
Abstract
Sex differences in cardiovascular disease and cardiac physiology have been reported in humans as well as in animal models. Premenopausal women have reduced cardiovascular disease compared to men, but the incidence of cardiovascular disease in women increases following menopause. Sex differences in cardiomyocytes likely contribute to the differences in male–female physiology and response to disease. Sex differences in the heart have been noted in electrophysiology, contractility, signaling, metabolism, and cardioprotection. These differences appear to be due, at least in part, to differences in gene and protein expression as well as in posttranslational protein modifications. This review will focus primarily on estrogen-mediated male–female differences in protein expression and signaling pathways in the heart and cardiac cells. It should be emphasized that these basic differences are not intrinsically beneficial or detrimental per se; the difference can be good or bad depending on the context and circumstances.
Similar content being viewed by others


Review
Introduction
Male–female differences in cardiovascular disease (CVD) are well documented. There are also sex differences in cardiac myocytes, and these differences likely contribute to the differences in male–female response to physiology and disease. The sex differences in mRNA/protein expression are thought to be due to differences in sex hormones, but differences due to having two X chromosomes versus an X and a Y chromosome [1] and epigenetic differences should also be considered. Although testosterone can clearly alter gene expression and contribute to sex differences, for simplicity, this review will focus on female sex hormones and primarily on the effects of estrogen. Even with limiting the discussion to the effect of estrogen, the mechanism(s) by which estrogen can regulate gene expression, protein levels, and posttranslational modifications (PTMs) is very complex. This review will focus primarily on estrogen-mediated male–female differences in protein expression and signaling pathways in cardiac cells.
Estrogen regulation of mRNA and protein levels
How does estrogen regulate gene expression?
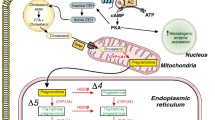
Estrogen can bind to classic estrogen receptors (ER), ERα and ERβ, which act as ligand-gated transcription factors that bind to DNA with the help of co-activators and co-repressors and alter DNA transcription (see [2] for more details). Some genes appear to have an estrogen response element (e.g., LPL [3]). Other genes such as endothelial nitric oxide synthase (eNOS) can be regulated indirectly by a sex-dependent microRNA (miRNA) cascade [4]. Estrogen can also alter cardiac gene expression indirectly. Estrogen-mediated changes in another organ could, via secreted factors or other similar mechanisms, lead to changes in cardiac tissue. The dosage of the X and Y chromosomes can also alter gene expression [1].
Estrogen can also activate rapid signaling pathways that can alter gene expression
Estrogen can bind to estrogen receptors at the plasma membrane and activate signaling pathways. ERα has been shown to be associated with the plasma membrane, and binding of estrogen leads to activation of PI3-kinase signaling. Palmitoylation of the ER is proposed to localize it to the plasma membrane. A splice variant of ERα has also been suggested to be responsible for rapid membrane-delimited estrogen signaling [5]. An orphan G protein-coupled receptor (GPR30 also known as GPER) has also been shown to bind estrogen, which leads to activation of signaling pathways such as PI3-kinase and ERK [6, 7]. Activation of plasma membrane receptors and the resultant activation of rapid signaling pathways can also alter gene expression, either by ligand-independent ER activation of gene expression secondary to phosphorylation of the ER or altering levels of co-activators and co-repressors or by activating ER-independent signals that alter gene expression.
Estrogen regulates mRNA and miRNA expression
Estrogen alteration of cell function can occur by its well-described action in altering transcription and in turn protein levels. Estrogen has been shown to regulate the expression of a large number of genes in the heart and cardiac cells. In general, two approaches are used to evaluate estrogen-regulated genes: a candidate approach and non-biased gene profiling approach. A candidate approach has been used to demonstrate estrogen regulation of expression of a number of genes including PGC-1α [8], connexin 43 (Cx43) [9, 10], adenine nucleotide translocator [11], heat shock proteins [12], and an inhibitor of calcineurin (MC1P1) [13]. Several studies have used a gene profiling approach and a few are mentioned to illustrate the approach. Gaborit examined the profile of 79 ion channel and transporter genes from non-failing male and female human hearts and found that female hearts exhibited decreased expression of several K+ channel subunits, as well as a decrease in Cx43 and phospholamban [14]. Ambrosi et al. also examined sex differences in genes involved in electrophysiology in the left atria and ventricles from failing and non-failing human hearts [15]. They found sex differences in ion channels in the left atria of failing hearts, but no sex differences were observed in the left ventricle. The authors suggest that the difference with the study of Gaborit et al. could be due in part to the age of the human donors. Studies have also examined changes in mRNA in ovariectomized (OVX) females with and without estrogen. Otsuki et al. [16] analyzed gene expression in the heart comparing OVX females with and without estrogen. They reported an increase in expression of seven genes and decreased expression of nine genes. It is difficult to draw conclusions from the list of genes that are regulated by estrogen. The genes regulated are likely to depend on the context of the cell, and thus, these estrogen-regulated genes may vary with disease, age, and sex. Indeed, estrogen has been shown to differentially regulate gene expression in males versus females.
