
Abstract
Background
Mesenchymal stromal cells (MSCs) and their extracellular vesicles (MSC-EVs) have demonstrated to elicit immunomodulatory and pro-regenerative properties that are beneficial for the treatment of chronic wounds. Thanks to different mediators, MSC-EVs have shown to play an important role in the proliferation, migration and cell survival of different skin cell populations. However, there is still a big bid to achieve the most effective, suitable and available source of MSC-EVs.
Methods
We isolated, characterized and compared medium-large EVs (m-lEVs) and small EVs (sEVs) obtained from hair follicle-derived MSCs (HF-MSCs) against the gold standard in regenerative medicine, EVs isolated from adipose tissue-derived MSCs (AT-MSCs).
Results
We demonstrated that HF-EVs, as well as AT-EVs, expressed typical MSC-EVs markers (CD9, CD44, CD63, CD81 and CD105) among other different functional markers. We showed that both cell types were able to increase human dermal fibroblasts (HDFs) proliferation and migration. Moreover, both MSC-EVs were able to increase angiogenesis in human umbilical vein endothelial cells (HUVECs) and protect HDFs exposed to a hyperglycemic environment from oxidative stress and cytotoxicity.
Conclusions
Taken together, HF-EVs demonstrated to exhibit comparable potential to that of AT-EVs as promising candidates in the treatment of chronic wounds.
Similar content being viewed by others


Background
Hair follicles (HFs) are skin appendages originated by the interactions of ectodermal-mesodermal cells in the early embryogenesis. HFs present a complex structure with a rich mixture of cell populations. It has been described that HFs contain different pools of stem cells—melanocyte, epithelial and mesenchymal stromal cells (MSCs)—that regulate hair growth, maintaining skin homeostasis and self-renewing processes continuously [1,2,3]. In the past decade, there has been an increasing interest in these hair follicle derived MSCs (HF-MSCs) due to their numerous advantages over other sources of MSCs—abundant availability of HFs that do not undergo functional and molecular changes in the human body and an easy collection not affected by gender or age—[2, 4, 5]. Furthermore, their role in skin homeostasis, hair growth and their pro-regenerative capacity are making these cells become one of the most suitable candidates for cell-based wound healing therapies [6,7,8,9,10]. Indeed, it has been observed that the direct administration of HF-MSCs can accelerate wound healing both in vitro and in vivo in diabetic mice models [11].
However, despite its demonstrated efficacy and safety, the use of cell therapy for chronic wound management have found important constraints through the harsh regulatory pathways and elevated production costs that impede its widespread use [7, 8, 12]. As a result, numerous researchers have studied the paracrine signaling of MSCs, which has been described as the main responsible for the therapeutic potential of these cells [13,14,15]. Among the bioactive cues that drive regenerative effects, extracellular vesicles (EVs) have gained special attention. In the form of membrane-surrounded units, EVs shuttle the messaging cargos of RNAs, DNAs and proteins to mediate intercellular communications [16]. Similarly to MSC-based therapies, MSCs-derived EVs (MSC-EVs) have demonstrated to elicit a wide variety of therapeutic effects in tissue repair and wound regeneration—immunomodulatory, proliferative, anti-apoptotic, pro-angiogenic effects etc.—[17,18,19]. Furthermore, EVs are more easily handled, characterized and stored than MSCs. In this regard, EVs-based therapies can be better standardized than cell-based therapies. Moreover, these therapies can be commercialized under a softer regulation pathway than cell-based therapies, being considered “biological medicines” [20].
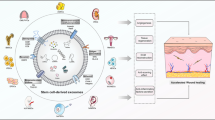
In this study, we report a complete characterization and functional comparison of EVs isolated from HF-MSCs (HF-EVs) against the gold standard in regenerative medicine, EVs derived from adipose tissue MSCs (AT-EVs) (Fig. 1). We have analyzed the MSC-EVs production profile, size, morphology and marker expression. Furthermore, we have compared the cell uptake and tested their potential in human dermal fibroblasts (HDFs)—one of the most important cell types involved in the wound healing process—and in human umbilical vein endothelial cells (HUVECs) to study the vascular behaviour of wounds. Following the MISEV 2018 guidelines [21], we have analyzed not only of the small EVs (sEVs) released by these cells, but also the medium-large EVs (m-lEVs) that are sometimes discarded, despite their demonstrated potential for regenerative medicine applications.

Scheme of the isolation method, characterization and functional assays of HF-EVs and AT-EVs
Materials and methods
EVs isolation and characterization
Cell culture conditions
HF-MSCs were isolated and characterized from HFs as previously described [22] and further cultured in Dulbecco’s modified Eagle’s medium (DMEM 49166-029, Gibco) supplemented with 10% of fetal bovine serum (FBS, Gibco) and 1% (v/v) penicillin/streptomycin (P/S, Gibco). AT-MSCs (ATCC® PCS-500–011™) were also cultured under the same conditions. Adult Human Dermal Fibroblasts (HDFs, ATCC® PCS-201-012™) were cultured in fibroblasts basal medium (ATCC PCS-201-030™) adding the fibroblasts growth kit-low serum (ATCC® PCS-201-041™) and 1% (v/v) penicillin/streptomycin (P/S, Gibco). Incubating conditions were 37ºC in a 5% CO2 atmosphere. Human umbilical vein endothelial cells (HUVECs, Lonza® C2517A) were cultured in the EGM™-2 Endothelial Cell Growth Medium-2 BulletKit™ (Lonza® CC-3162) with 1% (v/v) penicillin/streptomycin (P/S, Gibco). For EVs production, DMEM medium was used with 10% of EV-depleted FBS—by overnight ultracentrifugation 100,000×g (Beckman Coulter, Optima L-100XP)—. HF-MSCs and AT-MSCs were used at passages from 5 to 9 for EVs isolation. HDFs were used at passages 3–7. HUVECs were used at passages 3–7.
EVs isolation
All relevant data regarding the production, isolation and experimental section have been submitted to the EV-TRACK knowledgebase (EV-TRACK ID: EV210337) [23]. EVs were isolated and purified from the supernatant of HF-MSCs and AT-MSCs. At 70–80% of confluency, cells were washed thrice with PBS and the culture medium was replaced with EVs-depleted DMEM. After 72 h of production, the culture medium was collected and new medium was added. After three collections, cells were trypsinized, counted and further re-cultured. The collected medium was first centrifuged at 2000×g for 10 min at 4ºC to discard cell debris and then, freeze at -80ºC until the isolation and purification was given. All further centrifugation processes were performed at 4ºC. In brief, thawed culture supernatants were differentially centrifuged at 10,000×g for 30 min to obtain m-lEVs—pellet 10 K (P10K)—and the resulting supernatants at 100,000×g for 90 min—pellet 100 K (P100K)—to obtain sEVs. To reduce contaminating proteins, all pellets (P10K and P100K) were re-suspended in ice-cold PBS and ultracentrifuged again. Finally, both pellets were immediately re-suspended in 120 µL of ice-cold PBS and freeze at − 80ºC until use.
Nanoparticle tracking analysis
Particle concentration and size distribution within EV preparations was analyzed using the nanoparticle-tracking analysis (NTA), by measuring the rate of Brownian motion in a NanoSight LM10 system (Malvern Panalytical, Malvern, UK). The system was equipped with a fast video-capture and particle-tracking software. NTA post-acquisition settings were the same for all measurements. Each video was analyzed to give the mean, mode, and median vesicle size, as well as an estimate of the concentration. For measurement, original EVs suspension were diluted 1:100 with PBS and a volume of 500 µL were loaded on the camera, and 3 consecutive video recording of 40 s each were taken for every sample quantified.
Western blotting
PBS-resuspended EVs or cell lysates were mixed with 4 × NuPAGE LDS Sample Buffer (Thermo Fisher Scientific, Waltham, MA, USA). The samples were incubated for 5 min at 37 °C, 65 °C, and 95 °C, and separated on 4–12% precast gels (from Thermo Fisher Scientific, Waltham, MA, USA), at a concentration of 5 µg/lane (Bradford determination). The proteins were transferred into PVDF membranes with the iBLOT2 system (Thermo Fisher Scientific, Waltham, MA, USA). Antibodies employed were: mouse monoclonal antibody against CD9 #209,302 (R&D), Grp78 #40 (BD, East Rutherford, NJ, USA), CD13 #3D8 (Santa Cruz Biotechnology, Dallas, Tx, USA), EEA1 #14 (BD, East Rutherford, NJ, USA), CD63 #H5C6 (DSHB, Iowa City, IA, USA), LAMP1 #H4A3 (DSHB, Iowa City, IA, USA), alpha tubulin 1 #DM1A (Santa Cruz Biotechnology, Dallas, Tx, USA), CD81 #JS81 (BD, East Rutherford, NJ, USA), rabbit antibody against COX IV #4850 (Cell Signaling Technologies, Danvers, Ma, USA). All the primary antibodies were diluted 1:1000. Horseradish peroxidase (HRP)-conjugated secondary antibodies anti-mouse, rabbit and goat, were purchased from Jackson ImmunoResearch Lab, (West grove, PA, USA) to ensure minimal cross-reactivity across species.
