
Abstract
The adoptive transfer of natural killer (NK) cells is an emerging therapy in the field of immuno-oncology. In the last 3 decades, NK cells have been utilized to harness the anti-tumor immune response in a wide range of malignancies, most notably with early evidence of efficacy in hematologic malignancies. NK cells are dysfunctional in patients with hematologic malignancies, and their number and function are further impaired by chemotherapy, radiation, and immunosuppressants used in initial therapy and hematopoietic stem cell transplantation. Restoring this innate immune deficit may lead to improved therapeutic outcomes. NK cell adoptive transfer has proven to be a safe in these settings, even in the setting of HLA mismatch, and a deeper understanding of NK cell biology and optimized expansion techniques have improved scalability and therapeutic efficacy. Here, we review the use of NK cell therapy in hematologic malignancies and discuss strategies to further improve the efficacy of NK cells against these diseases.
Similar content being viewed by others
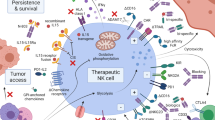

Background
Natural killer (NK) cells are cytotoxic lymphocytes that play a key role in recognizing malignant and virus infected cells and serve as a bridge between the innate and adaptive immune response. In hematologic malignancies, there is a qualitative and quantitative dysfunction of innate NK cells and defective NK cells at diagnosis portends a poor prognosis [1, 2]. For example, NK cell phenotypes at diagnosis of acute myeloid leukemia (AML) can be stratified into highly functional and dysfunctional groups with distinct transcriptional modifications in pathways involved in cytotoxicity, intracellular signaling, and metabolism [3]. Patients with this defective NK cell profile at diagnosis had a higher risk of relapse. In addition, patients with “hypomaturation” NK cell profiles have reduced overall and relapse-free survival [4, 5]. While the majority of clinical data supporting adoptive NK cell therapy to date is in adult myeloid malignancies, there is evidence to support the use of NK cells across a broad range of hematologic cancers, including multiple myeloma (MM). Pre-clinical data has been published for B and T cell lymphoblastic leukemia, non-Hodgkin lymphoma (NHL), and Hodgkin lymphoma (HL) [6,7,8]. In addition, while the graft-versus-leukemia (GVL) effect is historically thought to be more important in AML, there is mounting evidence that post-transplant immune recovery and NK cell alloreactivity confers lower risk of relapse in pediatric ALL and NHL [9,10,11,12]. Early clinical success and demonstration of safety of adoptive NK cell therapy in hematologic malignancies has led to more widespread use. As we learn more about the biology of NK cells and NK cell dysfunction in cancer, new strategies for successful NK cell therapy are emerging. This review focuses on the use of NK cell therapy to date in hematologic malignancies as well as barriers to success and future directions.
Advantages and early success of NK cell therapy
Although the ability of NK cells to recognize and kill leukemia cells was described 45 years ago, a clinically relevant role for NK cells in the treatment of leukemia was first demonstrated nearly 20 years later [13,14,15,16]. Early NK cell recovery after stem cell transplant and increased NK cells in the graft are associated with improved transplant outcomes in leukemia [17,18,19]. Additional evidence of NK cell-mediated GVL was demonstrated in the setting of HLA-mismatched hematopoietic stem cell transplant (HSCT) [20, 21]. Ruggeri et al. observed that patients with AML undergoing haploidentical HSCT had decreased relapse rates when HLA differences between the donor and recipient were present in the GVL direction in a missing-ligand model for NK cells [20]. This concept was termed “ligand–ligand mismatch” and similar studies confirmed the importance of NK cell alloreactivity in AML patients undergoing HSCT [20, 22,23,24]. Similarly, decreased relapse and increased survival were seen in patients receiving HLA-mismatched transplants in which the donor-recipient pair was also mismatched for KIR genes [25,26,27].
Supported by this early clinical evidence, adoptive NK cell therapy to augment the GVL effect was investigated. The earliest trials were performed with NK cells isolated from healthy donor leukapheresis products using immunomagnetic cell selection and overnight IL-2 activation [28,29,30]. Using this approach, Miller et al. demonstrated that infusion of haploidentical NK cells after chemotherapy could induce remission of poor-prognosis AML [31]. In a similar study, Rubnitz et al. reported the safety of KIR-mismatched NK cell infusion as post-remission consolidation therapy for children with AML, with no relapses reported in the 10 patients treated [32]. A similar approach has been used for adoptive transfer of NK cells in patients with refractory lymphoma and MM [33, 34]. Importantly, GVHD was not reported in any of these studies utilizing allogenic NK cells. Other studies using NK cells derived by this approach in the allogeneic HSCT setting in patients with lymphoid and myeloid malignancies have also demonstrated that NK cell infusions were safe and not associated with severe infusion reactions, GVHD, or graft rejection [35,36,37,38]. However, the response rates in these studies were variable (OS from 29% to 73%) and the NK cell doses produced by this approach were typically limited to a single dose of ≤107 /kg.
Advancements in NK cell sources and expansion methods have improved the potential for NK cell therapy by enabling repeated dosing with larger numbers of NK cells (Fig. 1). Expansion methods have been developed that use cytokines alone, or in combination with costimulatory antibodies or agonists [39, 40]. The development of irradiated feeder cells generated from autologous mononuclear cells, EBV-transformed lymphoblastoid cell lines, or tumor cell lines has led to improved NK cell maturation and proliferation ex vivo [41,42,43,44,45,46,47,48]. Feeder cells have also been genetically modified to express membrane-bound cytokines and co-stimulatory molecules such as 4-1BB, MICA, IL-15, and IL-21, which can result in > 1000-fold expansion over a period of weeks. NK cell growth may plateau, however, because of exhaustion and/or proliferative senescence attributed to shortened telomeres [46, 49, 50]. In our experience with irradiated K562 feeder cells expressing 4-1BB and membrane bound IL-21, the IL-21 leads to STAT3-mediated induction of telomerase reverse transcriptase and increased telomere length. NK cells expanded with this method are highly functional and do not show proliferative senescence, achieving an average 3000-fold expansion in 2 weeks and 20–80,000-fold in 3 weeks [7, 44].

Sources for expanded NK cells. Current NK cell therapies are derived from peripheral blood (PB) NK cells, umbilical cord/placental NK cells, NK cell lines, or induced pluripotent stem cells (iPSC). Isolated NK cells are expanded utilizing cytokine stimulation with or without the presence of feeder cells. Yellow lightning bolts indicate timepoints utilized for genetic alteration of the final expanded NK cell product
There have also been significant advances utilizing alternative allogeneic NK cell sources such as the umbilical cord blood, NK-like leukemia/lymphoma cell lines, and stem cell-derived NK cells, and there are advantages and disadvantages to each method (Fig. 1). While absolute lymphocyte numbers are low in UCB, it is relatively rich in NK cells compared to the peripheral blood, comprising 20–30% of the CB lymphocyte population [51]. The volume of the cord blood is limited, however, and freshly isolated CB NK cells are phenotypically immature and less cytotoxic with low expression of CD16 and activating receptors [51]. These quantitative and qualitative limitations of CB NK cells, however, can be overcome with cytokine stimulation and expansion, and expanded CB NK cells are highly functional against tumor targets [52,53,54]. A phase 1 study of cord blood-derived NK cells combined with autologous SCT in MM demonstrated the safety of NK cell doses up to 1 × 108/kg with no dose-limiting toxicity, no GVHD, and NK cells detectable in the peripheral blood for up to 26 days [55].
The NK cell lymphoma cell line, NK-92, has high proliferative capacity in culture, exerts anti-tumor cytotoxicity and is easily genetically modified using viral or non-viral transduction techniques [56]. While the infusion of a malignant cell line has the theoretical potential to cause malignancy itself, irradiation of the NK-92 cells prevents proliferation in vivo and there have been clinical trials demonstrating the safely of this method [57,58,59,60]. However, this limits in vivo persistence and eliminates in vivo expansion. Finally, there has been extensive study optimizing the generation of functional NK cells from embryonic stem cells and induced pluripotent stem cells (iPSC) [61, 62]. The advantages of this method include a highly uniform, standardized, end product, and relative ease of genetic engineering of these cells [63].
Clinical trials utilizing expanded NK cells have shown a high degree of safety and early evidence of benefit in both the transplant and non-transplant settings. Haploidentical IL-21-expanded NK cells delivered in the setting of haploidentical transplant with post-transplant cyclophosphamide led to a dramatic reduction in post-transplant relapse in patients with myeloid malignancies. Compared to historical matched controls, delivery of three infusions of NK cells (on days − 2, + 7, and +28) reduced leukemia relapse from 35 to 5%, respectively [64, 65]. Expanded NK cells have also been used safely in the setting of autologous SCT for MM in combination with lenalidomide [55]. Outside of the transplant setting, expanded NK cells in combination with chemotherapy yielded a 69% complete response rate in a phase I study of patients with relapsed/refractory AML [66]. In these early clinical trials, NK cells have been safe with no dose-limiting toxicity, cytokine release syndrome, or increase in rates of GVHD. Given these initial reports demonstrating safety and efficacy, there are currently dozens of clinical trials exploring the use of NK cells for hematologic malignancies.
Barriers to NK cell success and solutions
Pharmacokinetics: expansion and persistence of NK cells in vivo
In contrast to T and B cells that can persist for months to years, innate immune cells are believed to be relatively short-lived, lasting for days to weeks [67]. In addition to the already finite lifespan of NK cells, donor NK cells face the additional challenge of allo-rejection. The importance of cell persistence for the success of CAR T cell therapy in leukemia is well described [68]. Similarly, in vivo expansion and persistence of NK cells after adoptive transfer is likely to be important for clinical efficacy. Grzywacz et al. demonstrated that increased NK cell homing and persistence in the bone marrow 2–3 weeks after NK cell infusion for relapsed AML was important for clinical response [69]. Strategies to improve persistence and in vivo expansion of NK cells after adoptive transfer include pre-infusion lymphodepleting therapy, co-administration of cytokines, and repeated NK cell infusions. Recent studies revealing the ability of NK cells to develop immunological memory clearly demonstrate that the function and persistence of NK cells is enhanced by tumor cell or cytokine priming [70,71,72,73,74]. Cytokine primed or expanded NK cells, however, may become “addicted” to cytokine stimulation and undergo apoptosis upon cytokine withdrawal [75]. This highlights the need for endogenous or exogenous cytokine stimulation after adoptive transfer to facilitate persistence.
