in the rat aorta. Life Sci. 1997;60(6):391–6.
Venegas-Pont M, Sartori-Valinotti JC, Glover PH, Reckelhoff JF, Ryan MJ. Sexual dimorphism in the blood pressure response to angiotensin II in mice after angiotensin-converting enzyme blockade. American journal of hypertension. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. 2010 Jan;23(1):92-6.
Sullivan JC, Sasser JM, Pollock JS. Sexual dimorphism in oxidant status in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2007 Feb;292(2):R764–8.
Nevzati E, Shafighi M, Bakhtian KD, Treiber H, Fandino J, Fathi AR. Estrogen induces nitric oxide production via nitric oxide synthase activation in endothelial cells. Acta Neurochir Suppl. 2015;120:141–5.
MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, Sherman TS, et al. Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res. 1997 Sep;81(3):355–62.
Bhuiyan MS, Shioda N, Fukunaga K. Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. Am J Physiol Endocrinol Metab. 2007 Dec;293(6):E1606–14.
Octavia Y, Kararigas G, de Boer M, Chrifi I, Kietadisorn R, Swinnen M, et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J Cell Mol Med. 2017 Dec;21(12):3277–87.
Gragasin FS, Xu Y, Arenas IA, Kainth N, Davidge ST. Estrogen reduces angiotensin II-induced nitric oxide synthase and NAD(P) H oxidase expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2003 Jan 1;23(1):38–44.
Yu J, Zhao Y, Li B, Sun L, Huo H. 17beta-estradiol regulates the expression of antioxidant enzymes in myocardial cells by increasing Nrf2 translocation. J Biochem Mol Toxicol. 2012 Jul;26(7):264–9.
Barta T, Tosaki A, Haines D, Balla G, Lekli I. Endothelin-1-induced hypertrophic alterations and heme oxygenase-1 expression in cardiomyoblasts are counteracted by beta estradiol: in vitro and in vivo studies. Naunyn Schmiedeberg's Arch Pharmacol. 2018 Apr;391(4):371–83.
Posa A, Szabo R, Kupai K, Berko AM, Veszelka M, Szucs G, et al. Cardioprotective effect of selective estrogen receptor modulator raloxifene are mediated by heme oxygenase in estrogen-deficient rat. Oxidative Med Cell Longev. 2017;2017:2176749.
Posa A, Pavo I, Varga C. Heme oxygenase contributes to estradiol and raloxifene-induced vasorelaxation in estrogen deficiency. Int J Cardiol. 2015;189:252–4.
Hsu JT, Kan WH, Hsieh CH, Choudhry MA, Bland KI, Chaudry IH. Mechanism of salutary effects of estrogen on cardiac function following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. Crit Care Med. 2009 Aug;37(8):2338–44.
Szalay L, Shimizu T, Schwacha MG, Choudhry MA, Rue LW 3rd, Bland KI, et al. Mechanism of salutary effects of estradiol on organ function after trauma-hemorrhage: upregulation of heme oxygenase. Am J Physiol Heart Circ Physiol. 2005 Jul;289(1):H92–8.
Campos C, Casali KR, Baraldi D, Conzatti A, Araujo AS, Khaper N, et al. Efficacy of a low dose of estrogen on antioxidant defenses and heart rate variability. Oxidative Med Cell Longev. 2014;2014:218749.
Ribon-Demars A, Pialoux V, Boreau A, Marcouiller F, Lariviere R, Bairam A, et al. Protective roles of estradiol against vascular oxidative stress in ovariectomized female rats exposed to normoxia or intermittent hypoxia. Acta Physiol (Oxf). 2019 Feb;225(2):e13159.
Zhu X, Tang Z, Cong B, Du J, Wang C, Wang L, et al. Estrogens increase cystathionine-gamma-lyase expression and decrease inflammation and oxidative stress in the myocardium of ovariectomized rats. Menopause. 2013 Oct;20(10):1084–91.
El-Mas MM, Abdel-Rahman AA. Nongenomic effects of estrogen mediate the dose-related myocardial oxidative stress and dysfunction caused by acute ethanol in female rats. Am J Physiol Endocrinol Metab. 2014 Apr 1;306(7):E740–7.
Pfeilschifter J, Koditz R, Pfohl M, Schatz H. Changes in proinflammatory cytokine activity after menopause. Endocr Rev. 2002 Feb;23(1):90–119.
Sites CK, Toth MJ, Cushman M, L’Hommedieu GD, Tchernof A, Tracy RP, et al. Menopause-related differences in inflammation markers and their relationship to body fat distribution and insulin-stimulated glucose disposal. Fertil Steril. 2002 Jan;77(1):128–35.
Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, et al. Anorectic estrogen mimics leptin's effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 2007 Jan;13(1):89–94.
Ribas V, Nguyen MT, Henstridge DC, Nguyen AK, Beaven SW, Watt MJ, et al. Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am J Physiol Endocrinol Metab. 2010 Feb;298(2):E304–19.
Wallen WJ, Belanger MP, Wittnich C. Sex hormones and the selective estrogen receptor modulator tamoxifen modulate weekly body weights and food intakes in adolescent and adult rats. J Nutr. 2001 Sep;131(9):2351–7.
Morselli E, Fuente-Martin E, Finan B, Kim M, Frank A, Garcia-Caceres C, et al. Hypothalamic PGC-1alpha protects against high-fat diet exposure by regulating ERalpha. Cell Rep. 2014 Oct 23;9(2):633–45.
Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, et al. Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci U S A. 2007 Feb 13;104(7):2501–6.
Dahlman I, Vaxillaire M, Nilsson M, Lecoeur C, Gu HF, Cavalcanti-Proenca C, et al. Estrogen receptor alpha gene variants associate with type 2 diabetes and fasting plasma glucose. Pharmacogenet Genomics. 2008 Nov;18(11):967–75.
Okura T, Koda M, Ando F, Niino N, Ohta S, Shimokata H. Association of polymorphisms in the estrogen receptor alpha gene with body fat distribution. Int J Obes Relat Metab Disord. 2003 Sep;27(9):1020–7.
Lundholm L, Zang H, Hirschberg AL, Gustafsson JA, Arner P, Dahlman-Wright K. Key lipogenic gene expression can be decreased by estrogen in human adipose tissue. Fertil Steril. 2008 Jul;90(1):44–8.
Van Pelt RE, Gozansky WS, Schwartz RS, Kohrt WM. Intravenous estrogens increase insulin clearance and action in postmenopausal women. Am J Physiol Endocrinol Metab. 2003 Aug;285(2):E311–7.
Silvestri A, Gambacciani M, Vitale C, Monteleone P, Ciaponi M, Fini M, et al. Different effect of hormone replacement therapy, DHEAS and tibolone on endothelial function in postmenopausal women with increased cardiovascular risk. Maturitas. 2005 Apr 11;50(4):305–11.
Shivers KY, Amador N, Abrams L, Hunter D, Jenab S, Quinones-Jenab V. Estrogen alters baseline and inflammatory-induced cytokine levels independent from hypothalamic-pituitary-adrenal axis activity. Cytokine. 2015 Apr;72(2):121–9.
Amant C, Holm P, Xu Sh SH, Tritman N, Kearney M, Losordo DW. Estrogen receptor-mediated, nitric oxide-dependent modulation of the immunologic barrier function of the endothelium: regulation of fas ligand expression by estradiol. Circulation. 2001 Nov 20;104(21):2576–81.
Guzeloglu-Kayisli O, Halis G, Taskiran S, Kayisli UA, Arici A. DNA-binding ability of NF-kappaB is affected differently by ERalpha and ERbeta and its activation results in inhibition of estrogen responsiveness. Reprod Sci. 2008 May;15(5):493–505.
Santos RS, de Fatima LA, Frank AP, Carneiro EM, Clegg DJ. The effects of 17 alpha-estradiol to inhibit inflammation in vitro. Biol Sex Differ. 2017 Sep 6;8(1):30.
Mori M, Tsukahara F, Yoshioka T, Irie K, Ohta H. Suppression by 17beta-estradiol of monocyte adhesion to vascular endothelial cells is mediated by estrogen receptors. Life Sci. 2004 Jun 18;75(5):599–609.
Hildreth KL, Ozemek C, Kohrt WM, Blatchford PJ, Moreau KL. Vascular dysfunction across the stages of the menopausal transition is associated with menopausal symptoms and quality of life. Menopause (New York, NY). 2018 Sep;25(9):1011-9.
Virdis A, Ghiadoni L, Pinto S, Lombardo M, Petraglia F, Gennazzani A, et al. Mechanisms responsible for endothelial dysfunction associated with acute estrogen deprivation in normotensive women. Circulation. 2000 May 16;101(19):2258–63.
Sanada M, Higashi Y, Nakagawa K, Tsuda M, Kodama I, Kimura M, et al. Hormone replacement effects on endothelial function measured in the forearm resistance artery in normocholesterolemic and hypercholesterolemic postmenopausal women. The Journal of clinical endocrinology and metabolism. [Research Support, Non-U.S. Gov't]. 2002 Oct;87(10):4634-41.
Higashi Y, Sanada M, Sasaki S, Nakagawa K, Goto C, Matsuura H, et al. Effect of estrogen replacement therapy on endothelial function in peripheral resistance arteries in normotensive and hypertensive postmenopausal women. Hypertension. [Clinical Trial Research Support, Non-U.S. Gov't]. 2001 Feb;37(2 Pt 2):651-7.
Ylikorkala O, Orpana A, Puolakka J, Pyorala T, Viinikka L. Postmenopausal hormonal replacement decreases plasma levels of endothelin-1. J Clin Endocrinol Metab. 1995 Nov;80(11):3384–7.
Node K, Kitakaze M, Kosaka H, Minamino T, Sato H, Kuzuya T, et al. Roles of NO and Ca2+-activated K+ channels in coronary vasodilation induced by 17beta-estradiol in ischemic heart failure. FASEB J. 1997 Aug;11(10):793–9.
Raddino R, Manca C, Poli E, Bolognesi R, Visioli O. Effects of 17 beta-estradiol on the isolated rabbit heart. Arch Int Pharmacodyn Ther. 1986 May;281(1):57–65.
Ylikorkala O, Cacciatore B, Paakkari I, Tikkanen MJ, Viinikka L, Toivonen J. The long-term effects of oral and transdermal postmenopausal hormone replacement therapy on nitric oxide, endothelin-1, prostacyclin, and thromboxane. Fertil Steril. 1998 May;69(5):883–8.
Morey AK, Razandi M, Pedram A, Hu RM, Prins BA, Levin ER. Oestrogen and progesterone inhibit the stimulated production of endothelin-1. Biochem J. 1998 Mar 15;330(Pt 3):1097–105.