Bead-based multiplex flow cytometry assay
All isolated EVs were subjected to a surface-marker characterization by using a flow cytometry bead-based multiplex analysis (MACSPlex Exosome Kit, human, Miltenyi Biotec. 130-108-813). Samples were processed according to manufacturer’s protocol. Briefly, 2 µg of EVs were mixed with 120 µL of manufacturer’s buffer and then, 15 µL of the MACSPlex Exosome Capture Beads were added. Then, 15 µL of the detection antibody cocktail—5 µL of each MACSPlex exosome detection reagent CD9, CD63, CD81—were added. After that, samples were incubated 1 h at room temperature, protected from the light, on rotation—450 rpm. Next, 500 µL of MACSPlex buffer were added and samples were centrifuged at 3000×g for 5 min. Subsequently, 500 µL of supernatant were discarded and another 500 µL of buffer were added. Samples were incubated for 15 min at room temperature on rotation, protected from the light and then centrifuged 5 min at 3000×g. Finally, 500 µL of supernatants were discarded and approximately 150 µL of samples were re-suspended and used for the analysis. The flow cytometry analysis was performed using the MACSQuant® Analyzer 10 (Miltenyi Biotec) and results were processed with the MACSQuant Analyzer 10 software (Miltenyi Biotec). The 39 single bead populations were gated to determine the APC signal intensity on each bead population and the median fluorescence intensity (MFI) for each capture bead was measured. For each population, background was corrected by subtracting the respective MFI values from non-EVs controls that were treated exactly like the EVs-samples. Furthermore, values of the corresponding isotype control were also subtracted. Only positive markers are shown in the graphics.
Cryo-electron microscopy (cryo-EM)
EVs (3–5 µL) were spotted on glow-discharged lacey grids and cryo-fixed by plunge freezing at -180 °C in liquid ethane with a Vibrobot (FEI, The Netherlands). Grids were observed with a JEM-2200FS/CR TEM (JEOL, Japan), operating at 200 kV. Image measurements were performed with Image J software and between 80 and 120 single EVs were measured on each group for size and protein-decoration analysis.
CD63 immunostaining
Cells were grown onto 12 mm glass coverslips and fixed with 4% formaldehyde, and permeabilized with 0.1% saponin and 0.1% BSA in PBS. Incubation with primary and secondary antibodies were performed in a humid camera for 1 h each. Following two-10 min washes in PBS, cells were mounting with Fluoromount-G™ Mounting Medium with 2-(4-amidinophenyl)-1 h-indole-6-carboxamidine (DAPI) (Thermo Fisher Scientific, Waltham, MA, USA) for nuclei staining. Imaging of the slides were taken in a Leica SP8 confocal microscope.
EVs uptake and proliferation by HDFs
EVs staining
In order to stain EVs, Vybrant™ DiO Cell-Labeling Solution (Invitrogen, Cat. V22886, Thermo Fisher Scientific, Waltham, MA, USA) was used. Briefly, cultured HF-MSCs and AT-MSCs were washed with PBS and 50 µL of dye in 10 mL of culture medium were added. Cells were incubated for 30 min and after that, washed again thrice with PBS. Finally, EVs-depleted medium was added and a 72 h labeled-EVs production was obtained as previously described. As negative control, the same exact process was performed in flask without cells, to determine the possible carry-over of fluorescence by the culture medium.
EVs uptake by HDFs and flow cytometry
To perform capture experiments, 500,000 HDFs per well were seeded in 6 well plates and 12 mm slides, and incubated overnight with MSC-EVs stained as described above diluted in 2 mL of medium. Slides were fixed and directly observed in a Leica SP8 confocal microscopy, while HDFs in wells were washed twice with PBS and detached from the plate using TrypLE Select (1X) (Gibco, 12604013) and directly observed in a Cytoflex flow cytometer (Beckman Coulter Inc, Chicago, Il, USA), and data analysis was performed using the CytExpert v2.4 software (Beckman Coulter Inc, Chicago, IL, USA).
Proliferation assay
HDFs were seeded in 96 well plates—5000 cells/well—in 100 µL of complete medium. After 8 h of incubation, cells were washed with PBS and then cultured overnight with serum-free medium. After that, cells were washed with PBS and EVs were administered—1.5 × 1011 EVs/mL of P10K and 2 × 1011 and 4 × 1011 EVs/mL of P100K—in 100 µL of 1:5 complete medium in serum-free medium and incubated for 24 h. Complete medium was used as positive control. After that, cells were washed with PBS and 100 µL of CCK8 (Merck, Cat: 96992) 1:10 diluted in culture medium were added. After 4 h of incubation, absorbance was read with a plate reader (Infinite® 200 PRO series, TecanTrading AG, Männedorf, Switzerland) at 450 nm, using 650 nm as the reference wavelength.
Scratch assay
To evaluate whether EVs can promote migration of HDFs a scratch assay was performed. HDFs were seeded—70 µL at a density of 5 × 105 cells/mL—in each side of the 2 well IBIDI culture inserts® in 24-well plates (IBIDI, Cat. 80209) and subsequently were incubated for 8 h. Secondly, the culture medium was aspired and serum-free medium was added in order to minimize the cell proliferation. After an overnight incubation, culture inserts were extracted and the scratch was performed. Then, HDFs were washed with PBS, EVs were administered—2.5 × 109 EVs/mL of P10K and 1 × 1010 EVs/mL of P100K—in 300 µL of serum-free medium and cells were incubated for 48 h. At pre-defined time-points—0 h, 6 h, 12 h, 24 h, 36 h and 48 h—images of the closing area were taken by an optical microscope (Nikon, Japan). The percentage of migration in the area between wound edges was calculated by ImageJ software (National Institutes of Health, MA, USA).
Tube formation assay
To perform the tube formation assay 15 µL of Matrigel® matrix (Corning®, Cat. 356231) were added into the µ-Plate angiogenesis 96 well plates (IBIDI, Cat. 89646) and let polymerize for 30 min at 37 °C. HUVECs were seeded at a density of 4.5 × 105 cells/mL in 30 µL of complete medium, 15 µL of PBS and 25 µL of EVs—2 × 1011 EVs/mL of P10K and 3.5 × 1011 EVs/mL of P100K—in the matrigel bed. Medium with non-EVs control batches processed equally to the EVs batches was used as control. Complete medium was used as positive control. Following an incubation of 24 h, each well was photographed by an optical microscope (Nikon, Japan). After that, cells where stained with calcein AM (Invitrogen, “LIVE/DEAD™ Viability/Cytotoxicity Kit, for mammalian cells” Cat. L3224) and each well was photographed by a Nikon epi-fluorescence microscope equipped with a DSD2 confocal modulus (Nikon, Japan). The observed tubes were analyzed by using the ImageJ software.
Cell survival under hyperglycaemia assay
The effect of EVs on cell survival under a hyperglycaemic environment was studied in HDFs. Briefly, 10,000 cells were cultured in 96 well plates for 24 h. After that, HDFs were pre-treated with EVs—1 × 1011 EVs/mL of P10K and 3 × 1011 EVs/mL of P100K—for 6 h, rinsed with PBS and exposed to hyperglycaemic conditions of 150 mM glucose in HDFs serum-free culture medium. Cells pre-treated with non-EVs control batches processed equally to the EVs batches were used as control. Cells pre-treated with complete medium were used as a positive control. Results were normalized against cells grown under non-hyperglycaemic conditions. The metabolic activity was observed at 24 h, 48 h and 72 h by CCK8 assay as described earlier. Cell viability at 72 h was also observed with calcein AM/Ethidium homodimer (Invitrogen, “LIVE/DEAD™ Viability/Cytotoxicity Kit, for mammalian cells” Cat. L3224) and each well was photographed by a Nikon epi-fluorescence microscope equipped with a DSD2 confocal modulus (Nikon, Japan).