Early clinical trials with IL-2 activated haploidentical NK cells utilized lymphodepleting chemotherapy with cyclophosphamide and fludarabine as well as subcutaneous IL-2 to facilitate NK cell expansion and persistence [31, 32]. Miller et al. demonstrated that adequate lymphodepletion with high-dose cyclophosphamide and fludarabine led to in vivo NK cell expansion with persistence of donor NK cells for over 28 days in some patients [31]. There was an inverse correlation between lymphocyte count and endogenous serum IL-15 after lymphodepleting chemotherapy suggesting that successful NK cell expansion was secondary to both clearing of alloreactive recipient cells and IL-15 stimulation of NK cells. The administration of exogenous IL-2 not only promotes NK cell priming and proliferation but also selectively targets regulatory T cell expansion and is utilized to promote immune tolerance in autoimmune disease such as type 1 diabetes [76, 77]. The immunomodulatory effect of regulatory T cell expansion in response to exogenous IL-2 in cancer is not clear, however, and it is worth noting that the adoptive transfer of NK cells alone may increase the proportion of Tregs [78]. IL-15 stimulates CD8+ T cells, has a critical role in NK cell development, and promotes NK cell survival via expression of the anti-apoptotic factor Bcl-2 [79]. In addition to its role in NK cell development, IL-15 is critical for the survival of mature NK cells in vivo [79]. Using IL-15 in lieu of IL-2 in the setting of haploidentical NK-cell therapy led to improved NK cell expansion in vivo, however, still came with the unwanted side effects of cytokine release syndrome, neurologic toxicity, and NK cell exhaustion [80]. The IL-15 super agonist, ALT-803, was designed to extend the cytokine half-life and mimic the physiologic cell to cell trans-presentation of IL-15. A phase I trial of ALT-803 in relapsed AML/MDS post-transplant led to a more robust NK cell expansion than the traditional recombinant human IL-15 and a more tolerable toxicity profile with no cytokine release syndrome, GVHD, or other dose-limiting toxicities reported [81]. Clinical efficacy as monotherapy in this setting was limited, however, with only one patient out of 27 achieving a complete response. Clinical trials utilizing ALT-803 in combination with adoptive NK cell therapy for AML are underway (NCT02890758, NCT03050216). To overcome the issues with toxicity of exogenous cytokines, others have engineered NK cells to express novel receptors to enhance proliferation and function. Hematopoietic growth factors used to stimulate the erythropoietin receptor (EPOR) or thrombopoietin receptor (c-MPL) have demonstrated clinical safety. Viral transduction of NK-92 cells to express EPOR or c-MPL led to increased NK cell survival and cytotoxicity in response to TPO or EPO ligands [82].
Another strategy for improving NK cell persistence and clinical efficacy is repeated infusions of donor NK cells. While previous studies were limited by small NK cell numbers harvested from donor the peripheral blood, newer strategies for ex vivo NK cell expansion have allowed for repeated doses of high numbers of NK cells, reducing the need for in vivo expansion. Importantly, these NK cells also have a highly functional phenotype with improved cytotoxicity and cytokine secretion against a variety of cancers. NK cells expanded with IL-21-expressing irradiated feeder cells are highly metabolically active with a memory-like NK cell phenotype and increased expression of activating receptors and chemokine receptors associated with NK cell trafficking and persistence [83]. The persistence of expanded NK cells after adoptive transfer has not been extensively studied; however, a mouse model of IL-21 expanded NK cells suggests that expanded NK cells can survive for up 21 days without the need for exogenous cytokine stimulation [84]. Adoptive transfer at day 7 post-transplant correlated with increased number, function, and phenotype at day 28, suggestive of persistence [64]. These data suggest that in addition to improved NK cell numbers, expanded NK cells have anti-leukemic efficacy and potential for in vivo expansion and persistence.
Particularly in patients with urgent medical need such as in relapsed acute leukemias, an important consideration for adoptive NK cell therapy is the fast turnaround time needed to generate the therapeutic product. In this patient population, the time it takes to work up a donor, collect, and expand NK cells may allow for the leukemia tumor burden to grow out of control. In addition, NK cells from cancer patients are low in number and function and demonstrate limited expansion, providing insufficient numbers for effective autologous therapy [85]. For these reasons, the development of an allogenic NK cell bank for “off-the-shelf” therapy is desirable. Approaches include utilizing cord blood NK cells, iPSC-derived NK cells, and unrelated “optimal” donor peripheral blood NK cells. The latter strategy identifies donors who have HLA and KIR genotypes for optimal education, a high proportion of activating KIRs, and who have been exposed to CMV resulting in NKG2C+ “memory-like” NK cells. We have developed a universal-donor NK cell bank utilizing these “optimal” donors in collaboration with Be-the-Match Biotherapies (BTMB), and expanded NK cells from this bank are being utilized in a clinical trial for relapsed AML/MDS (NCT04220684). The use of these off-the-shelf NK cell sources will become increasingly important in the development of genetically engineered NK cell programs.
NK cell recognition of tumors: aberrant receptor/ligand expression
One advantage of NK cell therapy is the ability of NK cells to recognize tumors without the need for antigen presentation. In contrast to T cells, NK cell activation is tumor antigen-independent and is instead regulated by a balance of activating and inhibitory NK cell receptor signaling. Activating receptors recognize ligands on the surface of cancer or viral infected cells that signal danger, and inhibitory receptors are responsible for recognition of self. NK cell receptor classes include natural cytotoxicity receptors (NCR), C-type lectin receptors, and killer cell immunoglobulin-like receptors (KIRs). The presence of NK cell activating receptor stress ligands on the surface of tumor cells is crucial for NK cell recognition of these cells as abnormal. NK cell dysfunction via altered activating receptor expression or tumor downregulation of NK cell receptor ligands is a common method of tumor immune escape. For example, the absence of NKG2D and other NCR ligands on leukemic blasts allows them to escape NK cell surveillance [86, 87]. The DNAM-1 receptor/ligand axis is altered in patients with AML with both downregulation of DNAM-1 receptors on NK cells and low expression of DNAM-1 ligands (CD112/155) on AML clones leading to poor NK cell conjugation and killing [88, 89]. NK cells from patients with hematologic malignancies exhibit low expression of activating NCRs, including NKp46, DNAM-1, and NKG2D which impairs their effector function and predicts poor response to therapy [8, 90,91,92].
To address this, monoclonal antibodies to block inhibitory KIRs or stimulate NK cell activating receptors can tip the inhibition/activation balance in favor of NK cell activation. Inhibitory KIRs recognize HLA molecules, are distinguished by the number of extracellular immunoglobulin domains (2D or 3D), and are assigned an “L” to indicate that they have a long cytoplasmic tail containing immunoreceptor tyrosine-based inhibitory motifs (ITIMs). IPH2101 is an anti KIR2DL antibody that is being studied in AML, MM, B cell lymphoma with pre-clinical, and early clinical evidence of efficacy—particularly when combined with lenalidomide [93,94,95]. A follow-up phase II study in MM failed to demonstrate clinical efficacy, which may be secondary to blockade of normal NK cell education or licensing through inhibitory KIRs [96, 97]. Clinical trials utilizing fully licensed, expanded NK cells in combination with IPH2101 may elicit improved responses.
Stimulation of activating receptors is another way to tip the balance toward NK cell activation, and as mentioned above, tumors often downregulate NK cell receptor ligands to escape immune surveillance. NKG2D is a one of the most important activating receptors in the NK cell repertoire and recognizes cellular stress ligands MICA, MICB, and ULBP1-6. NKG2D also serves as a co-stimulatory receptor on cytotoxic T cells. Synthetic activation of the NKG2D receptor via NKG2D ligand and antitumor antibody fusion or NKG2D ligand/cytokine fusion is one way to overcome immune escape and facilitate NK cell-tumor interaction and may also serve a dual function in activation of T cells [98,99,100].
A key pathway responsible for NK cell recognition of tumors is via CD16 receptor recognition of antibody coated targets in a process called antibody dependent cellular cytotoxicity (ADCC). Binding of the crystallizable fragment (Fc) of IgG to the Fcγ receptor III (FcγRIIIa/CD16a) on NK cells creates a bridge between the NK cell and the tumor cell and leads to NK cell-mediated tumor lysis. This is an important mechanism of therapeutic efficacy for some anti-tumor monoclonal antibodies. In hematologic malignancies, rituximab (anti-CD20) and daratumumab (anti-CD38) are NK-cell-dependent antibodies that are widely utilized for B cell malignancies and MM, respectively. Combining monoclonal antibodies with adoptive NK cell therapy may further enhance tumor recognition by expanded NK cells.