Recent data have shown that estrogen can also regulate miRNA and that sex differences in miRNA can contribute to sex differences in mRNA levels. There are a number of studies showing estrogen regulation of miRNA in non-cardiac cells [17–19]. There are also a number of very recent studies showing a role for miRNA in the heart. A recent study has shown sexual dimorphic expression of a miRNA network in the healthy and hypertrophied heart [20]. They further showed that these effects are mediated by ERβ. miR-151 has been shown to be involved in susceptibility to arrhythmogenesis during myocardial infarction with estrogen deprivation [21]. Zhao et al. [22] showed that estrogen alters expression of miRNAs in the mouse aorta. They further showed that ERα mediates upregulation of miR-203 and that miR-203 is involved in vascular proliferation. miR-222 has been shown to be downregulated in female hearts and to play a role in regulation of nitric oxide synthase. These data support the concept that sex- and estrogen-dependent regulation of gene expression can be mediated, at least in part, by miRNA. This is an exciting area for future studies.
ERα and ERβ regulate expression of different genes and can regulate genes in opposite directions
Several studies have examined gene expression changes related to either ERα or ERβ. ERα and ERβ regulate different genes [23, 24]. For example, using OVX mice lacking either ERα or ERβ which were treated with estrogen for 1 week, O'Lone et al. showed that ERα and ERβ regulate different genes in the mouse aorta [25].
ERα and ERβ can also regulate gene expression in opposite directions [26–28]. It has been proposed that ERα and ERβ can counterbalance gene regulation in a yin/yang manner [26], for example, ERβ can repress genes that are upregulated by ERα and vice versa. Consistent with this concept, ERα increases Glut4 levels in the muscle, whereas ERβ suppresses Glut4 expression [27]. In breast cancer, an increase in ERα is thought to increase tumor cell proliferation while an increase in ERβ is thought to reduce it. ERβ increases while ERα decreases levels of iNOS in vascular smooth muscle cells [28]. Thus, the effect of estrogen on gene expression can vary depending on the relative proportion of ERα and ERβ.
The effect of estrogen can vary depending on the presence of co-activator and co-repressors
The differences in gene regulation between ERα and ERβ are usually attributed to differences in the binding of co-activators and co-repressors. Interestingly, estrogen treatment of male and female cardiac tissue can lead to opposite effects on gene expression, which is also attributed to difference in the availability of co-activators and co-repressors between male and female cardiomyocytes [29, 30]. For example, estrogen increases the progesterone receptor in females and decreases it in males [31]. Similarly, estrogen increases the myosin regulatory light chain in males and decreases it in females [30].
Although there are no clear reports of alteration of estrogen receptor co-activators or repressors in cardiac cells with myocardial infarction or caloric restriction, it is plausible that changes would occur. Furthermore, ER can activate transcription by interaction with transcription factors [2] such as AP1 [32], which have been reported to be altered in hypertrophy [33]. Thus, it is not surprising that there are sex differences in the changes in gene expression following myocardial infarction [34] and caloric restriction [35] in males versus females.
Protein and posttranslational modifications differ in males and females
In addition to altering mRNA levels, estrogen can activate signaling pathways [7, 36] that alter protein PTMs [37, 38], which can lead to additional estrogen-dependent changes in cardiac function. Lagranha et al. [37] examined proteomic differences in mitochondria from male versus female rats and found differences in phosphorylation of several proteins. Females had an increase in phosphorylation and activity of aldehyde dehydrogenase 2, an enzyme which has been shown to be cardioprotective. Females also had higher levels of phosphorylation of pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase than males. This increase in phosphorylation in females would be consistent with an estrogen-mediated increase in the PI3-kinase signaling pathway. Vatner and co-workers [39] also examined proteomic difference in monkey hearts as a function of age and sex. They found a decrease in levels of several metabolic enzymes in the aged male hearts, which they attributed to differences in ROS production in aged males versus females. McKee et al. examined the potential mechanism for the sex difference in progression of hypertrophic cardiomyopathy. They reported sex differences in cTnI phosphorylation [38]. Sex differences in posttranslational modifications of contractile proteins would be consistent with estrogen-mediated signaling pathways. These posttranslational modifications of contractile proteins could contribute to sex differences in excitation-contraction coupling.
Differences in S-nitrosylation (SNO), a protein modification that involves the addition of an NO moiety to protein sulfhydryl groups, have also been described [40, 41]. An increase in NOS has been reported in females, and this increase in NOS would be consistent with an increase in the NO-dependent PTM and SNO. An increase in protein SNO has been reported in hearts from female mice, and this increase in SNO is suggested to play a role in the reduced ischemia-reperfusion (I/R) injury in females, as inhibition of NOS with an inhibitor such as l-NAME blocks both cardioprotection and the increase in SNO in females [40, 41].