Ospina JA, Duckles SP, Krause DN. 17beta-estradiol decreases vascular tone in cerebral arteries by shifting COX-dependent vasoconstriction to vasodilation. Am J Physiol Heart Circ Physiol. 2003 Jul;285(1):H241–50.
Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res. 2005 Feb 18;96(3):376–83.
Huang A, Wu Y, Sun D, Koller A, Kaley G. Effect of estrogen on flow-induced dilation in NO deficiency: role of prostaglandins and EDHF. J Appl Physiol (1985). 2001 Dec;91(6):2561–6.
Huang A, Sun D, Wu Z, Yan C, Carroll MA, Jiang H, et al. Estrogen elicits cytochrome P450--mediated flow-induced dilation of arterioles in NO deficiency: role of PI3K-Akt phosphorylation in genomic regulation. Circ Res. 2004 Feb 6;94(2):245–52.
Morey AK, Pedram A, Razandi M, Prins BA, Hu RM, Biesiada E, et al. Estrogen and progesterone inhibit vascular smooth muscle proliferation. Endocrinology. 1997 Aug;138(8):3330–9.
Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, et al. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circulation research. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. 2002 May 31;90(10):1087-92.
Corcoran MP, Lichtenstein AH, Meydani M, Dillard A, Schaefer EJ, Lamon-Fava S. The effect of 17{beta}-estradiol on cholesterol content in human macrophages is influenced by the lipoprotein milieu. J Mol Endocrinol. 2011;47(1):109–17.
Choi HY, Park HC, Ha SK. Salt sensitivity and hypertension: a paradigm shift from kidney malfunction to vascular endothelial dysfunction. Electrolyte Blood Press. [Review]. 2015 Jun;13(1):7-16.
Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, et al. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. [Comparative Study Research Support, Non-U.S. Gov’t]. 1997 Dec 13;350(9093):1734-7.
Bigazzi R, Bianchi S, Baldari D, Sgherri G, Baldari G, Campese VM. Microalbuminuria in salt-sensitive patients. A marker for renal and cardiovascular risk factors. Hypertension. [Research Support, U.S. Gov't, P.H.S.]. 1994 Feb;23(2):195-9.
Miyoshi A, Suzuki H, Fujiwara M, Masai M, Iwasaki T. Impairment of endothelial function in salt-sensitive hypertension in humans. Am J Hypertens. 1997 Oct;10(10 Pt 1):1083–90.
Shukri MZ, Tan JW, Manosroi W, Pojoga LH, Rivera A, Williams JS, et al. Biological sex modulates the adrenal and blood pressure responses to angiotensin II. Hypertension. 2018 Jun;71(6):1083–90.
He J, Gu D, Chen J, Jaquish CE, Rao DC, Hixson JE, et al. Gender difference in blood pressure responses to dietary sodium intervention in the GenSalt study. Journal of hypertension. [Research Support, N.I.H., Extramural]. 2009 Jan;27(1):48-54.
Dubey RK, Oparil S, Imthurn B, Jackson EK. Sex hormones and hypertension. Cardiovascular research. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review]. 2002 Feb 15;53(3):688-708.
Hernandez Schulman I, Raij L. Salt sensitivity and hypertension after menopause: role of nitric oxide and angiotensin II. Am J Nephrol. [Research Support, U.S. Gov’t, Non-P.H.S. Review]. 2006;26(2):170-80.
Tominaga T, Suzuki H, Ogata Y, Matsukawa S, Saruta T. The role of sex hormones and sodium intake in postmenopausal hypertension. J Hum Hypertens. 1991 Dec;5(6):495–500.
Schulman IH, Aranda P, Raij L, Veronesi M, Aranda FJ, Martin R. Surgical menopause increases salt sensitivity of blood pressure. Hypertension. [Research Support, U.S. Gov’t, Non-P.H.S.]. 2006 Jun;47(6):1168-74.
Scuteri A, Lakatta EG, Anderson DE, Fleg JL. Transdermal 17 beta-oestradiol reduces salt sensitivity of blood pressure in postmenopausal women. J Hypertens. 2003 Dec;21(12):2419–20.
Harrison-Bernard LM, Schulman IH, Raij L. Postovariectomy hypertension is linked to increased renal AT1 receptor and salt sensitivity. Hypertension. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.]. 2003 Dec;42(6):1157-63.
Esslinger U, Garnier S, Korniat A, Proust C, Kararigas G, Muller-Nurasyid M, et al. Exome-wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS One. 2017;12(3):e0172995.
McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013 Jan;123(1):19–26.
Lalouel JM. Large-scale search for genes predisposing to essential hypertension. Am J Hypertens. 2003 Feb;16(2):163–6.
Waken RJ, de Las Fuentes L, Rao DC. A review of the genetics of hypertension with a focus on gene-environment interactions. Current hypertension reports. [Review]. 2017 Mar;19(3):23.
Gaignebet L, Kararigas G. En route to precision medicine through the integration of biological sex into pharmacogenomics. Clin Sci (Lond). 2017 Feb 01;131(4):329–42.
Sethi AA, Nordestgaard BG, Agerholm-Larsen B, Frandsen E, Jensen G, Tybjaerg-Hansen A. Angiotensinogen polymorphisms and elevated blood pressure in the general population: the Copenhagen City Heart Study. Hypertension. [Research Support, Non-U.S. Gov't]. 2001 Mar;37(3):875-81.
Chen W, Srinivasan SR, Li S, Boerwinkle E, Berenson GS. Gender-specific influence of NO synthase gene on blood pressure since childhood: the Bogalusa Heart Study. Hypertension. [Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.]. 2004 Nov;44(5):668-73.