It is worth noting the limitations of monoclonal antibodies and potential technologies to improve them. First, individual genetic polymorphisms of the CD16a have variable affinity to bind IgG [101, 102] and patients with the low affinity CD16 receptor may have suboptimal clinical responses to monoclonal antibody therapy [103]. In addition, the success of monoclonal antibody therapy is dependent on functional effector NK cells. With these concepts in mind, novel antibody constructs to simultaneously enhance the NK cell-tumor cell immune synapse and increase NK cell numbers are in development. Bi-specific and tri-specific killer engagers (BiKEs and TriKEs) are small molecule constructs composed of a single-chain variable fragment (scFv) (comprised of the heavy and variable light chains of an antibody connected by a short peptide) of one antibody linked to another scFv and/or cytokine. Most of the BiKEs/TriKEs in development include a high affinity anti-CD16 component to overcome the polymorphism differences in CD16 affinity mentioned above. The anti-CD16 component is combined with one or two tumor antigen-specific antibodies such as CD19/20 in B cell malignancies [104,105,106,107], CD33/CD123 in AML [108,109,110], CD30 in Hodgkin’s lymphoma [111], and HLA class II in lymphoma [112]. The newer generation of TriKEs incorporate cytokine stimulation to further enhance NK cell function upon antigen engagement. For example, Felices et al. engineered a TriKE that is composed of anti-CD16, anti-CD33, and an IL-15 moiety to drive NK cell activation, expansion, and persistence [113]. Finally, the role of immune checkpoints in NK cell regulation is described below and the addition of inhibitory receptor blockade/checkpoint blockade such as TGF-β inhibition to an NK cell engager may be an additional mechanism to improve NK cell ADCC [114].
Daratumumab is an FDA approved monoclonal antibody against CD38 that has changed the therapeutic landscape for MM with overall response rates of greater than 80% when combined with chemotherapy [115,116,117]. Pre-clinical and clinical reports have indicated that there may also be a role for daratumumab in T cell ALL and other CD19/22-negative hematologic malignancies [118, 119]. NK cells have high levels of CD38 on their surface and are depleted in patients treated with daratumumab as a result of NK-to-NK ADCC, referred to as “fratricide” [120]. CD38 negative or low NK cells are resistant to daratumumab-induced fratricide and have improved tumor cytotoxicity when combined with daratumumab compared to CD38+ NK cells [120]. To overcome NK cell fratricide induced by daratumumab, we generated CD38 knock out NK cells (CD38KO NK) using CRISPR/Cas9. These CD38KO NK cells are resistant to daratumumab-induced fratricide, have a superior metabolic profile, and improved ADCC against CD38 expressing MM [121]. Ongoing pre-clinical validation studies will determine if this method could be utilized across a wide range of CD38 expressing hematologic malignancies.
Combination drug therapies can increase NK cell receptor ligands on the surface of tumor cells and may be another way to improve NK cell recognition of tumors. For example, poly-ADP-ribose polymerase 1 (PARP1) plays a role in repressing expression of NKG2D ligands on AML cells and PARP inhibitors may be a therapeutic target to increase ligand expression and improve NK cell detection of leukemia stem cells [86]. In addition to improved NK cell receptor-ligand recognition, NK cells have improved cytotoxicity in combination with the PARP inhibitor olaparib against the breast, lung, and prostate carcinoma cells [122]. PARP inhibitors are also being utilized in myeloid malignancies, and their combination with adoptive NK cell therapy may further improve therapeutic efficacy.
Lenalidomide is an effective immunomodulatory treatment for MM, other B cell neoplasms, and MDS. In the context of NK cells, lenalidomide decreases the threshold for NK cell activation upon receptor stimulation, enhances antibody dependent cytotoxicity (ADCC), and upregulates receptor and ligand expression on NK cells and tumor cells, respectively [123,124,125]. Combination trials with lenalidomide and monoclonal antibodies have been promising in B cell lymphomas and MM, in part due to improved NK cell recognition of tumor targets via CD16 leading to ADCC [116, 126,127,128,129].
Finally, with the success of CAR T cell therapy in ALL and B cell lymphomas, there is a major push in the field to develop CAR-NK cell therapies. While T and NK cell effector functions are similar, a CAR NK cell has the added ability to recognize tumors through innate NK cell receptors, potentially preventing relapse due to antigen escape. Additionally, allogenic HLA-mismatched NK cells have been given safely without causing GVHD, highlighting the potential to produce universal donor or “off-the-shelf” CAR-NK cells to circumvent cost and timing constraints seen with manufacturing CAR-T cell therapy. Finally, NK cells are safe and cytokine release syndrome and neurologic toxicity have been minimal in NK cell trials to date. Historically, genetic modification of NK cells was unsuccessful due to NK cell resistance to viral transduction. In contrast to T cells, the innate function of NK cells as our first anti-viral defense renders them relatively resistant to traditional methods of gene modification through viral transduction. Viral transduction of peripheral blood NK cells with lentiviral vectors using a new baboon pseudo type has significantly increased high-transduction efficiency [130]. In addition, successful retroviral transduction has been demonstrated in the first phase I/II clinical trial utilizing cord blood derived CAR-NK cells, in which 11 patients with CLL or NHL were treated with a single dose of “off-the-shelf” CD19 CAR-NK cells [131]. The CAR-NK cells were equipped with CD19 CAR, IL-15, and an inducible caspase suicide gene. CAR-NK cells were well tolerated with no dose-limiting toxicity and no report of cytokine release syndrome with a response rate of 73%. CAR-NK cells expanded in vivo and were detectable for a least a year after infusion. Similar to data reported in CAR-T cell trials, patients who responded to therapy had a higher peak expansion of CAR-NK cells than those with no response. Alternative NK cell sources and newer methods of genetic engineering have enabled successful genetic modification of NK cells using non-viral methods. Non-viral transfection with electroporation and the use of transposons and CRISPR/Cas9 has led to improved efficiency and stable genomic insertion of NK cells [132]. Additional CAR NK cell targets being developed for hematologic malignancies include CD33, CD38, CD123, CD20, BCMA, CLL1, and FLT3 [58, 130, 133,134,135,136,137,138], and dual-target CARs such as CD19/20 or CD19/22.
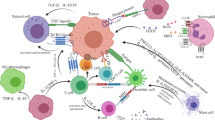
NK cell dysfunction within the tumor micro-environment
Hypoxia
Hypoxia in the TME drives angiogenesis and facilitates cancer progression and chemotherapy resistance largely through induction of the hypoxia-inducible factor (HIF) pathway as well as the PI3K/AKT/mTOR and NFκB pathways [139]. Within the bone marrow niche, pockets of hypoxia are essential for physiologic stem cell function and severe hypoxic conditions develop in the progression of hematologic malignancies including MM, leukemia, and lymphoma via similar mechanisms as solid tumors [140]. The anti-tumor immune response is dysfunctional under hypoxic conditions leading to T cell apoptosis, NK cell dysfunction, and promotion of Treg differentiation which further facilitates tumor survival [141, 142]. NK cell cytolytic function is impaired in hypoxic environments in part via decreased surface expression of activating receptors such as NKp46, NKp30, and NKG2D and CD16 [143,144,145]. Even after NK cell recognition of tumor cells, hypoxia-induced autophagy in cancer cells leads to degradation of granzyme B, rendering NK cells in hypoxic tumor microenvironments less cytotoxic [146].
NK cell dysfunction in the TME is also due to overexpression of the adenosine nucleotidase, CD73. Increased CD73 expression leads to extracellular accumulation of adenosine which has significant immunometabolic effects on NK cells [147] (Fig. 2). Adenosine stimulation of the A2A receptor on NK cells leads to negative effects on NK cell metabolism, cytokine production, and cytotoxicity [147]. A2AR blockade can restore NK cell function in the tumor environment [148] and targeting of the CD73/adenosine pathway may serve to boost the anti-tumor immune response as well as suppress tumor stem cell function [149].

Hypoxia. Hypoxia driven upregulation of CD73 leads to increased adenosine binding at the A2A receptor (A2AR). The A2AR inhibits NK cell function through the SOCS pathway via downregulation of activating receptors. Increased expression of the hypoxia inducible factor 1-alpha (HIF1α) in leukemic blasts results in upregulation of ADAM10 and subsequent cleavage of MIC-A—the canonical ligand for the NK cell activating receptor NKG2D
In addition to intrinsic NK cell dysfunction, hypoxia can also lead to tumor cell immune evasion via upregulation of checkpoint molecules and downregulation of NK cell ligands [150, 151]. The NKG2D ligand MICA is downregulated on tumor cells in the setting of hypoxia via HIF-1α induced expression of ADAM10 [150, 152] (Fig. 2). HIFs have also been shown to induce PD-L1 expression on the surface of tumor cells via interaction with PD-L1 gene promotors [151, 153]. Therapeutic blockade of the transcription factor HIF1 or ADAM10 may overcome hypoxia-induced dysfunction of NK cells [154].
Checkpoints (PD-1, TIM-3, TIGIT)
In addition to HLA class I-specific inhibitory receptors, NK cells express traditional immune checkpoint molecules such as PD-1, TIM-3, and TIGIT [155] (Fig. 3). While TIM-3 and TIGIT are expressed in healthy donor NK cells and are likely involved as conventional checkpoints for NK cells, the role of traditional T cell checkpoints like PD-1 in NK cell immune tolerance is not as well defined [156].

Immune checkpoints. Binding of leukemic cell-secreted galectin-9 at TIM-3, PD-L1 at PD-1, or TIGIT ligand at the TIGIT receptor inhibits NK cell cytotoxicity. Similarly, tumor cell expression of MHC Class 1 molecules leads to NK cell tolerance through interaction with inhibitory KIRs. Blockade of these immune checkpoint pathways reverses NK cell dysfunction in the tumor microenvironment
TIM-3 is constitutively expressed on NK cells from both healthy donors and cancer patients, and high expression of TIM-3 on NK cells has prognostic significance in solid tumors [157, 158]. In hematologic malignancies, Galactin-9 is overexpressed by tumor cells and the Tim-3/galactin-9 autocrine loop can lead to leukemic progression conferring an additional selective advantage to tumor expression of TIM-3 ligands [159]. High levels of Galectin-9 were seen in plasma of patients with AML and Galectin-9 secreted by AML blasts impairs NK cell killing of leukemic cells in culture [160]. Blockade of TIM-3 on NK cells isolated from cancer patients reversed tumor-associated NK cell exhaustion and restores cytolytic function [161].