Taken together, estrogen, by activation of nuclear receptors or by activation of rapid signaling pathways, can lead to changes in gene expression. These effects can be additive if the activation of nuclear receptors increases the expression of a signaling protein such as NOS or a kinase which can be activated by a rapid signaling pathway that is activated by estrogen. The next section will give a few examples of how these changes in gene expression and signaling can lead to sex differences in cardiac function.
Estrogen regulation of function
Calcium handling and electrophysiology
There are a number of documented sex differences in electrophysiology [42]. Women have a longer QT interval. The basis for this difference in QT interval is still somewhat debated, but estrogen has been shown to alter the levels of several ion channels that modulate QT interval. Sex differences in gene expression of several inwardly rectifying K+ channels have been reported [43–45]. Kv4.3 and Kv1.5 expression are regulated by estrogen [46]. Roden and co-workers have also shown sex differences in the late Na+ channels [47]. Sims et al. report that the long QT in women is due to an ERα-mediated increase in the L-type Ca2+ channel [48]. Iacobas et al. have analyzed the sex-dependent regulation of the gene network that regulates heart rhythm [49]. As mentioned previously, Ambrosi et al. have reported sex differences in genes involved in electrophysiology [15], and Gaborit has analyzed human tissue and found sex differences in several ion channels [14]. Gaborit et al. observed a sex-dependent isoform switch in the Na+/K+ ATPase; they found an increase in the α1 subunit in females and a decrease in the α3 subunit. Interestingly, Imahashi et al. have reported sex differences in Na+ levels during ischemia-reperfusion [50]. They found less of a rise in Na+ during ischemia in female hearts compared to male hearts. This sex difference in the rise in Na+ was increased if hearts were treated with isoproterenol prior to ischemia. Male–female differences in Ca2+ have also been observed. Treatment with isoproterenol causes less of an increase in the Ca2+ transient [51, 52] and less of an increase in SR Ca in females compared to males [52].
Sex differences in contractile proteins have also been reported. Patrizio et al. [53] have shown that estrogen regulates expression of the β-myosin heavy chain. McKee et al. have shown sex differences in myofilament function and in phosphorylation of TnI in mice with hypertrophic cardiomyopathy [38].
Metabolic differences
Sex differences in metabolism have been reported in humans as well as in animal models. Estrogen has been shown to increase insulin-stimulated glucose uptake in skeletal muscle [54], and ERα has been shown to be required for glucose uptake in the heart [55]. There are sex differences in insulin resistance and energy balance [56]. These effects of estrogen on metabolism are consistent with the Women's Health Initiative (WHI) finding that hormone replacement therapy (HRT) resulted in an improvement in insulin sensitivity [57]. Clegg et al. have reported sex differences in energy homeostasis [58]. Lewandowski's group [35] has shown male–female differences in triglyceride level following caloric restriction [35, 59]. Lyons et al. have shown sex differences in response to diabetes [60]. Using PET, Peterson has found that women have increased fatty acid metabolism compared to men. In contrast, Gabel has used 13C NMR and isopotomer analysis and found that compared to those from male mice, hearts from female mice have an increase in the ratio of carbohydrate metabolism to fatty acid oxidation [23]. The reason for this difference is unclear; it could be due to species differences and to differences in the relative levels of ERα versus ERβ in the different species.
Hormone replacement therapy and the Women's Health Initiative
Premenopausal women have reduced cardiovascular disease compared to men, but the incidence of cardiovascular disease in women increases following menopause. These observations led to the concept that hormone replacement therapy might reduce cardiovascular disease. However, in a large clinical trial, the WHI, HRT was not protective. To better understand why the HRT did not show protection, it is important to better understand the mechanism by which estrogen is protective in animal models. Several explanations have been proposed for the lack of protection of HRT in the WHI in contrast to the protection observed in premenopausal females. As reviewed elsewhere [61], the dosage, duration, type of estrogen, and mode of administration have been suggested to contribute to the lack of protection. One popular hypothesis is the timing hypothesis. The average age for initiating HRT in the WHI was 63 years of age; thus, many women in the WHI were 10 years or more past menopause. It has been proposed (see [61]) that the protective effects of estrogen are lost if estrogen is absent for many years. For example, it is suggested that changes might occur in the development of atherosclerotic plaques, etc. and that this might not be readily reversed with re-additon of estrogen. The WHI data have been re-analyzed to evaluate the timing hypothesis, and although some interpret the data as supporting the timing hypothesis [61, 62], others suggest that the re-analyzed data do not support the hypothesis [63–65] and others conclude that the data provided mixed support [66] and that further study is needed. Some additional trials have begun to more directly investigate the role of timing. Another explanation for the lack of HRT protection in the WHI is that age-related changes occur (with or without the continued presence of estrogen) and that these changes impair the protection by estrogen. For example, estrogen has been shown to upregulate nitric oxide synthase, and at least some of the protection afforded by estrogen is due to nitric oxide signaling. With age and oxidative stress, nitric oxide synthase can become ‘uncoupled’ and produce superoxide instead of nitric oxide. If nitric oxide synthase becomes uncoupled with age, this might contribute to the lack of beneficial effects of HRT in older women. In a variation on the timing hypothesis, it is interesting to speculate that these age-related changes might be slowed if estrogen is continuously present.