Kumar NN, Benjafield AV, Lin RC, Wang WY, Stowasser M, Morris BJ. Haplotype analysis of aldosterone synthase gene (CYP11B2) polymorphisms shows association with essential hypertension. J Hypertens. 2003 Jul;21(7):1331–7.
Rana BK, Insel PA, Payne SH, Abel K, Beutler E, Ziegler MG, et al. Population-based sample reveals gene-gender interactions in blood pressure in White Americans. Hypertension. [Research Support, N.I.H., Extramural]. 2007 Jan;49(1):96-106.
Sagnella GA, Rothwell MJ, Onipinla AK, Wicks PD, Cook DG, Cappuccio FP. A population study of ethnic variations in the angiotensin-converting enzyme I/D polymorphism: relationships with gender, hypertension and impaired glucose metabolism. J Hypertens. 1999 May;17(5):657–64.
Turner ST, Boerwinkle E, Sing CF. Context-dependent associations of the ACE I/D polymorphism with blood pressure. Hypertension. 1999 Oct;34(4 Pt 2):773–8.
Peter I, Shearman AM, Zucker DR, Schmid CH, Demissie S, Cupples LA, et al. Variation in estrogen-related genes and cross-sectional and longitudinal blood pressure in the Framingham Heart Study. Journal of hypertension. [Research Support, N.I.H., Extramural]. 2005 Dec;23(12):2193-200.
Deng HW, Li J, Li JL, Dowd R, Davies KM, Johnson M, et al. Association of estrogen receptor-alpha genotypes with body mass index in normal healthy postmenopausal Caucasian women. J Clin Endocrinol Metab. 2000 Aug;85(8):2748–51.
Nilsson M, Dahlman I, Ryden M, Nordstrom EA, Gustafsson JA, Arner P, et al. Oestrogen receptor alpha gene expression levels are reduced in obese compared to normal weight females. Int J Obes. 2007 Jun;31(6):900–7.
Jazbutyte V, Arias-Loza PA, Hu K, Widder J, Govindaraj V, von Poser-Klein C, et al. Ligand-dependent activation of ER {beta} lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovascular research. [Research Support, Non-U.S. Gov't]. 2008 Mar 1;77(4):774-81.
Chen C, Li Y, Chen F, Pan H, Shen H, Sun Z, et al. Estrogen receptor beta genetic variants and combined oral contraceptive use as relates to the risk of hypertension in Chinese women. Archives of medical research. [Research Support, Non-U.S. Gov't]. 2010 Nov;41(8):599-605.
Manosroi W, Tan JW, Rariy CM, Sun B, Goodarzi MO, Saxena AR, et al. The association of estrogen receptor-beta gene variation with salt-sensitive blood pressure. J Clin Endocrinol Metab. 2017 Nov 1;102(11):4124–35.
Nakayama T, Kuroi N, Sano M, Tabara Y, Katsuya T, Ogihara T, et al. Mutation of the follicle-stimulating hormone receptor gene 5′-untranslated region associated with female hypertension. Hypertension. [Research Support, Non-U.S. Gov’t]. 2006 Sep;48(3):512-8.
Cui C, Huang C, Liu K, Xu G, Yang J, Zhou Y, et al. Large-scale in silico identification of drugs exerting sex-specific effects in the heart. J Transl Med. 2018 Aug 29;16(1):236.
Huang C, Yang W, Wang J, Zhou Y, Geng B, Kararigas G, et al. The DrugPattern tool for drug set enrichment analysis and its prediction for beneficial effects of oxLDL on type 2 diabetes. J Genet Genomics. 2018 Jul 20;45(7):389–97.
Regitz-Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. [Review]. 2017 Jan;97(1):1–37.
Himmelmann A. Hypertension: an important precursor of heart failure. Blood Press. 1999;8(5-6):253–60.
Izzo R, Losi MA, Stabile E, Lonnebakken MT, Canciello G, Esposito G, et al. Development of left ventricular hypertrophy in treated hypertensive outpatients: the Campania salute network. Hypertension. 2017 Jan;69(1):136–42.
Gerdts E, Izzo R, Mancusi C, Losi MA, Manzi MV, Canciello G, et al. Left ventricular hypertrophy offsets the sex difference in cardiovascular risk (the Campania salute network). Int J Cardiol. 2018 May 1;258:257–61.
Aurigemma GP, Silver KH, McLaughlin M, Mauser J, Gaasch WH. Impact of chamber geometry and gender on left ventricular systolic function in patients > 60 years of age with aortic stenosis. Am J Cardiol. 1994 Oct 15;74(8):794–8.
Carroll JD, Carroll EP, Feldman T, Ward DM, Lang RM, McGaughey D, et al. Sex-associated differences in left ventricular function in aortic stenosis of the elderly. Circulation. 1992 Oct;86(4):1099–107.
Douglas PS, Otto CM, Mickel MC, Labovitz A, Reid CL, Davis KB. Gender differences in left ventricle geometry and function in patients undergoing balloon dilatation of the aortic valve for isolated aortic stenosis. NHLBI balloon Valvuloplasty registry. Br Heart J. 1995 Jun;73(6):548–54.
Gaignebet L, Kandula MM, Lehmann D. Knosalla C, Kreil DP, Kararigas G. Sex-specific human cardiomyocyte gene regulation in left ventricular pressure overload. Mayo Clin Proc; 2020 Jan 13.