PD-1 is only variably expressed on subsets of healthy donor NK cells but appears to be upregulated in tumor-associated NK cells [162]. Similar to its effect on T cells, stimulation of PD-1 on NK cells leads to impaired cytokine release and degranulation that may be secondary to impaired lytic granule polarization [155]. In HL and DLBCL, PD-1+ is elevated in both circulating and intra-tumoral NK cells compared to healthy controls [163]. In vitro PD-1 blockade with pembrolizumab enhances NK cell cytotoxicity via direct blockade of PD-1 on NK cells and indirectly by PD-1 inhibition of NK cell suppressive tumor-associated macrophages [163]. In addition, PD-L1 blockade may actually directly enhance PD-L1+ NK cell function and provide anti-tumor efficacy even in PD-L1 negative tumors [164]. Dong et al. demonstrated enhanced degranulation and cytokine production from PD-L1+ NK cells treated with an anti-PD-L1 antibody. PD-1/PD-L1 blockade is also being studied in AML, and multiple studies have reported high expression of PD-L1 on AML blasts [164]. While early clinical trials have hinted at efficacy of checkpoint blockade in AML/MDS, the role of NK cells in clinical responses to checkpoint blockade has yet to be defined [165, 166].
The co-inhibitory receptor T cell immunoglobulin and ITIM domain (TIGIT) is also expressed on NK cells [167]. NK cells isolated from patients with NHL have high expression of TIGIT compared to healthy controls [168]. Zhang et al. found high TIGIT expression on tumor infiltrating NK cells that exhibited an exhausted phenotype with impaired cytokine secretion and degranulation [169]. NK-cell specific deficiency of TIGIT prevented tumor metastasis and improved survival in a mouse model of melanoma. Blockade of TIGIT in vivo reversed the exhaustion of tumor infiltrating NK cells and slowed tumor growth, even in a T cell deficient SCID mouse model. Importantly, the therapeutic effects of anti-TIGIT therapy depended on the presence of NK cells. In a pre-clinical model of autologous stem cell transplant for MM, TIGIT blockade significantly prolonged disease control after transplant [170] and is currently being utilized in this clinical setting (NCT04150965).
Indoleamine 2,3-dioxygenase
Indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO) are intracellular enzymes responsible for tryptophan breakdown to kynurenine. Through stimulation of the aryl hydrocarbon receptor (AhR), the IDO/TDO/Kynurenine pathway promotes immune tolerance in the tumor microenvironment via suppression of NK cells and cytotoxic T cells and promotion of regulatory T cells [171] (Fig. 4). The role of tryptophan catabolism in cancer development and progression is an active area of research; however, it is clear that IDO is overexpressed in many different cancer types, including hematologic malignancies [172]. Functional IDO is overexpressed by AML blasts which correlates with increased regulatory T cells and predicts a poor prognosis [172,173,174]. IDO is also overexpressed Hodgkin and NHL and IDO expression in tumor tissue of DLBCL can stratify patients at risk for chemotherapy resistance and decreased survival [175,176,177,178,179]. In Hodgkin lymphoma, macrophages, dendritic cells, and endothelial cells in the TME express IDO and high IDO expression is associated with high-risk features with 5-year overall survival of 67.8% compared to patients with low IDO expression who have an OS of 91.7% [178]. Karihtala et al. found that although the percentage of tumor associated macrophages expressing IDO in HL samples was low, and high IDO expression was an independent poor prognostic factor [179]. Targeting the IDO/TDO/Kyurenin pathway is of clinical interest given the broad implication in tumor development and IDO/TDO inhibitors are actively being studied in the clinic. Selective IDO inhibitors, however, fail to prevent tryptophan metabolism by the TDO pathway and vice versa so targeting the downstream AHR pathway may be more efficient [180]. In AML, upregulation of AHR ligands by blasts hinders NK cell development and function via the micro-RNA, miR-29a/b1 [181]. The use of AHR antagonists has the potential to restore NK cell development and improve NK cell killing of AML blasts.

IDO. The intracellular enzyme Indoleamine 2,3-dioxygenase (IDO) catalyzes tryptophan to kynurenine, a ligand for the aryl hydrocarbon receptor (AHR). Upon kynurenine binding, AHR crosses the nuclear membrane and associates with the AHR nuclear translocator protein (ARNT). The AHR-ARNT complex binds to DNA promotor regions leading to differential expression of genes associated with immune tolerance of malignancy. In NK cells, AHR-ARNT activates transcription of miR-29b and inhibits NK cell maturation and function
Transforming growth factor-Beta
A key contributor to immunosuppression within the tumor microenvironment is transforming growth factor-beta (TGF-β) secreted by tumor cells and tumor-associated macrophages. The TGF-β family signaling pathway exerts diverse biological effects depending on the cell type and physiologic context and includes effects on cell proliferation, differentiation, communication, metabolism, and apoptosis. Within the context of cancer, TGF-β can have pro- and anti-tumorigenic effects functioning both as a tumor suppressor in pre-malignant cells and as a tumor promotor of cancer cell growth and metastasis [182]. In addition to direct effects on malignant cells, TGF-β acts as an immunosuppressive cytokine that inhibits T, B, and NK cell function [182]. Specifically, TGF-β-induced phosphorylation of SMAD2/SMAD3 in NK cells leads to decreased IFNγ production [183] and decreased anti-tumor cytotoxicity with phenotypic downregulation of the activating receptors NKG2D, NKp30, DNAM-1, TRAIL, and CD16 [184,185,186,187,188] (Fig. 5). The effect of TGF-β driven immune escape is well studied in pediatric and adult solid tumors [189,190,191,192,193,194,195,196,197]. In hematologic malignancies, aberrant TGF-β-SMAD signaling is implicated in ineffective hematopoiesis and leukemogenesis and evidence of TGF-β-mediated immune escape in leukemia/lymphoma is emerging [198,199,200].

TGF-β. TGF-β secreted by leukemic blasts binds to the TGF-β receptor on NK cells leading to phosphorylation, trimerization, and translocation of SMAD complexes across the nuclear membrane. The downstream epigenetic modifications inhibit NK cell function with decreased cytokine production, impaired cytotoxicity, and downregulation of NK cell activating receptors (NKG2D, NKp30, DNAM-1, TRAIL, and CD16)
Huang et al. investigated the effect of TGF-β on NK cell targeting of leukemia cells in vitro. The authors found that TGF-β significantly decreased NK cell killing of leukemia cell lines secondary to leukemia cell downregulation of the NK cell ligand CD48 as well as decreased ICAM-1 binding affinity resulting in impaired effector-target interaction [201]. In pediatric B-ALL, NK cell number and cytolytic function are significantly reduced at diagnosis compared to healthy controls [202]. Rouce et al. demonstrated that NK cells from the leukemia patients had an abnormal phenotype with increased expression of the inhibitory receptor NKG2A and reduced expression of the activating receptor NKp46 [202]. The authors found increased levels of TGF-β in the supernatants of ALL blast cultures and showed significant elevation in SMAD2/3 phosphorylation in NK cells isolated from patients with leukemia as well as NK cells co-cultured with ALL blasts. Importantly, blockade of TGF-β partially corrected the ALL-induced NK cell dysfunction highlighting a potential therapeutic target. In AML, defective NK cells at diagnosis are associated with increased risk of relapse and transcriptional analysis shows differential expression of TGF-β signaling pathways between highly functional NK cells and dysfunctional NK cells from patients with leukemia [3]. Decreased number and function of NK cells in the TME of lymphoma predicts a poor prognosis [203]. Similar to other solid tumors, TGF-β is expressed at high levels by both lymphoma cells and regulatory T cells within the lymphoma tumor microenvironment which is likely one of the immune escape mechanisms employed by lymphomas [204, 205]. Taken together, it is clear that similar to solid tumors, TGF-β plays a substantial role in tumor progression and immune evasion in hematologic malignancies.
Therapeutic antibodies and small molecule inhibitors targeting the TGF-β pathway are in development, but progress has been slow and focused primarily on solid tumors such as glioblastoma, pancreatic cancer, NSCLC, and hepatocellular carcinoma [206]. Galunisertib is a first in class oral inhibitor of the TGF-β receptor type 1 kinase that has shown some clinical efficacy as monotherapy or in combination with standard of care therapies [206]. In a phase II/III trial using galunisertib in low-intermediate risk MDS, 32% of transfusion-dependent patients had hematologic improvement with an acceptable safety profile. Interestingly, > 90% of patients had a > 20% reduction in plasma TGF-β levels and the authors found an increase in NK cell numbers during treatment with galunisertib. The use of galunisertib in combination with ex vivo expanded NK cells with antibody therapy reversed the TGF-β-induced suppression of cytotoxicity and led to reduction of tumor growth and improved survival in patient-derived xenografts of neuroblastoma [207]. Similar combination therapies utilizing TGF-β pathway inhibition combined with adoptive NK cell therapy have not been utilized in clinical trials to date but may enhance NK cell function in vivo.
In addition to direct TGF-β receptor blockade, NK cell engineering strategies have been utilized to overcome TGF-β inhibition of NK cells including TGF-β receptor knock out, the addition of dominant negative TGF-β receptors, and a TGF-β chimeric receptor with an intracellular NK cell activating domain [208,209,210]. Our lab utilized DNA-free Cas9 RNP editing of peripheral blood NK cells to successfully knock out the TGF-β receptor rendering them resistant to TGF-β-mediated suppression [208]. Yvon et al. genetically engineered cord blood NK cells using retroviral transduction to insert a dominant negative TGF-β receptor (DNRII) [210]. These DNRII-expressing NK cells exhibited normal expansion with irradiated feeder cells and had improved cytotoxicity of glioblastoma cells compared to non-transduced NK cells when exposed to TGF-β. Utilizing the same genetic modification platform, NK cells were engineered to express TGF-β receptors coupled with intracellular NK cell-specific activating domains to take advantage of receptor stimulation by TGF-β in the TME. The conversion of an inhibitory signal to an activating signal not only made these NK cells resistant to TGF-β but also led to increased NK cell activation and improved tumor control in a model of TGF-β secreting neuroblastoma [209].