Differences in ischemia-reperfusion injury
A number of animal studies have found a sex difference in cell death following ischemia and reperfusion. Females generally show less I/R injury compared to males [37, 67], although some studies do not observe reduced injury in females [68]. This discrepancy appears to be due to the extent of injury; a sex difference is usually apparent with more severe I/R injury. For example, with 20 min of ischemia, Cross et al. observed no sex difference in I/R injury, but a sex difference was revealed if contractility was increased prior to ischemia [69]. Estrogen appears to play a major role in this protection as OVX females show increased I/R injury and protection is restored with administration of estrogen [37]. Studies have been done, using either ERα or ERβ knockout mice, to investigate the relative role of ERα versus ERβ in reducing I/R injury. The results have been mixed with some studies suggesting a role for ERα [70–72] and others suggesting a role for ERβ [23, 24, 41, 73]. This discrepancy is likely due to complex and non-overlapping regulation of gene expression by ERα and ERβ. For example, in vascular cells, ERα has been shown to be important for protection from injury [74] whereas ERβ regulates vasodilation [75]. Differences in the models of I/R could result in differences in the relative levels of ERα versus ERβ or differences in co-activators and co-repressors, which, as discussed previously, can have profound effects on the response.
Plasma membrane-localized estrogen receptors are also important in cardioprotection. Treatment of a Langendorff perfused heart with a GPR30 agonist, G-1, has suggested a role for GPR30 in reducing I/R injury [7, 76]. An estrogen dendrimer conjugate (EDC), which is excluded from the nucleus, has been shown to reduce vascular injury, supporting a role for non-nuclear estrogen signaling [77]. The precise role of these different receptors and whether there is some redundancy will require further study.
Differences in hypertrophy and heart failure
Sex differences in the response to hypertrophy have been reported. In response to transaortic constriction (TAC), hypertrophy and heart failure develop more slowly in females compared to males [78] and more slowly in OVX females treated with estrogen compared to those treated with vehicle [79]. Studies with mice lacking either ERα or ERβ have generally suggested that ERβ is an important mediator of the protection as mice lacking ERβ develop hypertrophy similar to that observed in mice lacking estrogen [78]. There are also data showing that mice lacking GPR30 exhibit increased blood pressure [80]. Chronic treatment with the GPR30 activator G-1 has also been shown to attenuate heart failure [81]. Although ERβ appears to plan a key role in the sex differences in hypertrophy, the mechanism is likely to be more complicated and is an area that deserves future investigation.
Conclusions
There are clear sex differences in cardiac function, and these differences appear to be mediated at least in part by the action of estrogen on several unique estrogen receptors. The effects of estrogen differ depending on the relative levels of ERα and ERβ as well as on the complement of co-repressors and co-activators. The relative level of GRP30 is also likely to influence the response of the cell to estrogen. The relative levels of the estrogen receptors and the co-regulators can change with disease, age, and sex, thereby altering the response of the cell to estrogen. In addition to the role of estrogen, other sex hormones such as progesterone and testosterone and relative levels of the sex chromosomes are major contributors to sex differences. Future studies will be needed to better document how miRNA are regulated differently between males and females and to better elucidate the interactions between sex hormones and sex chromosomes. The interaction between signaling by ERα, ERβ, and GPR30 is also an important topic for future studies. A mechanistic understanding of the reasons for the discrepancy between reduced cardiovascular disease in premenopausal women and the lack of protection observed in the WHI is an important topic for future studies.