Reactive oxygen species (ROS) protection assay
Cell viability of HDFs under a ROS environment was studied in the protection assay. In brief, HDFs—1 × 105 cells/well—were seeded in a 96-well plate and allowed to adhere. After 24 h, cells were washed with PBS and the EVs pre-treatment—1 × 1011 EVs/mL of P10K and 3 × 1011 EVs/mL of P100K—was added for 2 h and 6 h respectively. After that, cells were washed and then incubated for 30 min under a ROS environment—100 µL of serum free medium with 15 mM of H2O2—. Then, cells were washed with PBS and incubated overnight in serum free medium to minimize the proliferation of the surviving cells. Finally, cells were washed with PBS and 100 µL of CCK8, 1:10 diluted in serum free culture medium, were added. After 4 h of incubation, the absorbance was read at 450 nm, using 650 nm as the reference wavelength. An alternative co-culture ROS assay was performed as follows. HDFs—1 × 105 cells/well—were seeded in a 96-well plate and allowed to adhere. After 24 h, cells were washed with PBS and then EVs were added in 100 µL of serum free medium with 625 µM of H2O2 for 4 h. After that, cells were washed with PBS and 100 µL of CCK8, 1:10 diluted in serum free culture medium, were added. After 4 h of incubation, absorbance was read. For all assays, medium with non-EVs control batches processed equally to the EVs batches was used as control and complete medium was used as positive control.
Statistical analysis
Results are expressed as the mean ± standard deviation. When normally distributed, results were analyzed through Student’s two-tailed t test, to compare between two independent groups, or through a one-way ANOVA test for multiple comparisons. Based on the Levene test for the homogeneity of variances, Bonferroni or Tamhane post-hoc analyses were applied. In contrast, Mann–Whitney’s non-parametric analysis was applied for non-normally distributed data. All the statistical computations were performed using SPSS 25.0 (SPSS®, Inc., Chicago, IL, USA).
Results
EVs isolation and characterization
As represented in Fig. 2a, Cryo-EM images showed that the majority of the obtained material was round-shaped and bi-membrane-bounded vesicular units. The diverse heterogeneity of the different populations of EVs can be appreciated by the distinct electron dense cargos and protein decorated / non-decorated membranes presented. For both cell types, NTA measurements showed sizes ranging from 100 nm to nearly 400 nm for the P100K EVs, and from 100 to 800 nm for the P10K (Fig. 2b).

EVs size, morphology and protein decoration analysis. a Cryo-EM images of EVs. Scale bars are 200 nm. b NTA size-profile of the isolated fractions. c Size profile and protein decoration analysis of the different fractions calculated by measuring Cryo-EM images. d Images of a non-decorated EV (I) and decorated EV (II) with their respective zoomed insets (right). Scale bars are 100 nm (left-sided images) and 20 nm (right-sided images)
We also analyzed and compared the morphology, size and protein decoration of the isolated EVs by using the cryo-EM images. We studied approximately 400 single EVs and the majority of the observed units had the aforementioned round shape morphology for both cell types. Notably, we also found a small population of EVs with irregular morphologies (Additional file 1: Fig. S1)—multilayered vesicles, long tubular, etc.—. The size distribution of EVs from cryo-EM images revealed populations of smaller size—< 50 nm—than by NTA, obtaining a higher proportion of sEVs for both cell types (Fig. 2c). Moreover, when we analyzed the presence or absence of membrane protein decoration, we found that the majority of EVs were non-decorated for both cell types (Fig. 2c, d). The decoration percentages of m-lEVs were 9.38% and 7.82% for AT-EVs and HF-EVs respectively, whereas in the case of sEVs the percentages were 16.20% and 14.28% for AT-EVs and HF-EVs respectively. However, we did not observe differences in the diameter of decorated vs non-decorated EVs between each EVs population (Additional file 1: Fig. S2).
When developing a EVs-based therapy it is crucial to achieve an adequate reproducibility in the batch-to-batch production. For such aim, we analyzed the mean and the mode size of the isolated EVs for the different obtained batches—every EVs isolation for each cell passage—. We did not find significant differences in any of the parameters for the different MSC-EVs (Fig. 3a, b). Furthermore, we obtained low relative standard deviations in the isolated batches for both cell types and pellets (Fig. 3a, b). As expected, we observed that the mean and mode sizes of EVs were lower for the P100K EVs than for the P10K EVs for both cell types. The overall deviations of the mean and mode size were also lower for the P100K compared to the P10K (Fig. 3a, b). In contrast, the efficiency of the obtained batches in terms of number of isolated EVs per final cell count showed significant differences between both cell types (Fig. 3c). Our results demonstrate that HF-MSCs produce less EVs than AT-MSCs for both P10K and P100K. Moreover, all cell viabilities at the end of every batch were adequate—between 95 and 98% for both cell types.

EVs batch-to batch analysis, western blotting and flow cytometry surface marker analysis. a Mean size of each EVs fraction for the different batches (n = 8). b Mode size of each EVs fraction for the different batches (n = 8). c Number of EVs compared to final cell count (n = 6). N.S. non-significant, *p < 0.05; **p < 0.005 between groups. d Western blotting analysis of EVs and non-EVs markers of three different batches for each cell type. e Multiplex bead-based flow cytometry assay for detection of EV surface markers (n = 3). f Comparison of EVs surface marker expression from (e): AT-P10K vs HF-P10K (I), AT-P100K vs HF-P100K (II), HF-P10K vs HF-P100K (III), and AT-P10K vs AT-P100K (IV). *p < 0.05; **p < 0.005; ***p < 0.001 between groups
Marker analysis by western blotting (Fig. 3d) depicted the expression of CD13, CD81 and LAMP1 in P10K and P100K pellets derived from both cell types. In contrast, only AT-EVs showed bands for the rest of EV markers, with CD63 in both P10K and P100K, and CD9 in P100K. In the particular case of CD63, we observed different patterns; HF-MSCs retained this tetraspanin inside, while AT-MSCs showed it on m-lEVs and sEVs. We should mention that both MSC sources contain intracellular CD63 as exhibited by immunofluorescence images (Additional file 1: Fig. S3). We have also analyzed the presence of non-vesicular markers, including COXIV—a mitochondrial—and GRP78—endoplasmic reticulum—proteins, which appeared mostly in the P10K. Finally, TUB1, Hsp70, and EEA1 were only detected in the cells, and not in the vesicular fractions.
EVs were also evaluated by flow cytometry for the expression of 37 surface markers. We analyzed three different batches and found populations positive for CD3, CD9, CD25, CD29, CD41b, CD44, CD49e, CD56, CD63, CD81, CD105, CD142, CD146, MCSP, HLA-ABC/MHC-I and SSEA-4 (Fig. 3e). However, only CD9, CD29, CD44, CD63, CD81, CD105, MCSP, HLA-ABC and SSEA-4 were positive for all pellets and cell types. Nevertheless, we found some significant differences between both cell types and pellets. As observed, HF-P10K expressed CD56—not in AT-P10K—and more CD44 and MCSP than AT-P10K; in contrast, AT-P10K expressed more SSEA-4 (Fig. 3f-I). Regarding the P100K, we observed the same differences concerning the expression of CD44, CD56—not expressed in AT-P100K—, MCSP and SSEA-4 as in the P10K. Additionally, we achieved a higher expression of CD29, CD41b—not expressed in AT-P100K—, CD81 and HLA-ABC in the HF-P100K EVs (Fig. 3f-II). We also compared the P10K EVs and P100K EVs of the same cell type. As shown in Fig. 3f-III we found that P10K HF-EVs expressed more CD49b but lower CD81 and CD41b. On the other hand, P10K AT-EVs expressed more CD105, CD49e, CD44 and CD29 but lower CD81 than P100K AT-EVs (Fig. 3f-IV).
EVs uptake and proliferation by human dermal fibroblasts
Uptake assay
After incubating HDF cells with EVs coming from fluorescence-labelled AT- and HF-MSCs, we observed a comparable uptake of EVs coming from both types of MSCs (Fig. 4). Although there is a higher percentage of positive events corresponding to AT-EVs preparations (Fig. 4a-I, II), the correction by number of EVs in the preparation, together with the variability of obtained data, make those changes non-significant (Fig. 4a-III). However, there were almost 10 times more capture events when the cells are incubated with P10K preparations, corresponding to m-lEVs, than 100 K preparations (sEVs) (Fig. 4a-III and Additional file 1: Fig. S4). This difference can be observed directly by immunofluorescence (Fig. 4b). A very low carry-over of fluorescence was observed in medium that was not conditioned by HDFs (Fig. 4a, c).