Finally, our lab developed a novel platform using TGF-β stimulation during expansion with IL-2 and irradiated feeder cells. Addition of TGF-β during expansion (TGF-β imprinting) does not affect their proliferation and paradoxically and results in hyperinflammatory NK cells that produce large amounts of IFNγ, TNFα, and GM-CSF when co-cultured with tumor cells even in the presence of TGF-β [211].
Conclusion and future perspectives
Cellular immunotherapy is on the forefront of progress in cancer research. The success of CD19 CAR T cells provided momentum to develop novel cell therapy strategies in both hematologic malignancies and solid tumors. NK cells provide an alternative cell source with a similar effector function but with decreased toxicity and the potential for an “off-the-shelf” model. Specifically, in contrast to T cells, the adoptive transfer of NK cells has not led to cytokine release syndrome or GVHD in the allogenic setting. In hematologic malignancies, innate NK cell tumor function is suppressed by cancer directed therapies and the tumor microenvironment and improving NK cell function is vital to the success of cancer treatment. Historically, most clinical trials of adoptive cell therapy have utilized products that are manufactured on a patient-by-patient basis. The high cost of manufacturing per patient delays in care while awaiting a product and variability in the final cell composition make “off-the-shelf” cell therapy highly desirable. Recent optimization of expansion techniques, validation of the safety and scalability of different NK cell sources, and the ability to safely use donor NK cells with minimal HLA matching will allow for true “off-the-shelf” NK cell therapy. To this end, NK cell banks are being developed and will be a timely and cost-effective way to standardize the use of NK cells in cancer therapy. In the future, restoring both the number and function of NK cells throughout a patient’s treatment course may become standard of care by using NK cells from established banks.
The broad applicability of NK cells stems from their ability to kill tumor targets without antigen presentation and pre-clinical and clinical data supporting the use of NK cells across a wide range of malignancies is being published. Particularly with the ubiquitous use of antibody therapy and the importance of NK cells for ADCC, adoptive transfer of NK cells will likely become a universal way to improve the efficacy of this therapeutic modality. In addition to their anti-tumor function, NK cells are important for viral surveillance and exploiting their natural cytotoxicity against viral infected cells may prove to be an additional benefit of the use of NK cells in immunocompromised patients. In vitro data suggests that NK cells may be effective against viral, bacterial, and fungal pathogens [212]. In the setting of haploidentical HSCT for AML, patients treated with NK cells had lower rates of viral reactivation including CMV and BK virus [64]. Clinical trials evaluating the efficacy of adoptive NK cell therapy for treatment of SARS-CoV-2 are underway and if successful may pave the way toward broad use of NK cells for infection indications.
While there is much optimism for the use of adoptive NK cell therapy in hematologic malignancies, tumor immune evasion remains a barrier to success and strategies to overcome these barriers are actively being investigated (Fig. 6). Specifically, progress made in our ability to genetically engineer NK cells opens up the field to seemingly unlimited therapeutic potential. Novel CAR targets and NK-specific CAR constructs are under development and will be the next major breakthrough for the success of NK cell therapy in hematologic malignancies. With the recent advances discussed in this review, we are on a cusp of an exciting time in the field of adoptive NK cell therapy.

NK cell therapy barriers and solutions. a Strategies to improve in vivo persistence of adoptive NK cells include use of lymphodepleting chemotherapy, repeated NK cell doses, exogenous cytokine stimulation, and cytokine secreting “armored” NK cells. b Tumor cell immune evasion can be overcome by antibodies directed at the tumor antigen to encourage NK cell ADCC, bispecific engagers interacting with NK activating receptors, or engineered chimeric antigen receptors. c Strategies to decrease immune evasion in the tumor microenvironment include (clockwise from the top left) genetic knock out/in of proteins to enhance NK function (ex. CD38 knock out), small molecule inhibitors or immunomodulatory drugs (ex. PARP inhibitors to upregulate NK activating receptors), priming the NK cells ex vivo for preservation of function in vivo (ex. TGFβ imprinting preserving cytolysis and cytokine secretion upon re-exposure to TGF β in the TME), and checkpoint blockade
Availability of data and materials
Not applicable—review manuscript.
Abbreviations
- NK:
-
Natural killer
- AML:
-
Acute myeloid leukemia
- NHL:
-
Non-Hodgkin lymphoma
- HL:
-
Hodgkin lymphoma
- GVL:
-
Graft-versus-leukemia
- HSCT:
-
Hematopoietic stem cell transplant
- NCR:
-
Natural cytotoxicity receptors
- KIR:
-
Killer cell immunoglobulin-like receptors
- PARP1:
-
Poly-ADP ribose polymerase I
- ITIMS:
-
Immunoreceptor tyrosine-based inhibitory motifs
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- Fc:
-
Crystallizable fragment
- BiKE:
-
Bi-specific killer engager
- TriKE:
-
Tri-specific killer engager
- HIF:
-
Hypoxia inducible factor
- IDO:
-
Indoleamine 2,3-dioxygenase
- TDO:
-
Tryptophan 2,3-dioxygenase
- AhR:
-
Aryl hydrocarbon receptor
- TGF- β:
-
Transforming growth factor beta
- DNRII:
-
Dominant negative TGF-β receptor
References
Kiladjian JJ, Bourgeois E, Lobe I, Braun T, Visentin G, Bourhis JH, Fenaux P, Chouaib S, Caignard A. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia. 2006;20(3):463–70. https://doi.org/10.1038/sj.leu.2404080.
Pizzolo G, Trentin L, Vinante F, Agostini C, Zambello R, Masciarelli M, Feruglio C, Dazzi F, Todeschini G, Chilosi M, Veneri D, Zanotti R, Benedetti F, Perona G, Semenzato G. Natural killer cell function and lymphoid subpopulations in acute non-lymphoblastic leukaemia in complete remission. Br J Cancer. 1988;58(3):368–72. https://doi.org/10.1038/bjc.1988.221.
Khaznadar Z, Boissel N, Agaugué S, Henry G, Cheok M, Vignon M, Geromin D, Cayuela JM, Castaigne S, Pautas C, Raffoux E, Lachuer J, Sigaux F, Preudhomme C, Dombret H, Dulphy N, Toubert A. Defective NK cells in acute myeloid leukemia patients at diagnosis are associated with blast transcriptional signatures of immune evasion. J Immunol. 2015;195(6):2580–90. https://doi.org/10.4049/jimmunol.1500262.
Chretien AS, Granjeaud S, Gondois-Rey F, Harbi S, Orlanducci F, Blaise D, et al. Increased NK cell maturation in patients with acute myeloid leukemia. Front Immunol. 2015;6:564.
Chretien AS, Fauriat C, Orlanducci F, Galseran C, Rey J, Bouvier Borg G, Gautherot E, Granjeaud S, Hamel-Broza JF, Demerle C, Ifrah N, Lacombe C, Cornillet-Lefebvre P, Delaunay J, Toubert A, Gregori E, Luche H, Malissen M, Arnoulet C, Nunes JA, Vey N, Olive D. Natural killer defective maturation is associated with adverse clinical outcome in patients with acute myeloid leukemia. Front Immunol. 2017;8:573. https://doi.org/10.3389/fimmu.2017.00573.
Ray AK, Somanchi SS, Dastgheyb N, Aquino-Lopez A, Cobanoglu ZE, Geier B, Lee DA. Expression of carcinoma, apoptosis, and cell-death-related genes are determinants for sensitivity of pediatric cancer cell lines to lysis by natural killer cells. Pediatr Blood Cancer. 2019;66(10):e27783. https://doi.org/10.1002/pbc.27783.
Lee DA. Cellular therapy: adoptive immunotherapy with expanded natural killer cells. Immunol Rev. 2019;290(1):85–99. https://doi.org/10.1111/imr.12793.
Chiu J, Ernst DM, Keating A. Acquired natural killer cell dysfunction in the tumor microenvironment of classic Hodgkin lymphoma. Front Immunol. 2018;9:267. https://doi.org/10.3389/fimmu.2018.00267.
Oevermann L, Michaelis SU, Mezger M, Lang P, Toporski J, Bertaina A, Zecca M, Moretta L, Locatelli F, Handgretinger R. KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood. 2014;124(17):2744–7. https://doi.org/10.1182/blood-2014-03-565069.
Babor F, Peters C, Manser AR, Glogova E, Sauer M, Potschger U, et al. Presence of centromeric but absence of telomeric group B KIR haplotypes in stem cell donors improve leukaemia control after HSCT for childhood ALL. Bone Marrow Transplant. 2019;54(11):1847–58. https://doi.org/10.1038/s41409-019-0543-z.
Yeshurun M, Weisdorf D, Rowe JM, Tallman MS, Zhang MJ, Wang HL, Saber W, de Lima M, Sandmaier BM, Uy G, Kamble RT, Cairo MS, Cooper BW, Cahn JY, Ganguly S, Camitta B, Verdonck LF, Dandoy C, Diaz MA, Savani BN, George B, Liesveld J, McGuirk J, Byrne M, Grunwald MR, Drobyski WR, Pulsipher MA, Abdel-Azim H, Prestidge T, Wieduwilt MJ, Martino R, Norkin M, Beitinjaneh A, Seo S, Nishihori T, Wirk B, Frangoul H, Bashey A, Mori S, Marks DI, Bachanova V. The impact of the graft-versus-leukemia effect on survival in acute lymphoblastic leukemia. Blood Adv. 2019;3(4):670–80. https://doi.org/10.1182/bloodadvances.2018027003.