References
Ji H, Zheng W, Wu X, Liu J, Ecelbarger CM, Watkins R, Arnold AP, Sandberg K: Sex chromosome effects unmasked in angiotensin II-induced hypertension. Hypertension 2010, 55(5):1275–1282. 10.1161/HYPERTENSIONAHA.109.144949
Murphy E: Estrogen signaling and cardiovascular disease. Circ Res 2011, 109(6):687–696. 10.1161/CIRCRESAHA.110.236687
Liu D, Deschamps A, Korach KS, Murphy E: Estrogen-enhanced gene expression of lipoprotein lipase in heart is antagonized by progesterone. Endocrinology 2008, 149(2):711–716. 10.1210/en.2007-0620
Evangelista AM, Deschamps AM, Liu D, Raghavachari N, Murphy E: miR-222 contributes to sex-dimorphic cardiac eNOS expression via ets-1. Physiol Genomics 2013, 45(12):493–498. 10.1152/physiolgenomics.00008.2013
Kim KH, Toomre D, Bender JR: Splice isoform estrogen receptors as integral transmembrane proteins. Mol Biol Cell 2011, 22(22):4415–4423. 10.1091/mbc.E11-05-0416
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER: A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307(5715):1625–1630. 10.1126/science.1106943
Deschamps AM, Murphy E: Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol 2009, 297(5):H1806-H1813. 10.1152/ajpheart.00283.2009
Hsieh YC, Yang S, Choudhry MA, Yu HP, Rue LW III, Bland KI, Chaudry IH: PGC-1 upregulation via estrogen receptors: a common mechanism of salutary effects of estrogen and flutamide on heart function after trauma-hemorrhage. Am J Physiol Heart Circ Physiol 2005, 289(6):H2665-H2672. 10.1152/ajpheart.00682.2005
Yu W, Dahl G, Werner R: The connexin43 gene is responsive to oestrogen. Proc Biol Sci 1994, 255(1343):125–132. 10.1098/rspb.1994.0018
Chung TH, Wang SM, Wu JC: 17beta-estradiol reduces the effect of metabolic inhibition on gap junction intercellular communication in rat cardiomyocytes via the estrogen receptor. J Mol Cell Cardiol 2004, 37(5):1013–1022. 10.1016/j.yjmcc.2004.08.003
Too CK, Giles A, Wilkinson M: Estrogen stimulates expression of adenine nucleotide translocator ANT1 messenger RNA in female rat hearts. Mol Cell Endocrinol 1999, 150(1–2):161–167.
Voss MR, Stallone JN, Li M, Cornelussen RN, Knuefermann P, Knowlton AA: Gender differences in the expression of heat shock proteins: the effect of estrogen. Am J Physiol Heart Circ Physiol 2003, 285(2):H687-H692.
Pedram A, Razandi M, Aitkenhead M, Levin ER: Estrogen inhibits cardiomyocyte hypertrophy in vitro: antagonism of calcineurin-related hypertrophy through induction of MCIP1. J Biol Chem 2005, 280(28):26339–26348. 10.1074/jbc.M414409200
Gaborit N, Andras V, Le Bouter S, Szuts V, Escande D, Nattel S, Demolombe S: Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J Mol Cell Cardiol 2010, 49(4):639–646. 10.1016/j.yjmcc.2010.06.005
Ambrosi CM, Yamada KA, Nerbonne JM, Efimov IR: Gender differences in electrophysiological gene expression in failing and non-failing human hearts. PLoS One 2013, 8(1):e54635. 10.1371/journal.pone.0054635
Otsuki M, Gao H, Dahlman-Wright K, Ohlsson C, Eguchi N, Urade Y, Gustafsson JA: Specific regulation of lipocalin-type prostaglandin D synthase in mouse heart by estrogen receptor beta. Mol Endocrinol 2003, 17(9):1844–1855. 10.1210/me.2003-0016
Pinho FG, Frampton AE, Nunes J, Krell J, Alshaker H, Jacob J, Pellegrino L, Roca-Alonso L, de Giorgio A, Harding V, Waxman J, Stebbing J, Pchejetski D, Castellano L: Downregulation of microRNA-515–5p by the estrogen receptor modulates sphingosine kinase 1 and breast cancer cell proliferation. Cancer Res 2013, 73(19):5936–5948. 10.1158/0008-5472.CAN-13-0158
Di Leva G, Piovan C, Gasparini P, Ngankeu A, Taccioli C, Briskin D, Cheung DG, Bolon B, Anderlucci L, Alder H, Nuovo G, Li M, Iorio MV, Galasso M, Santhanam R, Marcucci G, Perrotti D, Powell KA, Bratasz A, Garofalo M, Nephew KP, Croce CM: Estrogen mediated-activation of miR-191/425 cluster modulates tumorigenicity of breast cancer cells depending on estrogen receptor status. PLoS Genet 2013, 9(3):e1003311. 10.1371/journal.pgen.1003311
Kim K, Madak-Erdogan Z, Ventrella R, Katzenellenbogen BS: A MicroRNA196a2* and TP63 circuit regulated by estrogen receptor-alpha and ERK2 that controls breast cancer proliferation and invasiveness properties. Horm Cancer 2013, 4(2):78–91. 10.1007/s12672-012-0129-3
Queiros AM, Eschen C, Fliegner D, Kararigas G, Dworatzek E, Westphal C, Sanchez Ruderisch H, Regitz-Zagrosek V: Sex-and estrogen-dependent regulation of a miRNA network in the healthy and hypertrophied heart. Int J Cardiol 2013, 169(5):331–338. 10.1016/j.ijcard.2013.09.002
Zhang Y, Wang R, Du W, Wang S, Yang L, Pan Z, Li X, Xiong X, He H, Shi Y, Liu X, Yu S, Bi Z, Lu Y, Shan H: Downregulation of miR-151–5p contributes to increased susceptibility to arrhythmogenesis during myocardial infarction with estrogen deprivation. PLoS One 2013, 8(9):e72985. 10.1371/journal.pone.0072985
Zhao J, Imbrie GA, Baur WE, Iyer LK, Aronovitz MJ, Kershaw TB, Haselmann GM, Lu Q, Karas RH: Estrogen receptor-mediated regulation of microRNA inhibits proliferation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2012, 33(2):257–265.