EVs uptake by HDFs and proliferation assay. a EVs flow cytometry uptake: (I) Gating of the cell populations and percentage of positive events for the background control (Ctrl DIO), and the EV preparations of 10 K and 100 K for HF and AT cells. (II) The upper table shows the percentage of fluorescent cells after adding different preparations of EVs. Bottom table shows the number of vesicles per well added for each preparation. (III) The graphs present the percentage of positive events according to the flow cytometry study corrected by the number of EVs for each type of EVs. *p < 0.05, N.S. Non-significance. b Representative fluorescence micrographs of the capture of EVs and region of interest (ROI) for each preparation. The figure at the bottom show HDFs treated with the negative control corresponding to the carry-over of fluorescence of cell culture media never conditioned with HDFs, and untreated HDFs. c HDFs proliferation assay by CCK8 (n = 3). N.S. Non-significant, *p < 0.05; **p < 0.005; ***p < 0.001 against the control group; #p < 0.05 against the positive control group; &&&p < 0.001 between groups
Proliferation assay
It has been largely described that EVs promote the proliferation of different skin cell types such as HDFs among others [14]. When a chronic wound is given, the proliferative profile of the resident HDFs is impaired due to the impossibility to respond to the different cytokines and growth factors [24, 25]. In this regard, here, we compared the proliferative effect of the isolated MSC-EVs on HDFs. As shown in Fig. 4e, there is an important difference between the outcomes of P10K EVs as compared to the P100K EVs. P10K EVs showed an increase of approximately 60–70% of proliferation, as compared to the control, and AT-EVs, in addition, showed significant differences as compared to the positive control—complete medium—. In contrast, P100K EVs demonstrated a lower effect, which was dose-dependent. We did not notice significant differences between both cell types.
Scratch assay
In the scratch assay, we studied the pro-migratory effect of the MSC-EVs on HDFs. It is well-known that in chronic wound scenarios, there is also an impaired migration of these cells due to the harsh inflammatory environment and the impaired cell-to-cell communication [7, 24, 25]. In that sense, it is crucial to help these cells to increase their motility to favour the formation of granulation tissue ensuring an adequate closure of the wound. In this regard, we starved cells during the entire assay to minimize cell proliferation (Fig. 5a). Upon EVs treatment, we observed a faster wound closure of HDFs as compared to the control cells—treated with non-EVs control batches processed equally to the EVs batches—(Fig. 5b, c). A complete wound closure was achieved in all EVs-treated groups within 48 h, whereas the wound was still open in the control group (Fig. 5b). We did not observe significant differences between any of the EVs groups however, AT-P10K and HF-P10K obtained earlier significant differences—vs control group—as compared to their respective P100K EVs. We also measured the migration rate of the HDFs. As observed in the Fig. 5d only at the time-point of 8 h we achieved an increased migration rate in all groups as compared to the control group. Surprisingly, we did not obtain significant differences among all groups in the rest of the time-points. These results depicted that the main effect of EVs on the migration of HDFs was given between 4 and 8 h of treatment and maintained during the rest of the time-points.

Scratch assay. a Experimental setup. b Images of the treated and control groups at different time-points of the scratch assay (n = 3). c Percentages of wound closure at the different time-points. *p < 0.05; **p < 0.005; ***p < 0.001 against the control group. d Migration rate of HDFs at different time-points. *p < 0.05 of all groups against the control group. N.S. Non-significant
Tube formation assay
The biological process of angiogenesis involves the proliferation, migration and tube formation of endothelial cells. The formation of new vessels can determine the outcome of chronic wound healing because the newly formed vessels allow the oxygenation and nutrient supply to the wound site [7, 26]. To analyze the effect of the studied MSC-EVs on migration and tube formation of endothelial cells, EVs were incubated with HUVECs on a Matrigel® niche. As shown in Fig. 6a, the groups treated with EVs remarkably enhanced the number of formed tubes as compared to the control group—HUVECs treated with non-EVs control batches processed equally to the EVs batches—. We did not obtain significant differences between the EVs-treated groups and the positive control group—complete medium—. Neither were differences found within the groups treated with EVs—neither P10K nor P100K—.

Angiogenesis and antioxidant assays. a Number of formed tubes per well (n = 4). b Diameter length of the formed tubes. c Number of branching points per well. ***p < 0.001 against the control group, # p < 0.05 against the positive control group, N.S. non-significance. d Calcein-stained fluorescence and brightfield images of the formed vessels. Scale bars are 500 µm. e Cell survival under a H2O2-mediated ROS environment (I) after a 2 h pre-treatment with EVs (n = 4), (II) after a 6 h pre-treatment with EVs (n = 4) and (III) under a 4 h co-treatment of cells with EVs and H2O2 (n = 4). **p < 0.005; ***p < 0.001, n.s. non significance against control group, # p < 0.05 between groups
Furthermore, we studied the tube diameter length and the number of branching points—union points of formed tubes—(Fig. 6b, c). We did not observe differences in the tube diameter length between the different cell types, pellets and controls but, in accordance with the number of formed tubes, the treated groups obtained a higher number of branching points in comparison with the control group (Fig. 6d). Moreover, AT-P10K, HF-P10K and HF-P100K obtained a higher number of branching points as compared to the positive control.
Reactive oxygen species (ROS) protection assay
Having achieved biological effects on HDFs proliferation and migration, we then studied the protective effects of EVs against ROS, which are commonly present in chronic wound scenarios [27]. As can be seen in the Fig. 6e, we did not obtain significant differences in the EVs-treated groups as compared to the control group in any of the performed assays. We observed that a 2 h and 6 h pre-treatment of HDFs with complete medium—positive control—did not showed differences in the ROS apoptotic protection as compared to the control group (Fig. 6e-I, II). However, in a 4 h of co-culture with EVs and H2O2, only the positive control achieved better outcomes as compared to the rest of the groups. This may be expected because of the presence of ascorbic acid in the medium supplementation (Fig. 6e-III).
Cell survival under hyperglycaemia assay
Diabetic ulcers—a kind of chronic wounds—are characterized by a persistent inflammatory status with insufficient oxygen and nutrient accessibility caused by elevated glucose levels. As a result, a hypoxic environment with an elevated oxidative stress is formed, leading fibroblasts, as well as other skin cell types, to die [28, 29]. To study the cell survival of HDFs under a hyperglycemic environment we pre-treated HDFs with EVs for 6 h before exposing them to hyperglycemia. As observed in Fig. 7a, at 24 h we only obtained differences against the control group—HDFs treated with non-EVs control batches processed equally to the EVs batches—in the HF-P10K and positive control groups. However, after 48 h of hyperglycemic exposure, we achieved significant differences against the control in all the EVs-treated groups. Furthermore, AT-P10K and HF-P10K obtained higher outcomes as compared to the positive control group—complete medium—. We also observed that the EVs-treated groups did not obtain significant differences against the non-hyperglycemic group at this time point. Finally, at 72 h of hyperglycemia, we achieved significantly higher cell survival in the EVs-treated groups as compared to the control and positive control groups (Fig. 7b).

Cell survival under hyperglycemia assay. a Percentage of cell viability under hyperglycemia at 24 h, 48 h and 72 h (n = 4). *p < 0.05; **p < 0.005; ***p < 0.001 against the control group, ## p < 0.005, ### p < 0.001 against W/o glucose group; &p < 0.05; &&p < 0.005; &&&p < 0.001 between groups. b Calcein/ethidium stained images of the survival cells at 72 h of hyperglycemia. Scale bars are 500 µm
Discussion
While MSC-EVs have gained the attention of scientific community in the field of tissue engineering and regenerative medicine, there is still a race for the development of the most suitable and effective MSC-EVs therapy. AT-EVs have largely demonstrated their potential in chronic wound healing scenarios [28, 30,31,32,33,34,35,36]. However, to the best of our knowledge, to date there is no work characterizing and analyzing the therapeutic potential of HF-EVs in this field, or in any other. For such aim, we first analyzed the size and morphology of the isolated EVs. Both AT-EVs and HF-EVs had round-shaped bi-membrane morphologies and the size ranges varied between 50 and 700 nm for P10K EVs and between 30 and 400 nm for P100K EVs by using NTA and Cryo-EM images. These size results are consistent with those reported in the literature using UC as the isolation method for MSC-EVs [37,38,39,40].
There is a vast number of works analyzing MSC-EVs by electron microscopy [41,42,43,44]. However, a few works have visualized MSC-EVs by using cryo-EM and none of them has quantified the percentage of their protein decoration [33, 45,46,47]. This technique allows reducing sample damaging and the appearance of artefacts caused by fixation, dehydratation or the addition of heavy metals [48]. Our results depicted that, by using UC as the isolation method, only a small percentage of MSC-EVs presented proteins decorating their membrane—approximately 8% on P10K EVs and 15% on P100K EVs—. Further studies should be carried out to analyze if the percentage of protein decoration on MSC-EVs would be modified by using different isolation/purification methods. However, a seminal work by Rizzo et al. described that, by using UC as the isolation method, the 90% of the isolated EVs—obtained from C. Neoformans—presented protein decorated membranes [49]. Moreover, another work by Noble et al. mentioned that m-lEVs had more protein-membrane decoration than sEVs in the MCF10A cell line by using cryo-EM images [50]. However, they did not quantify the percentage of membrane-decorated EVs. These results may suggest that the protein decoration of EVs could be more influenced by the EV-source rather than the biogenesis or isolation method used. In addition, it has also been described the spontaneous formation of a “protein corona” around the surface of nanoparticles, viruses and EVs as a protein aggregation on membranes [51,52,53]. Thus, a more thorough analysis should be made to identify if these molecules are spontaneously formed or native components of EVs, and whether they influence the biological activity of these EVs. For example, some authors have argued that the presence of these protein coronas on different nanoparticles have a major influence on their bioavailability and biodistribution [52, 53].