Bachanova V, Weisdorf DJ, Wang T, Marsh SGE, Trachtenberg E, Haagenson MD, Spellman SR, Ladner M, Guethlein LA, Parham P, Miller JS, Cooley SA. Donor KIR B genotype improves progression-free survival of non-Hodgkin lymphoma patients receiving unrelated donor transplantation. Biol Blood Marrow Transplant. 2016;22(9):1602–7. https://doi.org/10.1016/j.bbmt.2016.05.016.
Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. 1975;16(2):230–9. https://doi.org/10.1002/ijc.2910160205.
Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. 1975;16(2):216–29. https://doi.org/10.1002/ijc.2910160204.
Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. 1975;5(2):117–21. https://doi.org/10.1002/eji.1830050209.
Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5(2):112–7. https://doi.org/10.1002/eji.1830050208.
Savani BN, Mielke S, Adams S, Uribe M, Rezvani K, Yong AS, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia. 2007;21(10):2145–52. https://doi.org/10.1038/sj.leu.2404892.
Yamasaki S, Henzan H, Ohno Y, Yamanaka T, Iino T, Itou Y, et al. Influence of transplanted dose of CD56+ cells on development of graft-versus-host disease in patients receiving G-CSF-mobilized peripheral blood progenitor cells from HLA-identical sibling donors. Bone Marrow Transplant. 2003;32(5):505–10. https://doi.org/10.1038/sj.bmt.1704165.
Kim DH, Sohn SK, Lee NY, Baek JH, Kim JG, Won DI, et al. Transplantation with higher dose of natural killer cells associated with better outcomes in terms of non-relapse mortality and infectious events after allogeneic peripheral blood stem cell transplantation from HLA-matched sibling donors. Eur J Haematol. 2005;75(4):299–308. https://doi.org/10.1111/j.1600-0609.2005.00514.x.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100. https://doi.org/10.1126/science.1068440.
Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, Urbani E, Negrin RS, Martelli MF, Velardi A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94(1):333–9. https://doi.org/10.1182/blood.V94.1.333.413a31_333_339.
Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, Maccario R, Bonetti F, Wojnar J, Martinetti M, Frassoni F, Giorgiani G, Bacigalupo A, Holowiecki J. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood. 2003;102(3):814–9. https://doi.org/10.1182/blood-2003-01-0091.
Ruggeri L, Mancusi A, Perruccio K, Burchielli E, Martelli MF, Velardi A. Natural killer cell alloreactivity for leukemia therapy. J Immunother. 2005;28(3):175–82. https://doi.org/10.1097/01.cji.0000161395.88959.1f.
Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. 2010;116(14):2411–9. https://doi.org/10.1182/blood-2010-05-283051.
Giebel S, Nowak I, Dziaczkowska J, Czerw T, Wojnar J, Krawczyk-Kulis M, Holowiecki J, Holowiecka-Goral A, Markiewicz M, Kopera M, Karolczyk A, Kyrcz-Krzemien S, Kusnierczyk P. Activating killer immunoglobulin-like receptor incompatibilities enhance graft-versus-host disease and affect survival after allogeneic hematopoietic stem cell transplantation. Eur J Haematol. 2009;83(4):343–56. https://doi.org/10.1111/j.1600-0609.2009.01280.x.
Symons HJ, Leffell MS, Rossiter ND, Zahurak M, Jones RJ, Fuchs EJ. Improved survival with inhibitory killer immunoglobulin receptor (KIR) gene mismatches and KIR haplotype B donors after nonmyeloablative, HLA-haploidentical bone marrow transplantation. Biol Blood Marrow Transplant. 2010;16(4):533–42. https://doi.org/10.1016/j.bbmt.2009.11.022.
Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Marsh SG, et al. Donor killer cell Ig-like receptor B haplotypes, recipient HLA-C1, and HLA-C mismatch enhance the clinical benefit of unrelated transplantation for acute myelogenous leukemia. J Immunol. 2014;192(10):4592–600. https://doi.org/10.4049/jimmunol.1302517.
Klingemann HG, Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy. 2004;6(1):15–22. https://doi.org/10.1080/14653240310004548.
Iyengar R, Handgretinger R, Babarin-Dorner A, Leimig T, Otto M, Geiger TL, Holladay MS, Houston J, Leung W. Purification of human natural killer cells using a clinical-scale immunomagnetic method. Cytotherapy. 2003;5(6):479–84. https://doi.org/10.1080/14653240310003558.
Meyer-Monard S, Passweg J, Siegler U, Kalberer C, Koehl U, Rovó A, Halter J, Stern M, Heim D, Alois Gratwohl JR, Tichelli A. Clinical-grade purification of natural killer cells in haploidentical hematopoietic stem cell transplantation. Transfusion. 2009;49(2):362–71. https://doi.org/10.1111/j.1537-2995.2008.01969.x.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7. https://doi.org/10.1182/blood-2004-07-2974.
Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, Pui CH, Leung W. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28(6):955–9. https://doi.org/10.1200/JCO.2009.24.4590.
Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother. 2010;59(11):1739–44. https://doi.org/10.1007/s00262-010-0896-z.
Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, Moreno A, DuPont B, Hsu KC, Baxter-Lowe LA, Cottler-Fox M, Shaughnessy Jr JD, Barlogie B, van Rhee F. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol. 2008;143(5):641–53. https://doi.org/10.1111/j.1365-2141.2008.07340.x.
Passweg JR, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kühne T, Favre G, Gratwohl A. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia. 2004;18(11):1835–8. https://doi.org/10.1038/sj.leu.2403524.
Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, Paulussen M, Gratwohl A, Klingebiel T, Bader P, Tichelli A, Schwabe D, Koehl U. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant. 2013;48(3):433–8. https://doi.org/10.1038/bmt.2012.162.
Koehl U, Kalberer C, Spanholtz J, Lee DA, Miller JS, Cooley S, Lowdell M, Uharek L, Klingemann H, Curti A, Leung W, Alici E. Advances in clinical NK cell studies: donor selection, manufacturing and quality control. Oncoimmunology. 2016;5(4):e1115178. https://doi.org/10.1080/2162402X.2015.1115178.
Rizzieri DA, Storms R, Chen DF, Long G, Yang Y, Nikcevich DA, Gasparetto C, Horwitz M, Chute J, Sullivan K, Hennig T, Misra D, Apple C, Baker M, Morris A, Green PG, Hasselblad V, Chao NJ. Natural killer cell-enriched donor lymphocyte infusions from a 3-6/6 HLA matched family member following nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2010;16(8):1107–14. https://doi.org/10.1016/j.bbmt.2010.02.018.
Heinze A, Grebe B, Bremm M, Huenecke S, Munir TA, Graafen L, et al. The synergistic use of IL-15 and IL-21 for the generation of NK cells from CD3/CD19-depleted grafts improves their ex vivo expansion and cytotoxic potential against neuroblastoma: perspective for optimized immunotherapy post haploidentical stem cell transplantation. Front Immunol. 2019;10:2816.
Li X, He C, Liu C, Ma J, Ma P, Cui H, et al. Expansion of NK cells from PBMCs using immobilized 4-1BBL and interleukin-21. Int J Oncol. 2015;47(1):335–42. https://doi.org/10.3892/ijo.2015.3005.
Lim O, Lee Y, Chung H, Her JH, Kang SM, Jung MY, Min B, Shin H, Kim TM, Heo DS, Hwang YK, Shin EC. GMP-compliant, large-scale expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells in vitro and in vivo. PLoS One. 2013;8(1):e53611. https://doi.org/10.1371/journal.pone.0053611.
Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, Childs R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11(3):341–55. https://doi.org/10.1080/14653240902807034.
Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. Plos One. 2013;8(10):e76781. https://doi.org/10.1371/journal.pone.0076781.
Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH, Champlin RE, Cooper LJN, Lee DA. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. Plos One. 2012;7(1):e30264. https://doi.org/10.1371/journal.pone.0030264.
Harada H, Watanabe S, Saijo K, Ishiwata I, Ohno T. A Wilms tumor cell line, HFWT, can greatly stimulate proliferation of CD56+ human natural killer cells and their novel precursors in blood mononuclear cells. Exp Hematol. 2004;32(7):614–21. https://doi.org/10.1016/j.exphem.2004.03.011.
Fujisaki H, Kakuda H, Imai C, Mullighan CG, Campana D. Replicative potential of human natural killer cells. Br J Haematol. 2009;145(5):606–13. https://doi.org/10.1111/j.1365-2141.2009.07667.x.
Jiang B, Wu X, Li XN, Yang X, Zhou Y, Yan H, Wei AH, Yan W. Expansion of NK cells by engineered K562 cells co-expressing 4-1BBL and mMICA, combined with soluble IL-21. Cell Immunol. 2014;290(1):10–20. https://doi.org/10.1016/j.cellimm.2014.04.011.
Phan MT, Lee SH, Kim SK, Cho D. Expansion of NK cells using genetically engineered K562 feeder cells. Methods Mol Biol. 2016;1441:167–74. https://doi.org/10.1007/978-1-4939-3684-7_14.
Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, Schaap N, Dolstra H. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. Plos One. 2011;6(6):e20740. https://doi.org/10.1371/journal.pone.0020740.
Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, Eldridge P, Leung WH, Campana D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–7. https://doi.org/10.1158/0008-5472.CAN-08-3712.
Dalle JH, Menezes J, Wagner E, Blagdon M, Champagne J, Champagne MA, Duval M. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr Res. 2005;57(5 Pt 1):649–55. https://doi.org/10.1203/01.PDR.0000156501.55431.20.
Ayello J, van de Ven C, Fortino W, Wade-Harris C, Satwani P, Baxi L, Simpson LL, Sanger W, Pickering D, Kurtzberg J, Cairo MS. Characterization of cord blood natural killer and lymphokine activated killer lymphocytes following ex vivo cellular engineering. Biol Blood Marrow Transplant. 2006;12(6):608–22. https://doi.org/10.1016/j.bbmt.2006.01.009.