Gabel SA, Chen J, Petranka JG, Yamamura K, Walker VR, London RE, Korach KS, Steenbergen C, Murphy : Estrogen receptor beta mediates gender differences in ischemia reperfusion injury. J Mol Cell Cardiol 2003, 35(6):A16-A.
Nikolic I, Liu D, Bell JA, Collins J, Steenbergen C, Murphy E: Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J Mol Cell Cardiol 2007, 42(4):769–780. 10.1016/j.yjmcc.2007.01.014
O'Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, Hansen U: Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol Endocrinol 2007, 21(6):1281–1296. 10.1210/me.2006-0497
Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C: Estrogen receptor (ER)-beta reduces ER alpha-regulated gene transcription, supporting a “ying yang” relationship between ER alpha and ER beta in mice. Mol Endocrinol 2003, 17(2):203–208. 10.1210/me.2002-0206
Barros RP, Machado UF, Warner M, Gustafsson JA: Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci USA 2006, 103(5):1605–1608. 10.1073/pnas.0510391103
Tsutsumi S, Zhang X, Takata K, Takahashi K, Karas RH, Kurachi H, Mendelsohn ME: Differential regulation of the inducible nitric oxide synthase gene by estrogen receptors 1 and 2. J Endocrinol 2008, 199(2):267–273. 10.1677/JOE-07-0292
Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, Stypmann J, Roepcke S, Brokat S, Mahmoodzadeh S, Brozova E, Davidson MM, Ruiz Noppinger P, Grohe C, Regitz-Zagrosek V: Sex-specific pathways in early cardiac response to pressure overload in mice. J Mol Med (Berl) 2008, 86(9):1013–1024. 10.1007/s00109-008-0385-4
Kararigas G, Bito V, Tinel H, Becher E, Baczko I, Knosalla C, Albrecht-Kupper B, Sipido KR, Regitz-Zagrosek V: Transcriptome characterization of estrogen-treated human myocardium identifies myosin regulatory light chain interacting protein as a sex-specific element influencing contractile function. J Am Coll Cardiol 2012, 59(4):410–417. 10.1016/j.jacc.2011.09.054
Kararigas G, Becher E, Mahmoodzadeh S, Knosalla C, Hetzer R, Regitz-Zagrosek V: Sex-specific modification of progesterone receptor expression by 17beta-oestradiol in human cardiac tissues. Biol Sex Differ 2010, 1(1):2. 10.1186/2042-6410-1-2
Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL: Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J Biol Chem 2001, 276(17):13615–13621.
von Harsdorf R, Edwards JG, Shen YT, Kudej RK, Dietz R, Leinwand LA, Nadal-Ginard B, Vatner SF: Identification of a cis-acting regulatory element conferring inducibility of the atrial natriuretic factor gene in acute pressure overload. J Clin Invest 1997, 100(5):1294–1304. 10.1172/JCI119643
Chen Q, Williams R, Healy CL, Wright CD, Wu SC, O'Connell TD: An association between gene expression and better survival in female mice following myocardial infarction. J Mol Cell Cardiol 2010, 49(5):801–811. 10.1016/j.yjmcc.2010.08.002
Banke NH, Yan L, Pound KM, Dhar S, Reinhardt H, De Lorenzo MS, Vatner SF, Lewandowski ED: Sexual dimorphism in cardiac triacylglyceride dynamics in mice on long term caloric restriction. J Mol Cell Cardiol 2012, 52(3):733–740. 10.1016/j.yjmcc.2011.11.014
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK: Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407(6803):538–541. 10.1038/35035131
Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E: Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res 2010, 106(11):1681–1691. 10.1161/CIRCRESAHA.109.213645
McKee LA, Chen H, Regan JA, Behunin SM, Walker JW, Walker JS, Konhilas JP: Sexually dimorphic myofilament function and cardiac troponin I phosphospecies distribution in hypertrophic cardiomyopathy mice. Arch Biochem Biophys 2013, 535(1):39–48. 10.1016/j.abb.2012.12.023
Yan L, Ge H, Li H, Lieber SC, Natividad F, Resuello RR, Kim SJ, Akeju S, Sun A, Loo K, Peppas AP, Rossi F, Lewandowski ED, Thomas AP, Vatner SF, Vatner DE: Gender-specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol 2004, 37(5):921–929. 10.1016/j.yjmcc.2004.06.012
Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E: Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res 2006, 98(3):403–411. 10.1161/01.RES.0000202707.79018.0a
Lin J, Steenbergen C, Murphy E, Sun J: Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation 2009, 120(3):245–254. 10.1161/CIRCULATIONAHA.109.868729
Parks RJ, Howlett SE: Sex differences in mechanisms of cardiac excitation-contraction coupling. Pflugers Arch 2013, 465(5):747–763. 10.1007/s00424-013-1233-0
Yang PC, Clancy CE: Effects of sex hormones on cardiac repolarization. J Cardiovasc Pharmacol 2010, 56(2):123–129. 10.1097/FJC.0b013e3181d6f7dd
Yang PC, Clancy CE: Gender-based differences in cardiac diseases. J Biomed Res 2011, 25(2):81–89. 10.1016/S1674-8301(11)60010-9
Drici MD, Burklow TR, Haridasse V, Glazer RI, Woosley RL: Sex hormones prolong the QT interval and downregulate potassium channel expression in the rabbit heart. Circulation 1996, 94(6):1471–1474. 10.1161/01.CIR.94.6.1471
Saito T, Ciobotaru A, Bopassa JC, Toro L, Stefani E, Eghbali M: Estrogen contributes to gender differences in mouse ventricular repolarization. Circ Res 2009, 105(4):343–352. 10.1161/CIRCRESAHA.108.190041
Lowe JS, Stroud DM, Yang T, Hall L, Atack TC, Roden DM: Increased late sodium current contributes to long QT-related arrhythmia susceptibility in female mice. Cardiovasc Res 2012, 95(3):300–307. 10.1093/cvr/cvs160
Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G: Sex, age, and regional differences in L-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long QT type 2. Circ Res 2008, 102(9):e86-e100. 10.1161/CIRCRESAHA.108.173740
Iacobas DA, Iacobas S, Thomas N, Spray DC: Sex-dependent gene regulatory networks of the heart rhythm. Funct Integr Genomics 2010, 10(1):73–86. 10.1007/s10142-009-0137-8
Imahashi K, London RE, Steenbergen C, Murphy E: Male/female differences in intracellular Na + regulation during ischemia/reperfusion in mouse heart. J Mol Cell Cardiol 2004, 37(3):747–753. 10.1016/j.yjmcc.2004.06.010
Curl CL, Wendt IR, Kotsanas G: Effects of gender on intracellular. Pflugers Arch 2001, 441(5):709–716. 10.1007/s004240000473
Chen J, Petranka J, Yamamura K, London RE, Steenbergen C, Murphy E: Gender differences in sarcoplasmic reticulum calcium loading after isoproterenol. Am J Physiol Heart Circ Physiol 2003, 285(6):H2657-H2662.
Patrizio M, Musumeci M, Piccone A, Raggi C, Mattei E, Marano G: Hormonal regulation of beta-myosin heavy chain expression in the mouse left ventricle. J Endocrinol 2013, 216(3):287–296. 10.1530/JOE-12-0201
Gorres BK, Bomhoff GL, Morris JK, Geiger PC: In vivo stimulation of oestrogen receptor alpha increases insulin-stimulated skeletal muscle glucose uptake. J Physiol 2011, 589(Pt 8):2041–2054.
Arias-Loza PA, Kreissl MC, Kneitz S, Kaiser FR, Israel I, Hu K, Frantz S, Bayer B, Fritzemeier KH, Korach KS, Pelzer T: The estrogen receptor-alpha is required and sufficient to maintain physiological glucose uptake in the mouse heart. Hypertension 2012, 60(4):1070–1077. 10.1161/HYPERTENSIONAHA.111.190389
Geer EB, Shen W, Strohmayer E, Post KD, Freda PU: Body composition and cardiovascular risk markers after remission of Cushing's disease: a prospective study using whole-body MRI. J Clin Endocrinol Metab 2012, 97(5):1702–1711. 10.1210/jc.2011-3123
Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, Allen C, Bassford T, Burke G, Torrens J, Howard BV, Investigat WsHI: Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women's Health Initiative Hormone Trial. Diabetologia 2004, 47(7):1175–1187.
Shi HF, Seeley RJ, Clegg DJ: Sexual differences in the control of energy homeostasis. Front Neuroendocrinol 2009, 30(3):396–404. 10.1016/j.yfrne.2009.03.004
Banke NH, Pound KM, DeLorenzo MS, Yan L, Reinhardt HA, Vatner DE, Vatner SF, Lewandowski ED: Gender distinguishes myocardial triacylglyceride dynamics in response to long term caloric restriction in mice. Circulation 2009, 120(18):S893-S.