Kararigas G, Dworatzek E, Petrov G, Summer H, Schulze TM, Baczko I, et al. Sex-dependent regulation of fibrosis and inflammation in human left ventricular remodelling under pressure overload. Eur J Heart Fail. 2014 Nov;16(11):1160–7.
Petrov G, Dworatzek E, Schulze TM, Dandel M, Kararigas G, Mahmoodzadeh S, et al. Maladaptive remodeling is associated with impaired survival in women but not in men after aortic valve replacement. JACC Cardiovasc Imaging. 2014 Nov;7(11):1073–80.
Villari B, Campbell SE, Schneider J, Vassalli G, Chiariello M, Hess OM. Sex-dependent differences in left ventricular function and structure in chronic pressure overload. Eur Heart J. 1995 Oct;16(10):1410–9.
Gerdts E, Zabalgoitia M, Bjornstad H, Svendsen TL, Devereux RB. Gender differences in systolic left ventricular function in hypertensive patients with electrocardiographic left ventricular hypertrophy (the LIFE study). Am J Cardiol. 2001 Apr 15;87(8):980–3.
Devereux RB, Bella JN, Palmieri V, Oberman A, Kitzman DW, Hopkins PN, et al. Left ventricular systolic dysfunction in a biracial sample of hypertensive adults: the hypertension genetic epidemiology network (HyperGEN) study. Hypertension. 2001 Sep;38(3):417–23.
Gerdts E, Okin PM, de Simone G, Cramariuc D, Wachtell K, Boman K, et al. Gender differences in left ventricular structure and function during antihypertensive treatment: the losartan intervention for endpoint reduction in hypertension study. Hypertension. 2008 Apr;51(4):1109–14.
De Simone G, Devereux RB, Chinali M, Roman MJ, Barac A, Panza JA, et al. Sex differences in obesity-related changes in left ventricular morphology: the strong heart study. J Hypertens. 2011 Jul;29(7):1431–8.
Light KC, Hinderliter AL, West SG, Grewen KM, Steege JF, Sherwood A, et al. Hormone replacement improves hemodynamic profile and left ventricular geometry in hypertensive and normotensive postmenopausal women. Journal of hypertension. [Clinical Trial Randomized Controlled Trial Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. 2001 Feb;19(2):269-78.
Miya Y, Sumino H, Ichikawa S, Nakamura T, Kanda T, Kumakura H, et al. Effects of hormone replacement therapy on left ventricular hypertrophy and growth-promoting factors in hypertensive postmenopausal women. Hypertension Res. 2002 Mar;25(2):153–9.
Sharkey LC, Holycross BJ, Park S, Shiry LJ, Hoepf TM, McCune SA, et al. Effect of ovariectomy and estrogen replacement on cardiovascular disease in heart failure-prone SHHF/mcc- fa cp rats. J Mol Cell Cardiol. 1999 Aug;31(8):1527–37.
Kararigas G, Fliegner D, Gustafsson JA, Regitz-Zagrosek V. Role of the estrogen/estrogen-receptor-beta axis in the genomic response to pressure overload-induced hypertrophy. Physiol Genomics. 2011 Apr 27;43(8):438–46.
Kararigas G, Nguyen BT, Jarry H. Estrogen modulates cardiac growth through an estrogen receptor alpha-dependent mechanism in healthy ovariectomized mice. Mol Cell Endocrinol. 2014 Feb 15;382(2):909–14.
Kararigas G, Nguyen BT, Zelarayan LC, Hassenpflug M, Toischer K, Sanchez-Ruderisch H, et al. Genetic background defines the regulation of postnatal cardiac growth by 17beta-estradiol through a beta-catenin mechanism. Endocrinology. 2014 Jul;155(7):2667–76.
Duft K, Schanz M, Pham H, Abdelwahab A, Schriever C, Kararigas G, et al. 17beta-estradiol-induced interaction of estrogen receptor alpha and human atrial essential myosin light chain modulates cardiac contractile function. Basic Res Cardiol. 2017 Jan;112(1):1.
Mahmoodzadeh S, Pham TH, Kuehne A, Fielitz B, Dworatzek E, Kararigas G, et al. 17beta-estradiol-induced interaction of ERalpha with NPPA regulates gene expression in cardiomyocytes. Cardiovasc Res. 2012 Dec 1;96(3):411–21.
Nguyen BT, Kararigas G, Jarry H. Dose-dependent effects of a genistein-enriched diet in the heart of ovariectomized mice. Genes Nutr. 2012 Oct 30;8(4):383–90.
Nguyen BT, Kararigas G, Wuttke W, Jarry H. Long-term treatment of ovariectomized mice with estradiol or phytoestrogens as a new model to study the role of estrogenic substances in the heart. Planta Med. 2012 Jan;78(1):6–11.
Schubert C, Raparelli V, Westphal C, Dworatzek E, Petrov G, Kararigas G, et al. Reduction of apoptosis and preservation of mitochondrial integrity under ischemia/reperfusion injury is mediated by estrogen receptor beta. Biol Sex Differ. 2016;7:53.
Scheuer J, Malhotra A, Schaible TF, Capasso J. Effects of gonadectomy and hormonal replacement on rat hearts. Circ Res. 1987 Jul;61(1):12–9.
Kararigas G, Becher E, Mahmoodzadeh S, Knosalla C, Hetzer R, Regitz-Zagrosek V. Sex-specific modification of progesterone receptor expression by 17beta-oestradiol in human cardiac tissues. Biol Sex Differ. 2010;1(1):2.