To ensure the reproducibility and quality of EV-based treatments, the establishment of a reliable and standardized isolation method for EVs is crucial. For such purpose, we studied the batch-to-batch reproducibility of the isolated EVs. Despite mean and mode size results had adequate reproducibility between batches for both pellets and cell types, results depicted that the number of EVs regarding the final cell count had greater variations. These results may suggest that AT-MSCs produce naturally larger quantities of EVs than HF-MSCs in the studied conditions. The characterization of the EVs protein markers by western blotting showed the presence of CD13—a marker of stemness associated to regeneration properties—in both cell lines [54], as well as the presence of CD81 in all the preparations. The results for CD63 presented a different trafficking of this molecule, since AT-MSCs released more protein in EVs, while HF-MSCs cells kept most of the protein intracellularly. This retention, together with the absence of CD9 in HF-EVs, the slightly increase of CD81 in the sEVs population secreted by HF-MSCs, and the retention of LAMP1 in HF-MSCs, suggest that both cell lines had a different balance of endosomal/ectosomal mechanism of EVs biogenesis [55]. Nevertheless, further studies inhibiting those routes would be necessary to clarify their relative importance. We also observed the presence of COXIV, a protein associated to mitochondria. The presence of mitochondrial proteins in some populations of EVs has been described in melanoma preparations [56], among other cell types, giving rise to the recently coined term “mitovesicles” [57].
The surface marker analysis of EVs by flow cytometry depicted significant differences in the expression profile of different proteins between both cell types and pellets. Using this technique, we demonstrated the presence of the transmembrane proteins CD9, CD63, and CD81 in all EVs. Interestingly, the results observed for those markers differ from western blot analysis. The reasons behind may lie in the technical differences between both approaches; bead capture only measures exposed epitopes, while western blotting shows the contribution for all the protein content of the preparation. Western blot quantification has also information regarding the size of the protein. Additional file 1: Fig. S5 shows the presence of bands detected by the antibody with higher strength in HF-EVs, below 37 kDa, which certainly can compensate the differences observed in the western blot between 37 and 50 kDa.
Another highly expressed marker in all of our EVs was SSEA-4, an embryonic stem cell marker popularly known as a “stemness” marker. It has been described the presence of this molecule in EVs from bone marrow MSCs (BM-MSCs), AT-MSCs [58, 59] as well as in other MSCs [60, 61]. Based on our findings, we observed the expression of SSEA-4 in sEVs and m-lEVs derived from both HF-MSCs and AT-MSCs. Furthermore, we also noticed the expression of CD105, a characteristic MSCs marker [62]. Specifically, we observed more expression in P10K EVs than in P100K EVs, for both cell types. In terms of functionality, AT-EVs and HF-EVs expressed several adhesion molecules such as CD29, CD44 and CD49e—mostly in P10K EVs—, which facilitate their homing to inflamed and injured tissues such as chronic wounds [63]. Another interesting marker found slightly represented in both P10K EVs—but not in P100K—was CD146 or melanoma cell adhesion molecule (MCAM), a molecule involved in permeability, monocyte transmigration and angiogenesis that is present in many tumors and endothelial cells [64, 65]. Another melanoma-associated marker found in all EVs—but especially found in HF-EVs—was MCSP. This molecule is a highly expressed marker in benign and malign melanomas and melanoma-derived EVs [66, 67]. However, the expression of MCSP in healthy tissues has also been described [66]. Ghali et al. demonstrated that HF-MSCs, among other hair follicle surrounding cells, express MCSP [68]. Thus, our results depicted that the EVs shuttled by HF-MSCs also express MCSP. Tissue factor (TF), also known as CD142 is recognized as one of the most important players in the coagulation process [69, 70]. Furthermore, it has been described that this marker also plays a role in wound re-epithelialization and necrosis [70]. The fact that principally P10K HF-EVs expressed this marker may suggests a more important role for HF-EVs than AT-EVs in the earliest phases of the wound healing process. Another receptor surprisingly found only in HF-P100K was CD41b or integrin αIIβ. This marker is a well-known collagen-binding integrin found in MSCs, as well as in many other cell types such as platelets. [71,72,73]. Finally, the expression of CD56 was exclusively found in HF-EVs. The presence of CD56 has been commonly found in NK cells and EVs—with immunomodulatory properties [74, 75]—and in neural lineage cells [76]. However, It has been also described the presence of CD56 in HFs and this has been related to the maintenance of the hair follicle immune privilege [76,77,78]. Thus, the role of CD56 in HF-EVs should be investigated to clarify whether the presence of this molecule in HF-EVs may be related to immunomodulatory properties.
When we analyzed the HDFs uptake of EVs, we found no differences in the P10K and P100K uptake between both cell types. On the other hand, we found that these cells were able to better internalize m-lEVs than sEVs. Indeed, we observed an almost tenfold fluorescence increase in HDFs after they captured m-lEVs as compared to sEVs. Although different aspects can influence this result, such as the number of membrane bound fluorochrome per vesicle in both m-lEV and sEVs, it is noteworthy that P10K EVs were able to induce a higher increase in cell proliferation than P100Ks in both cell types. More studies should be made in this regard to clarify whether these effects are related to a higher internalization or to intrinsic properties of the different EV populations. Furthermore, an additional analysis should be carried out in order the test the internalization efficacy of HDFs in the presence of other cell types such as phagocytes, keratinocytes etc. Regardless of that, our results confirmed that both m-lEVs and sEVs were able to increase proliferation of fibroblasts, as previously described in MSCs [35, 79]. Interestingly, we found that P10Ks were able to increase the proliferation of HDFs up to 25–40% more than P100Ks. It has been previously described the proliferative effect of m-lEVs on AT-MSCs [35], however, we have not found any work comparing the proliferative potency of sEVs vs m-lEVs in AT-MSCs neither in HF-MSCs. Promoting the migration of fibroblasts has also been an important requirement for every developing therapy intended for chronic wounds [7]. Besides the proliferative effects of MSC-EVs on different cell types, it has been also described that MSC-EVs can also promote the migration of fibroblasts, among other cell types [80]. In this work, we demonstrated that HF-EVs and AT-EVs alike were able to promote the migration of HDFs. Furthermore, we found that the effect observed on HDFs migration was achieved between 4–8 h—regardless of cell and pellet type—8 h and was maintained until the end of the assay.
Numerous studies have demonstrated that the oxidative stress increases tissue inflammation resulting in a stagnation of the wound healing process [81, 82]. In addition, a dysfunction of the vasculature, along with limited neovascularization and angiogenic processes, creates hypoxic conditions in chronic wounds [7, 83]. Some works have demonstrated that EVs derived from different cell populations can protect diverse cell types against ROS-induced apoptosis [84, 85]. In this regard, different MSC-EVs obtained from bone marrow or umbilical cord have demonstrated ROS-protective effects on cardiac stem cells and HaCaT cells [86, 87]. In contrast, only few researchers have described protective effects against ROS-induced apoptosis by means of AT-EVs on cardiomyocytes [88], NIH3T3 fibroblasts [28] and HDFs [89]. Here, we did not achieve such protective effects in any of the studied conditions with any of our pellet or cell types. In keeping with these results, Matsuoka et al. only observed a 10% of viability increase in the EVs-treated groups when compared to the control group. However, they did not employ a EVs-depleted medium in the assay. Based on all these findings, a more thorough analysis is warranted to clarify the role of MSC-EVs on the viability of HDFs under H2O2 oxidative stress.