Boissel L, Tuncer HH, Betancur M, Wolfberg A, Klingemann H. Umbilical cord mesenchymal stem cells increase expansion of cord blood natural killer cells. Biol Blood Marrow Transplant. 2008;14(9):1031–8. https://doi.org/10.1016/j.bbmt.2008.06.016.
Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, de Vries J, Boerman O, Schaap N, van der Voort R, Spanholtz J, Dolstra H. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rg(null) mice. Plos One. 2013;8(6):e64384. https://doi.org/10.1371/journal.pone.0064384.
Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, Muftuoglu M, Liu E, Orlowski RZ, Cooper L, Lee D, Parmar S, Cao K, Sobieiski C, Saliba R, Hosing C, Ahmed S, Nieto Y, Bashir Q, Patel K, Bollard C, Qazilbash M, Champlin R, Rezvani K, Shpall EJ. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177(3):457–66. https://doi.org/10.1111/bjh.14570.
Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91.
Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, Farah R, Dorritie KA, Raptis A, Lim SH, Wang H, Lapteva N, Mei Z, Butterfield LH, Rooney CM, Whiteside TL. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19(10):1225–32. https://doi.org/10.1016/j.jcyt.2017.07.008.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W, Chen G, Zhu X, Wu D, Yu J. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15(12):1563–70. https://doi.org/10.1016/j.jcyt.2013.06.017.
Williams BA, Law AD, Routy B, den Hollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. 2017;8(51):89256–68. https://doi.org/10.18632/oncotarget.19204.
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–83. https://doi.org/10.5966/sctm.2012-0084.
Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, Kaufman DS. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113(24):6094–101. https://doi.org/10.1182/blood-2008-06-165225.
Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg KJ. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin Immunopathol. 2019;41(1):59–68. https://doi.org/10.1007/s00281-018-0721-x.
Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, Rondon G, Chen J, Soebbing D, Kaur I, Gulbis A, Ahmed S, Rezvani K, Shpall EJ, Lee DA, Champlin RE. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130(16):1857–68. https://doi.org/10.1182/blood-2017-05-785659.
Ciurea SO, Soebbing D, Rondon G, Cao K, Alatrash G, Ahmed S, et al. Interim results of a phase 2 clinical trial using Mb-IL21 ex vivo expanded NK cells to enhance graft versus leukemia effect for patients with myeloid malignancies after haploidentical transplantation. Blood. 2017;130(Supplement 1):3179.
Silla L, Paz A, Filho V, Hamerschlak N, Bittencourt R, Lee D. CD56 bright /CD16 bright NK-cell adoptive immunotherapy in patients with concurrent CNS disease and relapsed or refractory (R/R) AML. J Clin Oncol. 2020;38:3025.
Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, Griffin GE, Taylor GP, Tough DF, Beverley PCL, Macallan DC. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121(2):258–65. https://doi.org/10.1111/j.1365-2567.2007.02573.x.
Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, Waldron E, Chakraborty A, Awasthi R, Levine BL, Melenhorst JJ, Grupp SA, June CH, Lacey SF. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130(21):2317–25. https://doi.org/10.1182/blood-2017-06-786129.
Grzywacz B, Moench L, McKenna D Jr, Tessier KM, Bachanova V, Cooley S, et al. Natural killer cell homing and persistence in the bone marrow after adoptive immunotherapy correlates with better leukemia control. J Immunother. 2019;42(2):65–72. https://doi.org/10.1097/CJI.0000000000000250.
Zwirner NW, Domaica CI. Cytokine regulation of natural killer cell effector functions. Biofactors. 2010;36(4):274–88. https://doi.org/10.1002/biof.107.
Pal M, Schwab L, Yermakova A, Mace EM, Claus R, Krahl AC, Woiterski J, Hartwig UF, Orange JS, Handgretinger R, André MC. Tumor-priming converts NK cells to memory-like NK cells. Oncoimmunology. 2017;6(6):e1317411. https://doi.org/10.1080/2162402X.2017.1317411.
Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, Cooper MA, Fehniger TA. Cytokine activation induces human memory-like NK cells. Blood. 2012;120(24):4751–60. https://doi.org/10.1182/blood-2012-04-419283.
Berrien-Elliott MM, Wagner JA, Fehniger TA. Human cytokine-induced memory-like natural killer cells. J Innate Immun. 2015;7(6):563–71. https://doi.org/10.1159/000382019.
Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351–65. https://doi.org/10.1084/jem.20120944.
Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, Lapteva N, McKenna D, Wagner JE, Blazar BR, Tolar J. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol Blood Marrow Transplant. 2014;20(8):1252–7. https://doi.org/10.1016/j.bbmt.2014.05.004.
Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107(6):2409–14. https://doi.org/10.1182/blood-2005-06-2399.
Shevach EM. Application of IL-2 therapy to target T regulatory cell function. Trends Immunol. 2012;33(12):626–32. https://doi.org/10.1016/j.it.2012.07.007.
Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115(21):4293–301. https://doi.org/10.1182/blood-2009-05-222190.
Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100(10):3633–8. https://doi.org/10.1182/blood-2001-12-0293.
Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019;3(13):1970–80. https://doi.org/10.1182/bloodadvances.2018028332.
Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, DeFor TE, Vivek S, Peck L, DiPersio JF, Cashen AF, Kyllo R, Musiek A, Schaffer A, Anadkat MJ, Rosman I, Miller D, Egan JO, Jeng EK, Rock A, Wong HC, Fehniger TA, Miller JS. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131(23):2515–27. https://doi.org/10.1182/blood-2017-12-823757.
Chanswangphuwana C, Allan DSJ, Chakraborty M, Reger RN, Childs RW. Augmentation of NK cell proliferation and anti-tumor immunity by transgenic expression of receptors for EPO or TPO. Mol Ther. 2021;29(1):47–59. https://doi.org/10.1016/j.ymthe.2020.09.023.
Ojo EO, Sharma AA, Liu R, Moreton S, Checkley-Luttge MA, Gupta K, Lee G, Lee DA, Otegbeye F, Sekaly RP, de Lima M, Wald DN. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. 2019;9(1):14916. https://doi.org/10.1038/s41598-019-51287-6.
Vahedi F, Nham T, Poznanski SM, Chew MV, Shenouda MM, Lee D, Ashkar AA. Ex vivo expanded human NK cells survive and proliferate in humanized mice with autologous human immune cells. Sci Rep. 2017;7(1):12083. https://doi.org/10.1038/s41598-017-12223-8.
Pierson BA, Miller JS. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood. 1996;88(6):2279–87. https://doi.org/10.1182/blood.V88.6.2279.bloodjournal8862279.
Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T, Falcone M, Nievergall E, Dörfel D, Hanns P, Passweg JR, Lutz C, Schwaller J, Zeiser R, Blazar BR, Caligiuri MA, Dirnhofer S, Lundberg P, Kanz L, Quintanilla-Martinez L, Steinle A, Trumpp A, Salih HR, Lengerke C. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572(7768):254–9. https://doi.org/10.1038/s41586-019-1410-1.
Nowbakht P, Ionescu MC, Rohner A, Kalberer CP, Rossy E, Mori L, et al. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood. 2005;105(9):3615–22. https://doi.org/10.1182/blood-2004-07-2585.
Sanchez-Correa B, Gayoso I, Bergua JM, Casado JG, Morgado S, Solana R, Tarazona R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol Cell Biol. 2012;90(1):109–15. https://doi.org/10.1038/icb.2011.15.
Kearney CJ, Ramsbottom KM, Voskoboinik I, Darcy PK, Oliaro J. Loss of DNAM-1 ligand expression by acute myeloid leukemia cells renders them resistant to NK cell killing. Oncoimmunology. 2016;5(8):e1196308. https://doi.org/10.1080/2162402X.2016.1196308.
Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood. 2002;99(10):3661–7. https://doi.org/10.1182/blood.V99.10.3661.
Stringaris K, Sekine T, Khoder A, Alsuliman A, Razzaghi B, Sargeant R, Pavlu J, Brisley G, de Lavallade H, Sarvaria A, Marin D, Mielke S, Apperley JF, Shpall EJ, Barrett J, Rezvani K. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica. 2014;99(5):836–47. https://doi.org/10.3324/haematol.2013.087536.
Pazina T, MacFarlane AWt, Bernabei L, Dulaimi E, Kotcher R, Yam C, et al. Alterations of NK cell phenotype in the disease course of multiple myeloma Cancers (Basel). 2021;13(2):226.
Benson DM, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood. 2012;120(22):4324–33. https://doi.org/10.1182/blood-2012-06-438028.
Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, Etienne A, Andre P, Romagne F, Benson D, Dombret H, Olive D. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012;120(22):4317–23. https://doi.org/10.1182/blood-2012-06-437558.
Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, Fuseri N, Bonnafous C, Czerwinski D, Rajapaksa A, Waller E, Ugolini S, Vivier E, Romagné F, Levy R, Bléry M, André P. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123(5):678–86. https://doi.org/10.1182/blood-2013-08-519199.
Korde N, Carlsten M, Lee MJ, Minter A, Tan E, Kwok M, Manasanch E, Bhutani M, Tageja N, Roschewski M, Zingone A, Costello R, Mulquin M, Zuchlinski D, Maric I, Calvo KR, Braylan R, Tembhare P, Yuan C, Stetler-Stevenson M, Trepel J, Childs R, Landgren O. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica. 2014;99(6):e81–3. https://doi.org/10.3324/haematol.2013.103085.
Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, Landgren O, Childs RW. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. 2016;22(21):5211–22. https://doi.org/10.1158/1078-0432.CCR-16-1108.