Kararigas G, Bito V, Tinel H, Becher E, Baczko I, Knosalla C, et al. Transcriptome characterization of estrogen-treated human myocardium identifies myosin regulatory light chain interacting protein as a sex-specific element influencing contractile function. J Am Coll Cardiol. 2012 Jan 24;59(4):410–7.
Kararigas G, Fliegner D, Forler S, Klein O, Schubert C, Gustafsson JA, et al. Comparative proteomic analysis reveals sex and estrogen receptor beta effects in the pressure overloaded heart. J Proteome Res. 2014 Dec 5;13(12):5829–36.
Sanchez-Ruderisch H, Queiros AM, Fliegner D, Eschen C, Kararigas G, Regitz-Zagrosek V. Sex-specific regulation of cardiac microRNAs targeting mitochondrial proteins in pressure overload. Biol Sex Differ. 2019 Feb 6;10(1):8.
Murphy E, Steenbergen C. Estrogen regulation of protein expression and signaling pathways in the heart. Biol Sex Differ. 2014 Mar 10;5(1):6.
Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. 2010 Jun;298(6):R1597–606.
Hein S, Hassel D, Kararigas G. The zebrafish (Danio rerio) is a relevant model for studying sex-specific effects of 17beta-estradiol in the adult heart. Int J Mol Sci. 2019 Dec;13:20(24) 6287.
Queiros AM, Eschen C, Fliegner D, Kararigas G, Dworatzek E, Westphal C, et al. Sex- and estrogen-dependent regulation of a miRNA network in the healthy and hypertrophied heart. Int J Cardiol. 2013 Nov 20;169(5):331–8.
van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation. 2001 Sep 18;104(12):1419–23.
Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology. [Research Support, N.I.H., Extramural Research Support, U.S. Gov't, Non-P.H.S.]. 2008 Jul;149(7):3361-9.
Babiker FA, De Windt LJ, van Eickels M, Thijssen V, Bronsaer RJ, Grohe C, et al. 17beta-estradiol antagonizes cardiomyocyte hypertrophy by autocrine/paracrine stimulation of a guanylyl cyclase a receptor-cyclic guanosine monophosphate-dependent protein kinase pathway. Circulation. 2004 Jan 20;109(2):269–76.
Babiker FA, Lips D, Meyer R, Delvaux E, Zandberg P, Janssen B, et al. Estrogen receptor beta protects the murine heart against left ventricular hypertrophy. Arterioscler Thromb Vasc Biol. 2006 Jul;26(7):1524–30.
Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, et al. Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. 2005 Feb;288(2):H469–76.
Peter I, Shearman AM, Vasan RS, Zucker DR, Schmid CH, Demissie S, et al. Association of estrogen receptor beta gene polymorphisms with left ventricular mass and wall thickness in women. Am J Hypertens. 2005 Nov;18(11):1388–95.
Benetos A, Rudnichi A, Safar M, Guize L. Pulse pressure and cardiovascular mortality in normotensive and hypertensive subjects. Hypertension. 1998 Sep;32(3):560–4.
Cox RH. Age-related changes in arterial wall mechanics and composition of NIA Fischer rats. Mech Ageing Dev. 1983 Sep;23(1):21–36.
Cattell MA, Anderson JC, Hasleton PS. Age-related changes in amounts and concentrations of collagen and elastin in normotensive human thoracic aorta. Clin Chim Acta. 1996 Feb 9;245(1):73–84.
Bruel A, Oxlund H. Changes in biomechanical properties, composition of collagen and elastin, and advanced glycation endproducts of the rat aorta in relation to age. Atherosclerosis. 1996 Dec 20;127(2):155–65.
Marque V, Kieffer P, Atkinson J, Lartaud-Idjouadiene I. Elastic properties and composition of the aortic wall in old spontaneously hypertensive rats. Hypertension. 1999 Sep;34(3):415–22.
Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998 May 21;393(6682):276–80.
Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003 Jan;130(2):411–23.
Altinbas L, Bormann N, Lehmann D, Jeuthe S, Wulsten D, Kornak U, et al. Assessment of bones deficient in fibrillin-1 microfibrils reveals pronounced sex differences. Int J Mol Sci. 2019 Dec;1:20(23) 6059.
Bhushan R, Altinbas L, Jager M, Zaradzki M, Lehmann D, Timmermann B, et al. An integrative systems approach identifies novel candidates in Marfan syndrome-related pathophysiology. J Cell Mol Med. 2019 Apr;23(4):2526–35.
Ramirez F, Carta L, Lee-Arteaga S, Liu C, Nistala H, Smaldone S. Fibrillin-rich microfibrils - structural and instructive determinants of mammalian development and physiology. Connect Tissue Res. 2008;49(1):1–6.
Smaldone S, Ramirez F. Fibrillin microfibrils in bone physiology. Matrix Biol. 2016 May-Jul;52-54:191–7.
Milewicz DM, Urban Z, Boyd C. Genetic disorders of the elastic fiber system. Matrix Biol. 2000 Nov;19(6):471–80.
Dasouki M, Markova D, Garola R, Sasaki T, Charbonneau NL, Sakai LY, et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007 Nov 15;143A(22):2635–41.
Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, et al. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet. 2002 Sep 1;11(18):2113–8.
Le VP, Knutsen RH, Mecham RP, Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ Physiol. 2011 Jul;301(1):H221–9.
Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, et al. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003 Nov;112(9):1419–28.