It has been largely described the angiogenic potential of different MSC-EVs such as those produced by BM-MSCs, umbilical cord MSCs (UC-MSCs) or AT-MSCs, among others. Such effect has been attributed to the transfer of pro-angiogenic proteins—VEGF, PDGF, EGF, FGF etc.—and mi-RNAs including miR-31or miR-125 among others [90,91,92]. In this work, we found pro-angiogenic properties not only in AT-EVs but also in HF-EVs with both assayed pellets. Once again, we did not observe differences among groups in the size or morphology of the formed vessels. The need for increased blood flow and oxygenation reaches a high importance in diabetic chronic ulcers. The hyperglycemic microenvironment associated with diabetes challenges cell survival in diabetic wounds, causing oxidative stress and delaying the healing. For example, it has been described that hyperglycemia destabilizes HIF-1α and impairs its function at a concentration of 30 mM [28]. Our results depicted that with only a 6 h of pre-treatment, all EVs were able to protect and increase cell survival of HDFs under a cytotoxic hyperglycemic environment after 24 h of treatment and at least until 72 h of assay. Interestingly, our results were even better than those obtained by the positive control group—complete medium—. Here, we found that both pellets from AT-MSCs and HF-MSCs were able to reduce oxidative stress and increase HDFs metabolism and viability under a hyperglycemic condition.
Conclusions
In conclusion, in this work, we describe for the first time to our knowledge, a complete characterization and functional comparison of m-lEVs and sEVs obtained from HF-MSCs against those obtained from AT-MSCs. Here, we made an in vitro screening of different MSC-EVs populations in different bio-relevant assays in order to analyze the efficacy of the formulations. We demonstrated the presence of the classical EV-markers among many others involved in biological processes not only in sEVs but also in m-lEVs. In this regard, HF-EVs showed a better “pro-regenerative” marker profile than AT-EVs for both m-lEVs and sEVs. We also analyzed the batch-to-batch production, the morphology and the protein decoration of these EVs depicting some differences between both cell types and pellets. Furthermore, we demonstrated that HF-EVs, equal than AT-EVs, were able to induce migration and proliferation in HDFs, as well as angiogenesis in HUVECs. However, we found that m-lEVs obtained better outcomes increasing HDFs proliferation. We did not observe protective effects against H2O2-induced oxidative stress in HDFs, but did prove that HF-EVs could protect HDFs from cell death and oxidative stress under hyperglycemic conditions to the same extent as AT-EVs. Altogether, we here postulate a promising source of EVs—not only sEVs but also m-lEVs—obtained from HF-MSCs with a great potential for chronic wound treatments and regenerative medicine therapies in general.
Data availability
Raw data is available from the corresponding author upon reasonable request.
Abbreviations
- MSCs:
-
Mesenchymal stromal cells
- EVs:
-
Extracellular vesicles
- HFs:
-
Hair follicles
- AT:
-
Adipose tissue
- HDFs:
-
Human dermal fibroblasts
- HUVECs:
-
Human umbilical vein endothelial cells
- NTA:
-
Nanoparticle tracking analysis
- WB:
-
Western blot
- Cryo-EM:
-
Cryo electron microscopy
- DAPI:
-
2-(4-Amidinophenyl)-1 h-indole-6-carboxamidine
- CCK8:
-
Cell counting kit 8
- ROS:
-
Reactive oxygen species
References
Martino PA, Heitman N, Rendl M. The dermal sheath: an emerging component of the hair follicle stem cell niche. Exp Dermatol. 2021. https://doi.org/10.1111/exd.14204.
Wang B, Liu XM, Liu ZN, Wang Y, Han X, Lian AB, et al. Human hair follicle-derived mesenchymal stem cells: isolation, expansion, and differentiation. World J Stem Cells. 2020. https://doi.org/10.4252/wjsc.v12.i6.462.
Gentile P, Garcovich S. Advances in regenerative stem cell therapy in androgenic alopecia and hair loss: WNT pathway, growth-factor, and mesenchymal stem cell signaling impact analysis on cell growth and hair follicle development. Cells. 2019;8:466.
Zhou D, Cheng H, Liu J, Zhang L. Establishment of human hair follicle mesenchymal stem cells with overexpressed human hepatocyte growth factor. Iran J Basic Med Sci. 2017. https://doi.org/10.22038/IJBMS.2017.8834.
Wang Y, Liu J, Tan X, Li G, Gao Y, Liu X, et al. Induced pluripotent stem cells from human hair follicle mesenchymal stem cells. Stem Cell Rev Rep. 2013. https://doi.org/10.1007/s12015-012-9420-5.
Gonzales KAU, Fuchs E. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev Cell. 2017;43:387–401.
Las Heras K, Igartua M, Santos-Vizcaino E, Hernandez RM. Chronic wounds: current status, available strategies and emerging therapeutic solutions. J Control Release. 2020;328:532–50.
Han Y, Li X, Zhang Y, Han Y, Chang F, Ding J. Mesenchymal stem cells for regenerative medicine. Cells. 2019;8:886.
Gentile P, Garcovich S. systematic review of platelet-rich plasma use in androgenetic alopecia compared with minoxidil((R)), finasteride((R)), and adult stem cell-based therapy. Int J Mol Sci. 2020;21:2702.
Gentile P, Scioli MG, Bielli A, De Angelis B, De Sio C, De Fazio D, et al. Platelet-rich plasma and micrografts enriched with autologous human follicle mesenchymal stem cells improve hair re-growth in androgenetic alopecia. Biomol Pathway Anal Clin Eval Biomed. 2019;7:27.
Ma D, Kua JE, Lim WK, Lee ST, Chua AW. In vitro characterization of human hair follicle dermal sheath mesenchymal stromal cells and their potential in enhancing diabetic wound healing. Cytotherapy. 2015. https://doi.org/10.1016/j.jcyt.2015.04.001.
Heras KL, Igartua M, Santos-Vizcaino E, Hernandez RM. Cell-based dressings: a journey through chronic wound management. Biomater Adv. 2022. https://doi.org/10.1016/j.bioadv.2022.212738.
Baraniak PR, McDevitt TC. Stem cell paracrine actions and tissue regeneration. Regen Med. 2010. https://doi.org/10.2217/rme.09.74.
Nagelkerke A, Ojansivu M, van der Koog L, Whittaker TE, Cunnane EM, Silva AM, et al. Extracellular vesicles for tissue repair and regeneration: evidence, challenges and opportunities. Adv Drug Deliv Rev. 2021;175:113775.
Munoz-Perez E, Gonzalez-Pujana A, Igartua M, Santos-Vizcaino E, Hernandez RM. Mesenchymal stromal cell secretome for the treatment of immune-mediated inflammatory diseases: latest trends in isolation, content optimization and delivery avenues. Pharmaceutics. 2021;13:1082.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977.
Wiklander OPB, Brennan MA, Lotvall J, Breakefield XO, El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. 2019;11:eaav8521.
Cabral J, Ryan AE, Griffin MD, Ritter T. Extracellular vesicles as modulators of wound healing. Adv Drug Deliv Rev. 2018;129:394–406.
Tsiapalis D, O’Driscoll L. Mesenchymal stem cell derived extracellular vesicles for tissue engineering and regenerative medicine applications. Cells. 2020;9:991.
Silva AKA, Morille M, Piffoux M, Arumugam S, Mauduit P, Larghero J, et al. Development of extracellular vesicle-based medicinal products: a position paper of the group “Extracellular Vesicle translatiOn to clinicaL perspectiVEs - EVOLVE France.” Adv Drug Deliv Rev. 2021;179:114001.
Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018. https://doi.org/10.1080/20013078.2018.1535750.
Hernaez-Estrada B, Gonzalez-Pujana A, Cuevas A, Izeta A, Spiller KL, Igartua M, et al. Human hair follicle-derived mesenchymal stromal cells from the lower dermal sheath as a competitive alternative for immunomodulation. Biomedicines. 2022;10:253.
EV-TRACK Consortium, Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017. https://doi.org/10.1038/nmeth.4185.
Wall IB, Moseley R, Baird DM, Kipling D, Giles P, Laffafian I, et al. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J Invest Dermatol. 2008. https://doi.org/10.1038/jid.2008.114.
Zou ML, Teng YY, Wu JJ, Liu SY, Tang XY, Jia Y, et al. Fibroblasts: heterogeneous cells with potential in regenerative therapy for scarless wound healing. Front Cell Dev Biol. 2021. https://doi.org/10.3389/fcell.2021.713605.
Martin P. Wound healing–aiming for perfect skin regeneration. Science. 1997. https://doi.org/10.1126/science.276.5309.75.
Dunnill C, Patton T, Brennan J, Barrett J, Dryden M, Cooke J, et al. Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int Wound J. 2017. https://doi.org/10.1111/iwj.12557.
Shiekh PA, Singh A, Kumar A. Exosome laden oxygen releasing antioxidant and antibacterial cryogel wound dressing OxOBand alleviate diabetic and infectious wound healing. Biomaterials. 2020;249:120020.
Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. Dis Model Mech. 2014. https://doi.org/10.1242/dmm.016782.