Lyons MR, Peterson LR, McGill JB, Herrero P, Coggan AR, Saeed IM, Recklein C, Schechtman KB, Gropler RJ: Impact of sex on the heart's metabolic and functional responses to diabetic therapies. Am J Physiol Heart Circ Physiol 2013, 305(11):H1584-H1591. 10.1152/ajpheart.00420.2013
Yang XP, Reckelhoff JF: Estrogen, hormonal replacement therapy and cardiovascular disease. Curr Opin Nephrol Hypertens 2011, 20(2):133–138. 10.1097/MNH.0b013e3283431921
Harman SM, Vittinghoff E, Brinton EA, Budoff MJ, Cedars MI, Lobo RA, Merriam GR, Miller VM, Naftolin F, Pal L, Santoro N, Taylor HS, Black DM: Timing and duration of menopausal hormone treatment may affect cardiovascular outcomes. Am J Med 2011, 124(3):199–205. 10.1016/j.amjmed.2010.09.021
Manson JE, Chlebowski RT, Stefanick ML, Aragaki AK, Rossouw JE, Prentice RL, Anderson G, Howard BV, Thomson CA, LaCroix AZ, Wactawski-Wende J, Jackson RD, Limacher M, Margolis KL, Wassertheil-Smoller S, Beresford SA, Cauley JA, Eaton CB, Gass M, Hsia J, Johnson KC, Kooperberg C, Kuller LH, Lewis CE, Liu S, Martin LW, Ockene JK, O'Sullivan MJ, Powell LH, Simon MS, et al.: Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA 2013, 310(13):1353–1368. 10.1001/jama.2013.278040
Prentice RL, Manson JE, Langer RD, Anderson GL, Pettinger M, Jackson RD, Johnson KC, Kuller LH, Lane DS, Wactawski-Wende J, Brzyski R, Allison M, Ockene J, Sarto G, Rossouw JE: Benefits and risks of postmenopausal hormone therapy when it is initiated soon after menopause. Am J Epidemiol 2009, 170(1):12–23. 10.1093/aje/kwp115
Barrett-Connor E: Hormones and heart disease in women: the timing hypothesis. Am J Epidemiol 2007, 166(5):506–510. 10.1093/aje/kwm214
Toh S, Hernandez-Diaz S, Logan R, Rossouw JE, Hernan MA: Coronary heart disease in postmenopausal recipients of estrogen plus progestin therapy: does the increased risk ever disappear? A randomized trial. Ann Intern Med 2010, 152(4):211–217. 10.7326/0003-4819-152-4-201002160-00005
Bae S, Zhang L: Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. J Pharmacol Exp Ther 2005, 315(3):1125–1135. 10.1124/jpet.105.090803
Przyklenk K, Ovize M, Bauer B, Kloner RA: Gender does not influence acute myocardial infarction in adult dogs. Am Heart J 1995, 129(6):1108–1113. 10.1016/0002-8703(95)90390-9
Cross HR, Murphy E, Steenbergen C: Ca(2+) loading and adrenergic stimulation reveal male/female differences in susceptibility to ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2002, 283(2):H481-H489.
Wang M, Crisostomo P, Wairiuko GM, Meldrum DR: Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol 2006, 290(6):H2204-H2209. 10.1152/ajpheart.01219.2005
Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR: Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol 2000, 278(5):H1640-H1647.
Booth EA, Marchesi M, Kilbourne EJ, Lucchesi BR: 17Beta-estradiol as a receptor-mediated cardioprotective agent. J Pharmacol Exp Ther 2003, 307(1):395–401. 10.1124/jpet.103.054205
Hsieh YC, Choudhry MA, Yu HP, Shimizu T, Yang S, Suzuki T, Chen J, Bland KI, Chaudry IH: Inhibition of cardiac PGC-1alpha expression abolishes ERbeta agonist-mediated cardioprotection following trauma-hemorrhage. FASEB J 2006, 20(8):1109–1117. 10.1096/fj.05-5549com
Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME: Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res 2002, 90(10):1087–1092. 10.1161/01.RES.0000021114.92282.FA
Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA, Mendelsohn ME: Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 2002, 295(5554):505–508. 10.1126/science.1065250
Bopassa JC, Eghbali M, Toro L, Stefani E: A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2010, 298(1):H16-H23. 10.1152/ajpheart.00588.2009
Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW: Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest 2010, 120(7):2319–2330. 10.1172/JCI38291
Skavdahl M, Steenbergen C, Clark J, Myers P, Demianenko T, Mao L, Rockman HA, Korach KS, Murphy E: Estrogen receptor-beta mediates male–female differences in the development of pressure overload hypertrophy. Am J Physiol Heart Circ Physiol 2005, 288(2):H469-H476.
van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA: 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation 2001, 104(12):1419–1423. 10.1161/hc3601.095577
Martensson UE, Salehi SA, Windahl S, Gomez MF, Sward K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grande PO, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson BO, Ohlsson C, Olde B, Leeb-Lundberg LM: Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 2009, 150(2):687–698. 10.1210/en.2008-0623
Kang S, Liu Y, Sun D, Zhou C, Liu A, Xu C, Hao Y, Li D, Yan C, Sun H: Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS One 2012, 7(10):e48185. 10.1371/journal.pone.0048185
Acknowledgements
EM was supported by ZIA HL002065 and ZIA HL006059. CS was supported by 5R01 HL039752.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EM wrote the review, and CS read, edited, and made additions to the review. Both authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Murphy, E., Steenbergen, C. Estrogen regulation of protein expression and signaling pathways in the heart. Biol Sex Differ 5, 6 (2014). https://doi.org/10.1186/2042-6410-5-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2042-6410-5-6