Avolio A, Jones D, Tafazzoli-Shadpour M. Quantification of alterations in structure and function of elastin in the arterial media. Hypertension. 1998 Jul;32(1):170–5.
Messerli FH, Frohlich ED, Ventura HO. Arterial compliance in essential hypertension. J Cardiovasc Pharmacol. 1985;7(Suppl 2):S33–5.
Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003 Jan 7;107(1):139–46.
Kawasaki T, Sasayama S, Yagi S, Asakawa T, Hirai T. Non-invasive assessment of the age related changes in stiffness of major branches of the human arteries. Cardiovasc Res. 1987 Sep;21(9):678–87.
Tsai SS, Lin YS, Hwang JS, Chu PH. Vital roles of age and metabolic syndrome-associated risk factors in sex-specific arterial stiffness across nearly lifelong ages: possible implication of menopause and andropause. Atherosclerosis. 2017 Mar;258:26–33.
Gatzka CD, Kingwell BA, Cameron JD, Berry KL, Liang YL, Dewar EM, et al. Gender differences in the timing of arterial wave reflection beyond differences in body height. Journal of hypertension. [Research Support, Non-U.S. Gov’t]. 2001 Dec;19(12):2197-203.
Guajardo I, Ayer A, Johnson AD, Ganz P, Mills C, Donovan C, et al. Sex differences in vascular dysfunction and cardiovascular outcomes: the cardiac, endothelial function, and arterial stiffness in ESRD (CERES) study. Hemodial Int. 2017 Mar;08.
Coutinho T, Borlaug BA, Pellikka PA, Turner ST, Kullo IJ. Sex differences in arterial stiffness and ventricular-arterial interactions. J Am Coll Cardiol. 2013 Jan 8;61(1):96–103.
Russo C, Jin Z, Palmieri V, Homma S, Rundek T, Elkind MS, et al. Arterial stiffness and wave reflection: sex differences and relationship with left ventricular diastolic function. Hypertension. 2012 Aug;60(2):362–8.
Tomiyama H, Yamashina A, Arai T, Hirose K, Koji Y, Chikamori T, et al. Influences of age and gender on results of noninvasive brachial-ankle pulse wave velocity measurement--a survey of 12517 subjects. Atherosclerosis. 2003 Feb;166(2):303–9.
Waddell TK, Dart AM, Gatzka CD, Cameron JD, Kingwell BA. Women exhibit a greater age-related increase in proximal aortic stiffness than men. J Hypertens. 2001 Dec;19(12):2205–12.
Staessen JA, van der Heijden-Spek JJ, Safar ME, Den Hond E, Gasowski J, Fagard RH, et al. Menopause and the characteristics of the large arteries in a population study. J Hum Hypertens. 2001 Aug;15(8):511–8.
Zaydun G, Tomiyama H, Hashimoto H, Arai T, Koji Y, Yambe M, et al. Menopause is an independent factor augmenting the age-related increase in arterial stiffness in the early postmenopausal phase. Atherosclerosis. 2006 Jan;184(1):137–42.
Gompel A, Boutouyrie P, Joannides R, Christin-Maitre S, Kearny-Schwartz A, Kunz K, et al. Association of menopause and hormone replacement therapy with large artery remodeling. Fertil Steril. [Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t]. 2011 Dec;96(6):1445-50.
da Costa LS, de Oliveira MA, Rubim VS, Wajngarten M, Aldrighi JM, Rosano GM, et al. Effects of hormone replacement therapy or raloxifene on ambulatory blood pressure and arterial stiffness in treated hypertensive postmenopausal women. The American journal of cardiology. [Clinical Trial Randomized Controlled Trial]. 2004 Dec 01;94(11):1453-6.
Dworatzek E, Baczko I, Kararigas G. Effects of aging on cardiac extracellular matrix in men and women. Proteomics Clin Appl. 2016 Jan;10(1):84–91.
Qiu H, Tian B, Resuello RG, Natividad FF, Peppas A, Shen YT, et al. Sex-specific regulation of gene expression in the aging monkey aorta. Physiol Genomics. 2007 Apr 24;29(2):169–80.
Qiu H, Depre C, Ghosh K, Resuello RG, Natividad FF, Rossi F, et al. Mechanism of gender-specific differences in aortic stiffness with aging in nonhuman primates. Circulation. 2007 Aug 7;116(6):669–76.
Mugge A, Riedel M, Barton M, Kuhn M, Lichtlen PR. Endothelium independent relaxation of human coronary arteries by 17 beta-oestradiol in vitro. Cardiovasc Res. [Comparative Study]. 1993 Nov;27(11):1939-42.
Han G, Ma H, Chintala R, Fulton DJ, Barman SA, White RE. Essential role of the 90-kilodalton heat shock protein in mediating nongenomic estrogen signaling in coronary artery smooth muscle. J Pharmacol Exp Ther. 2009 Jun;329(3):850–5.
Sullivan TR Jr, Karas RH, Aronovitz M, Faller GT, Ziar JP, Smith JJ, et al. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J Clin Invest. 1995 Nov;96(5):2482–8.
Karas RH, Hodgin JB, Kwoun M, Krege JH, Aronovitz M, Mackey W, et al. Estrogen inhibits the vascular injury response in estrogen receptor beta-deficient female mice. Proc Natl Acad Sci U S A. 1999 Dec 21;96(26):15133–6.
Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010 Jun 12;375(9731):2073–81.
Zhang QL, Koenig W, Raum E, Stegmaier C, Brenner H, Rothenbacher D. Epidemiology of chronic kidney disease: results from a population of older adults in Germany. Prev Med. 2009 Feb;48(2):122–7.