Shafei S, Khanmohammadi M, Heidari R, Ghanbari H, Taghdiri Nooshabadi V, Farzamfar S, et al. Exosome loaded alginate hydrogel promotes tissue regeneration in full-thickness skin wounds: An in vivo study. J Biomed Mater Res A. 2020. https://doi.org/10.1002/jbm.a.36835.
Wang C, Wang M, Xu T, Zhang X, Lin C, Gao W, et al. Engineering bioactive self-healing antibacterial exosomes hydrogel for promoting chronic diabetic wound healing and complete skin regeneration. Theranostics. 2019. https://doi.org/10.7150/thno.29766.
An Y, Lin S, Tan X, Zhu S, Nie F, Zhen Y, et al. Exosomes from adipose-derived stem cells and application to skin wound healing. Cell Prolif. 2021. https://doi.org/10.1111/cpr.12993.
Choi JS, Cho WL, Choi YJ, Kim JD, Park HA, Kim SY, et al. Functional recovery in photo-damaged human dermal fibroblasts by human adipose-derived stem cell extracellular vesicles. J Extracell Vesicles. 2019. https://doi.org/10.1080/20013078.2019.1565885.
Pomatto M, Gai C, Negro F, Cedrino M, Grange C, Ceccotti E, et al. Differential therapeutic effect of extracellular vesicles derived by bone marrow and adipose mesenchymal stem cells on wound healing of diabetic ulcers and correlation to their cargoes. Int J Mol Sci. 2021;22:3851.
Ren S, Chen J, Duscher D, Liu Y, Guo G, Kang Y, et al. Microvesicles from human adipose stem cells promote wound healing by optimizing cellular functions via AKT and ERK signaling pathways. Stem Cell Res Ther. 2019. https://doi.org/10.1186/s13287-019-1152-x.
Gentile P, Garcovich S. Concise Review: adipose-derived stem cells (ASCs) and adipocyte-secreted exosomal microRNA (A-SE-miR) modulate cancer growth and proMote wound repair. J Clin Med. 2019;8:855.
Lee JH, Ha DH, Go HK, Youn J, Kim HK, Jin RC, et al. Reproducible large-scale isolation of exosomes from adipose tissue-derived mesenchymal stem/stromal cells and their application in acute kidney injury. Int J Mol Sci. 2020;21:4774.
Franquesa M, Hoogduijn MJ, Ripoll E, Luk F, Salih M, Betjes MG, et al. Update on controls for isolation and quantification methodology of extracellular vesicles derived from adipose tissue mesenchymal stem cells. Front Immunol. 2014. https://doi.org/10.3389/fimmu.2014.00525.
Fafian-Labora J, Morente-Lopez M, Sanchez-Dopico MJ, Arntz OJ, van de Loo FAJ, De Toro J, et al. Influence of mesenchymal stem cell-derived extracellular vesicles in vitro and their role in ageing. Stem Cell Res Ther. 2020. https://doi.org/10.1186/s13287-019-1534-0.
Konoshenko MY, Lekchnov EA, Vlassov AV, Laktionov PP. Isolation of extracellular vesicles: general methodologies and latest trends. Biomed Res Int. 2018. https://doi.org/10.1155/2018/8545347.
Nakazaki M, Morita T, Lankford KL, Askenase PW, Kocsis JD. Small extracellular vesicles released by infused mesenchymal stromal cells target M2 macrophages and promote TGF-beta upregulation, microvascular stabilization and functional recovery in a rodent model of severe spinal cord injury. J Extracell Vesicles. 2021. https://doi.org/10.1002/jev2.12137.
Born LJ, Chang KH, Shoureshi P, Lay F, Bengali S, Hsu ATW, et al. HOTAIR-loaded mesenchymal stem/stromal cell extracellular vesicles enhance angiogenesis and wound healing. Adv Healthc Mater. 2021. https://doi.org/10.1002/adhm.202002070.
Tao SC, Guo SC, Li M, Ke QF, Guo YP, Zhang CQ. Chitosan wound dressings incorporating exosomes derived from MicroRNA-126-overexpressing synovium mesenchymal stem cells provide sustained release of exosomes and heal full-thickness skin defects in a diabetic rat model. Stem Cells Transl Med. 2017. https://doi.org/10.5966/sctm.2016-0275.
Ti D, Hao H, Tong C, Liu J, Dong L, Zheng J, et al. LPS-preconditioned mesenchymal stromal cells modify macrophage polarization for resolution of chronic inflammation via exosome-shuttled let-7b. J Transl Med. 2015. https://doi.org/10.1186/s12967-015-0642-6.
Priglinger E, Strasser J, Buchroithner B, Weber F, Wolbank S, Auer D, et al. Label-free characterization of an extracellular vesicle-based therapeutic. J Extracell Vesicles. 2021. https://doi.org/10.1002/jev2.12156.
Lee KS, Lee J, Kim HK, Yeom SH, Woo CH, Jung YJ, et al. Extracellular vesicles from adipose tissue-derived stem cells alleviate osteoporosis through osteoprotegerin and miR-21-5p. J Extracell Vesicles. 2021. https://doi.org/10.1002/jev2.12152.
Monguio-Tortajada M, Roura S, Galvez-Monton C, Pujal JM, Aran G, Sanjurjo L, et al. Nanosized UCMSC-derived extracellular vesicles but not conditioned medium exclusively inhibit the inflammatory response of stimulated T cells: implications for nanomedicine. Theranostics. 2017. https://doi.org/10.7150/thno.16154.
Orlov I, Myasnikov AG, Andronov L, Natchiar SK, Khatter H, Beinsteiner B, et al. The integrative role of cryo electron microscopy in molecular and cellular structural biology. Biol Cell. 2017. https://doi.org/10.1111/boc.201600042.
Rizzo J, Wong SSW, Gazi AD, Moyrand F, Chaze T, Commere PH, et al. Cryptococcus extracellular vesicles properties and their use as vaccine platforms. J Extracell Vesicles. 2021. https://doi.org/10.1002/jev2.12129.
Noble JM, Roberts LM, Vidavsky N, Chiou AE, Fischbach C, Paszek MJ, et al. Direct comparison of optical and electron microscopy methods for structural characterization of extracellular vesicles. J Struct Biol. 2020;210:107474.
Toth EA, Turiak L, Visnovitz T, Cserep C, Mazlo A, Sodar BW, et al. Formation of a protein corona on the surface of extracellular vesicles in blood plasma. J Extracell Vesicles. 2021. https://doi.org/10.1002/jev2.12140.
Ezzat K, Pernemalm M, Palsson S, Roberts TC, Jarver P, Dondalska A, et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat Commun. 2019. https://doi.org/10.1038/s41467-019-10192-2.
Xiao W, Gao H. The impact of protein corona on the behavior and targeting capability of nanoparticle-based delivery system. Int J Pharm. 2018;552:328–39.
Lin MJ, Li S, Yang LJ, Ye DY, Xu LQ, Zhang X, et al. Plasma membrane vesicles of human umbilical cord mesenchymal stem cells ameliorate acetaminophen-induced damage in HepG2 cells: a novel stem cell therapy. Stem Cell Res Ther. 2020. https://doi.org/10.1186/s13287-020-01738-z.
Mathieu M, Nevo N, Jouve M, Valenzuela JI, Maurin M, Verweij FJ, et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat Commun. 2021. https://doi.org/10.1038/s41467-021-24384-2.
Jang SC, Crescitelli R, Cvjetkovic A, Belgrano V, Olofsson Bagge R, Sundfeldt K, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles. 2019. https://doi.org/10.1080/20013078.2019.1635420.
D’Acunzo P, Perez-Gonzalez R, Kim Y, Hargash T, Miller C, Alldred MJ, et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci Adv. 2021. https://doi.org/10.1126/sciadv.abe5085.
Cavallo C, Merli G, Borzi RM, Zini N, D’Adamo S, Guescini M, et al. Small Extracellular Vesicles from adipose derived stromal cells significantly attenuate in vitro the NF-kappaB dependent inflammatory/catabolic environment of osteoarthritis. Sci Rep. 2021. https://doi.org/10.1038/s41598-020-80032-7.
Gonzalez-Cubero E, Gonzalez-Fernandez ML, Gutierrez-Velasco L, Navarro-Ramirez E, Villar-Suarez V. Isolation and characterization of exosomes from adipose tissue-derived mesenchymal stem cells. J Anat. 2021. https://doi.org/10.1111/joa.13365.
Adamo A, Brandi J, Caligola S, Delfino P, Bazzoni R, Carusone R, et al. Extracellular vesicles mediate mesenchymal stromal cell-dependent regulation of B Cell PI3K-AKT signaling pathway and actin cytoskeleton. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.00446.
Lv FJ, Tuan RS, Cheung KM, Leung VY. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014. https://doi.org/10.1002/stem.1681.