Rothenbacher D, Klenk J, Denkinger M, Karakas M, Nikolaus T, Peter R, et al. Prevalence and determinants of chronic kidney disease in community-dwelling elderly by various estimating equations. BMC Public Health. 2012 May 10;12:343.
Kastarinen M, Juutilainen A, Kastarinen H, Salomaa V, Karhapaa P, Tuomilehto J, et al. Risk factors for end-stage renal disease in a community-based population: 26-year follow-up of 25,821 men and women in eastern Finland. J Intern Med. 2010 Jun;267(6):612–20.
Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta-analysis. Journal of the American Society of Nephrology : JASN. [Meta-Analysis]. 2000 Feb;11(2):319-29.
Cattran DC, Reich HN, Beanlands HJ, Miller JA, Scholey JW, Troyanov S. The impact of sex in primary glomerulonephritis. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. [Comparative Study Research Support, Non-U.S. Gov’t]. 2008 Jul;23(7):2247-53.
Evans M, Fryzek JP, Elinder CG, Cohen SS, McLaughlin JK, Nyren O, et al. The natural history of chronic renal failure: results from an unselected, population-based, inception cohort in Sweden. American journal of kidney diseases : the official journal of the National Kidney Foundation. [Research Support, Non-U.S. Gov't]. 2005 Nov;46(5):863-70.
Eriksen BO, Ingebretsen OC. The progression of chronic kidney disease: a 10-year population-based study of the effects of gender and age. Kidney Int. 2006 Jan;69(2):375–82.
Lu H, Lei X, Klaassen C. Gender differences in renal nuclear receptors and aryl hydrocarbon receptor in 5/6 nephrectomized rats. Kidney Int. 2006 Dec;70(11):1920–8.
Fanelli C, Delle H, Cavaglieri RC, Dominguez WV, Noronha IL. Gender differences in the progression of experimental chronic kidney disease induced by chronic nitric oxide inhibition. Biomed Res Int. 2017;2017:2159739.
Robert R, Ghazali DA, Favreau F, Mauco G, Hauet T, Goujon JM. Gender difference and sex hormone production in rodent renal ischemia reperfusion injury and repair. J Inflamm (Lond). 2011 Jun 9;8:14.
Lima-Posada I, Portas-Cortes C, Perez-Villalva R, Fontana F, Rodriguez-Romo R, Prieto R, et al. Gender differences in the acute kidney injury to chronic kidney disease transition. Sci Rep. 2017 Sep 25;7(1):12270.
Hinojosa-Laborde C, Lange DL, Haywood JR. Role of female sex hormones in the development and reversal of dahl hypertension. Hypertension. 2000 Jan;35(1 Pt 2):484–9.
Stehman-Breen CO, Gillen D, Gipson D. Prescription of hormone replacement therapy in postmenopausal women with renal failure. Kidney Int. 1999 Dec;56(6):2243–7.
Kwan G, Neugarten J, Sherman M, Ding Q, Fotadar U, Lei J, et al. Effects of sex hormones on mesangial cell proliferation and collagen synthesis. Kidney Int. 1996 Oct;50(4):1173–9.
Lei J, Silbiger S, Ziyadeh FN, Neugarten J. Serum-stimulated alpha 1 type IV collagen gene transcription is mediated by TGF-beta and inhibited by estradiol. Am J Phys. 1998 Feb;274(2):F252–8.
Silbiger S, Lei J, Neugarten J. Estradiol suppresses type I collagen synthesis in mesangial cells via activation of activator protein-1. Kidney Int. 1999 Apr;55(4):1268–76.
Silbiger S, Lei J, Ziyadeh FN, Neugarten J. Estradiol reverses TGF-beta1-stimulated type IV collagen gene transcription in murine mesangial cells. Am J Phys. 1998 Jun;274(6):F1113–8.
Zdunek M, Silbiger S, Lei J, Neugarten J. Protein kinase CK2 mediates TGF-beta1-stimulated type IV collagen gene transcription and its reversal by estradiol. Kidney Int. [Research Support, Non-U.S. Gov't]. 2001 Dec;60(6):2097-108.
Weinstein JR, Anderson S. The aging kidney: physiological changes. Adv Chronic Kidney Dis. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S. Review]. 2010 Jul;17(4):302-7.
Diwan V, Small D, Kauter K, Gobe GC, Brown L. Gender differences in adenine-induced chronic kidney disease and cardiovascular complications in rats. Am J Physiol Renal Physiol. 2014 Dec 1;307(11):F1169–78.
Kararigas G, Seeland U. Barcena de Arellano ML, Dworatzek E, Regitz-Zagrosek V. why the study of the effects of biological sex is important. Commentary. Ann Ist Super Sanita. 2016 Apr-Jun;52(2):149–50.
NAMS. The 2017 hormone therapy position statement of the North American Menopause Society. Menopause. 2017 Jul;24(7):728–53.
Acknowledgements
Not applicable.
Funding
GK acknowledges support from the DZHK (German Centre for Cardiovascular Research) and the BMBF (German Ministry for Education and Research).
Author information
Authors and Affiliations
Contributions
G.K. conceived the work. A.R.S. and G.K. wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sabbatini, A.R., Kararigas, G. Estrogen-related mechanisms in sex differences of hypertension and target organ damage. Biol Sex Differ 11, 31 (2020). https://doi.org/10.1186/s13293-020-00306-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13293-020-00306-7