Qiu G, Zheng G, Ge M, Wang J, Huang R, Shu Q, et al. Functional proteins of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res Ther. 2019. https://doi.org/10.1186/s13287-019-1484-6.
Harrell CR, Jankovic MG, Fellabaum C, Volarevic A, Djonov V, Arsenijevic A, et al. Molecular mechanisms responsible for anti-inflammatory and immunosuppressive effects of mesenchymal stem cell-derived factors. Adv Exp Med Biol. 2019. https://doi.org/10.1007/5584_2018_306.
Harhouri K, Kebir A, Guillet B, Foucault-Bertaud A, Voytenko S, Piercecchi-Marti MD, et al. Soluble CD146 displays angiogenic properties and promotes neovascularization in experimental hind-limb ischemia. Blood. 2010. https://doi.org/10.1182/blood-2009-06-229591.
Wang Z, Xu Q, Zhang N, Du X, Xu G, Yan X. CD146, from a melanoma cell adhesion molecule to a signaling receptor. Signal Transduct Target Ther. 2020. https://doi.org/10.1038/s41392-020-00259-8.
de Bruyn M, Rybczynska AA, Wei Y, Schwenkert M, Fey GH, Dierckx RA, et al. Melanoma-associated chondroitin sulfate proteoglycan (MCSP)-targeted delivery of soluble TRAIL potently inhibits melanoma outgrowth in vitro and in vivo. Mol Cancer. 2010. https://doi.org/10.1186/1476-4598-9-301.
Wang J, Wuethrich A, Sina AA, Lane RE, Lin LL, Wang Y, et al. Tracking extracellular vesicle phenotypic changes enables treatment monitoring in melanoma. Sci Adv. 2020. https://doi.org/10.1126/sciadv.aax3223.
Ghali L, Wong ST, Tidman N, Quinn A, Philpott MP, Leigh IM. Epidermal and hair follicle progenitor cells express melanoma-associated chondroitin sulfate proteoglycan core protein. J Invest Dermatol. 2004;122:433–42.
Hoffman M. The tissue factor pathway and wound healing. Semin Thromb Hemost. 2018. https://doi.org/10.1055/s-0037-1606181.
Inbal A, Lubetsky A, Krapp T, Castel D, Shaish A, Dickneitte G, et al. Impaired wound healing in factor XIII deficient mice. Thromb Haemost. 2005. https://doi.org/10.1160/TH05-04-0291.
Brahmer A, Neuberger E, Esch-Heisser L, Haller N, Jorgensen MM, Baek R, et al. Platelets, endothelial cells and leukocytes contribute to the exercise-triggered release of extracellular vesicles into the circulation. J Extracell Vesicles. 2019. https://doi.org/10.1080/20013078.2019.1615820.
Jung SM, Moroi M. Platelets interact with soluble and insoluble collagens through characteristically different reactions. J Biol Chem. 1998. https://doi.org/10.1074/jbc.273.24.14827.
Popov C, Radic T, Haasters F, Prall WC, Aszodi A, Gullberg D, et al. Integrins alpha2beta1 and alpha11beta1 regulate the survival of mesenchymal stem cells on collagen I. Cell Death Dis. 2011. https://doi.org/10.1038/cddis.2011.71.
Poznanski SM, Ashkar AA. Shining light on the significance of NK cell CD56 brightness. Cell Mol Immunol. 2018. https://doi.org/10.1038/s41423-018-0163-3.
Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci USA. 2006;103:5941–6.
Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the immune system: more than a marker for cytotoxicity? Front Immunol. 2017. https://doi.org/10.3389/fimmu.2017.00892.
Beljaards RC, Kirtschig G. Expression of neural cell adhesion molecule (CD56) in basal and squamous cell carcinoma. Dermatol Surg. 2008. https://doi.org/10.1111/j.1524-4725.2008.34327.x.
Ito T, Ito N, Saatoff M, Hashizume H, Fukamizu H, Nickoloff BJ, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. 2008;128:1196–206.
Shabbir A, Cox A, Rodriguez-Menocal L, Salgado M, Van Badiavas E. Mesenchymal stem cell exosomes induce proliferation and migration of normal and chronic wound fibroblasts, and enhance angiogenesis in vitro. Stem Cells Dev. 2015. https://doi.org/10.1089/scd.2014.0316.
Manchon E, Hirt N, Bouaziz JD, Jabrane-Ferrat N, Al-Daccak R. Stem cells-derived extracellular vesicles: potential therapeutics for wound healing in chronic inflammatory skin diseases. Int J Mol Sci. 2021;22:3130.
Schafer M, Werner S. Oxidative stress in normal and impaired wound repair. Pharmacol Res. 2008. https://doi.org/10.1016/j.phrs.2008.06.004.
James TJ, Hughes MA, Cherry GW, Taylor RP. Evidence of oxidative stress in chronic venous ulcers. Wound Repair Regen. 2003;11:172–6.
Sen CK. Wound healing essentials: let there be oxygen. Wound Repair Regen. 2009. https://doi.org/10.1111/j.1524-475X.2008.00436.x.
Bodega G, Alique M, Puebla L, Carracedo J, Ramirez RM. Microvesicles: ROS scavengers and ROS producers. J Extracell Vesicles. 2019. https://doi.org/10.1080/20013078.2019.1626654.
Xiao J, Pan Y, Li XH, Yang XY, Feng YL, Tan HH, et al. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016. https://doi.org/10.1038/cddis.2016.181.
Shi B, Wang Y, Zhao R, Long X, Deng W, Wang Z. Bone marrow mesenchymal stem cell-derived exosomal miR-21 protects C-kit+ cardiac stem cells from oxidative injury through the PTEN/PI3K/Akt axis. PLoS ONE. 2018. https://doi.org/10.1371/journal.pone.0191616.
Zhao G, Liu F, Liu Z, Zuo K, Wang B, Zhang Y, et al. MSC-derived exosomes attenuate cell death through suppressing AIF nucleus translocation and enhance cutaneous wound healing. Stem Cell Res Ther. 2020. https://doi.org/10.1186/s13287-020-01616-8.
Liu Z, Xu Y, Wan Y, Gao J, Chu Y, Li J. Exosomes from adipose-derived mesenchymal stem cells prevent cardiomyocyte apoptosis induced by oxidative stress. Cell Death Discov. 2019. https://doi.org/10.1038/s41420-019-0159-5.
Matsuoka T, Takanashi K, Dan K, Yamamoto K, Tomobe K, Shinozuka T. Effects of mesenchymal stem cell-derived exosomes on oxidative stress responses in skin cells. Mol Biol Rep. 2021. https://doi.org/10.1007/s11033-021-06473-z.
Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. Extracellular vesicles in angiogenesis. Circ Res. 2017. https://doi.org/10.1161/CIRCRESAHA.117.309681.
Merino-Gonzalez C, Zuniga FA, Escudero C, Ormazabal V, Reyes C, Nova-Lamperti E, et al. Mesenchymal stem cell-derived extracellular vesicles promote angiogenesis: potencial clinical application. Front Physiol. 2016. https://doi.org/10.3389/fphys.2016.00024.
Bian X, Ma K, Zhang C, Fu X. Therapeutic angiogenesis using stem cell-derived extracellular vesicles: an emerging approach for treatment of ischemic diseases. Stem Cell Res Ther. 2019. https://doi.org/10.1186/s13287-019-1276-z.
Acknowledgements
Authors also acknowledge the electron microscopy platform of CIC bioGUNE and ICTS “NANBIOSIS” (Drug Formulation Unit, U10) for their contribution.
Funding
This research was funded by the Basque Government (Consolidated Groups, IT907-16 and PRE_2018_1_0412).
Author information
Authors and Affiliations
Contributions
RMH, JMFP, ESV, KLEH and FR contributed to methodology and conceptualization; KLH, FR and CGV contributed to investigation; KLH and FR contributed to data analysis and curation; KLH and FR contributed to writing—original draft; ESV, RMH, MI, FR and JMFP contributed to correction and edition of the manuscript; and RMH, MI and JMFP contributed to funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Hair follicles were obtained from occipital scalps of routine hair transplant procedures with the application of FUE technique (Clínica Dermatológica Ercilla, Spain). All patients signed an informed consent under an approved protocol (M10_2019_053, M30_2019_054) by the research ethics committee of the Basque Country.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Las Heras, K., Royo, F., Garcia-Vallicrosa, C. et al. Extracellular vesicles from hair follicle-derived mesenchymal stromal cells: isolation, characterization and therapeutic potential for chronic wound healing. Stem Cell Res Ther 13, 147 (2022). https://doi.org/10.1186/s13287-022-02824-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-022-02824-0