Kellner C, Hallack D, Glorius P, Staudinger M, Mohseni Nodehi S, de Weers M, van de Winkel JGJ, Parren PWHI, Stauch M, Valerius T, Repp R, Humpe A, Gramatzki M, Peipp M. Fusion proteins between ligands for NKG2D and CD20-directed single-chain variable fragments sensitize lymphoma cells for natural killer cell-mediated lysis and enhance antibody-dependent cellular cytotoxicity. Leukemia. 2012;26(4):830–4. https://doi.org/10.1038/leu.2011.288.
von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, Simhadri VR, Draube A, Reiser M, Purr I, Hallek M, Engert A. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood. 2006;107(5):1955–62. https://doi.org/10.1182/blood-2005-05-2177.
Ding H, Yang X, Wei Y. Fusion proteins of NKG2D/NKG2DL in cancer immunotherapy. Int J Mol Sci. 2018;19(1):177.
Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, Dimmock EA, Kortsaris A, Mitsiades C, Anderson KC, Fox EA, Treon SP. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc {gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007;110(7):2561–4. https://doi.org/10.1182/blood-2007-01-070656.
Congy-Jolivet N, Bolzec A, Ternant D, Ohresser M, Watier H, Thibault G. Fc gamma RIIIa expression is not increased on natural killer cells expressing the Fc gamma RIIIa-158V allotype. Cancer Res. 2008;68(4):976–80. https://doi.org/10.1158/0008-5472.CAN-07-6523.
Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–7. https://doi.org/10.1200/JCO.2003.05.013.
Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, Vallera DA, Miller JS, Bachanova V. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. 2019;3(6):897–907. https://doi.org/10.1182/bloodadvances.2018029371.
Kellner C, Bruenke J, Horner H, Schubert J, Schwenkert M, Mentz K, Barbin K, Stein C, Peipp M, Stockmeyer B, Fey GH. Heterodimeric bispecific antibody-derivatives against CD19 and CD16 induce effective antibody-dependent cellular cytotoxicity against B-lymphoid tumor cells. Cancer Lett. 2011;303(2):128–39. https://doi.org/10.1016/j.canlet.2011.01.020.
Schubert I, Kellner C, Stein C, Kugler M, Schwenkert M, Saul D, et al. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. MAbs. 2011;3(1):21–30. https://doi.org/10.4161/mabs.3.1.14057.
Glorius P, Baerenwaldt A, Kellner C, Staudinger M, Dechant M, Stauch M, Beurskens FJ, Parren PWHI, JGJ W, Valerius T, Humpe A, Repp R, Gramatzki M, Nimmerjahn F, Peipp M. The novel tribody [(CD20)(2)xCD16] efficiently triggers effector cell-mediated lysis of malignant B cells. Leukemia. 2013;27(1):190–201. https://doi.org/10.1038/leu.2012.150.
Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, Spellman S, Haagenson MD, Lenvik AJ, Litzow MR, Epling-Burnette PK, Blazar BR, Weiner LM, Weisdorf DJ, Vallera DA, Miller JS. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. 2014;123(19):3016–26. https://doi.org/10.1182/blood-2013-10-533398.
Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, Zhang B, Lenvik AJ, Panoskaltsis-Mortari A, Verneris MR, Tolar J, Cooley S, Weisdorf DJ, Blazar BR, Miller JS. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440–50. https://doi.org/10.1158/1078-0432.CCR-15-2710.
Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, Ross JA, Luo X, Weisdorf DJ, Walcheck B, Vallera DA, Miller JS. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. 2013;19(14):3844–55. https://doi.org/10.1158/1078-0432.CCR-13-0505.
Ellwanger K, Reusch U, Fucek I, Wingert S, Ross T, Muller T, et al. Redirected optimized cell killing (ROCK(R)): a highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. MAbs. 2019;11(5):899–918. https://doi.org/10.1080/19420862.2019.1616506.
Bruenke J, Fischer B, Barbin K, Schreiter K, Wachter Y, Mahr K, Titgemeyer F, Niederweis M, Peipp M, Zunino SJ, Repp R, Valerius T, Fey GH. A recombinant bispecific single-chain Fv antibody against HLA class II and FcgammaRIII (CD16) triggers effective lysis of lymphoma cells. Br J Haematol. 2004;125(2):167–79. https://doi.org/10.1111/j.1365-2141.2004.04893.x.
Felices M, Lenvik TR, Kodal B, Lenvik AJ, Hinderlie P, Bendzick LE, Schirm DK, Kaminski MF, McElmurry R, Geller MA, Eckfeldt CE, Vallera DA, Miller JS. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engager. Cancer Immunol Res. 2020;8(9):1139–49. https://doi.org/10.1158/2326-6066.CIR-19-0837.
Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. 2017;31:64–75. https://doi.org/10.1016/j.smim.2017.07.011.
Blair HA. Daratumumab: a review in relapsed and/or refractory multiple myeloma. Drugs. 2017;77(18):2013–24. https://doi.org/10.1007/s40265-017-0837-7.
Plesner T, Arkenau HT, Gay F, Minnema MC, Boccadoro M, Moreau P, Cavenagh J, Perrot A, Laubach JP, Krejcik J, Ahmadi T, de Boer C, Chen D, Chiu C, Schecter JM, Richardson PG. Enduring efficacy and tolerability of daratumumab in combination with lenalidomide and dexamethasone in patients with relapsed or relapsed/refractory multiple myeloma (GEN503): final results of an open-label, phase 1/2 study. Br J Haematol. 2019;186(3):e35–e9. https://doi.org/10.1111/bjh.15879.
Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S, Lokhorst HM, Voorhees PM, Richardson PG, Chari A, Sasser AK, Axel A, Feng H, Uhlar CM, Wang J, Khan I, Ahmadi T, Nahi H. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 2016;128(1):37–44. https://doi.org/10.1182/blood-2016-03-705210.
Vogiatzi F, Winterberg D, Lenk L, Buchmann S, Cario G, Schrappe M, Peipp M, Richter-Pechanska P, Kulozik AE, Lentes J, Bergmann AK, Valerius T, Frielitz FS, Kellner C, Schewe DM. Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood. 2019;134(8):713–6. https://doi.org/10.1182/blood.2019000904.
Bride KL, Vincent TL, Im SY, Aplenc R, Barrett DM, Carroll WL, Carson R, Dai Y, Devidas M, Dunsmore KP, Fuller T, Glisovic-Aplenc T, Horton TM, Hunger SP, Loh ML, Maude SL, Raetz EA, Winter SS, Grupp SA, Hermiston ML, Wood BL, Teachey DT Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood. 2018;131(9):995–9, 999, doi: https://doi.org/10.1182/blood-2017-07-794214.
Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, Yu J. Fratricide of NK cells in daratumumab therapy for multiple myeloma overcome by ex vivo-expanded autologous NK cells. Clin Cancer Res. 2018;24(16):4006–17. https://doi.org/10.1158/1078-0432.CCR-17-3117.
Naeimi Kararoudi M, Nagai Y, Elmas E, Pereira MSF, Ali SA, Imus PH, et al. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020;136(21):2416–27. https://doi.org/10.1182/blood.2020006200.
Fenerty KE, Padget M, Wolfson B, Gameiro SR, Su Z, Lee JH, Rabizadeh S, Soon-Shiong P, Hodge JW. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J Immunother Cancer. 2018;6(1):133. https://doi.org/10.1186/s40425-018-0445-4.
Giuliani M, Janji B, Berchem G. Activation of NK cells and disruption of PD-L1/PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget. 2017;8(14):24031–44. https://doi.org/10.18632/oncotarget.15234.
Gribben JG, Fowler N, Morschhauser F. Mechanisms of action of lenalidomide in B-cell non-Hodgkin lymphoma. J Clin Oncol. 2015;33(25):2803–11. https://doi.org/10.1200/JCO.2014.59.5363.
Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood. 2015;126(1):50–60. https://doi.org/10.1182/blood-2015-01-625004.
Delfau-Larue MH, Boulland ML, Beldi-Ferchiou A, Feugier P, Maisonneuve H, Casasnovas RO, Lemonnier F, Pica GM, Houot R, Ysebaert L, Tilly H, Eisenmann JC, le Gouill S, Ribrag V, Godmer P, Glaisner S, Cartron G, Xerri L, Salles GA, Fest T, Morschhauser F. Lenalidomide/rituximab induces high molecular response in untreated follicular lymphoma: LYSA ancillary RELEVANCE study. Blood Adv. 2020;4(14):3217–23. https://doi.org/10.1182/bloodadvances.2020001955.
Leonard JP, Trneny M, Izutsu K, Fowler NH, Hong X, Zhu J, Zhang H, Offner F, Scheliga A, Nowakowski GS, Pinto A, Re F, Fogliatto LM, Scheinberg P, Flinn IW, Moreira C, Cabeçadas J, Liu D, Kalambakas S, Fustier P, Wu C, Gribben JG, for the AUGMENT Trial Investigators. AUGMENT: a phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol. 2019;37(14):1188–99. https://doi.org/10.1200/JCO.19.00010.
Stevens WBC, Bakunina K, Cuijpers M, Chamuleau M, Beeker A, Fijnheer R, Hebart H, Visser HPJ, Doorduijn JK, Linton K, Dreyling M, de Jong D, Kersten MJ. HOVON110/ReBeL study: results of the phase I part of a randomized phase I/II study of lenalidomide, rituximab with or without bendamustine in patients with relapsed/refractory follicular lymphoma. Hemasphere. 2020;4(1):e325. https://doi.org/10.1097/HS9.0000000000000325.
Zinzani PL, Pellegrini C, Gandolfi L, Stefoni V, Quirini F, Derenzini E, Broccoli A, Argnani L, Pileri S, Baccarani M. Combination of lenalidomide and rituximab in elderly patients with relapsed or refractory diffuse large B-cell lymphoma: a phase 2 trial. Clin Lymphoma Myeloma Leuk. 2011;11(6):462–6. https://doi.org/10.1016/j.clml.2011.02.001.
Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, Roméro H, Selleri S, Béland K, Guiot M, Tremblay-Laganière C, Dicaire R, Barreiro L, Lee DA, Verhoeyen E, Haddad E. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front Immunol. 2019;10:2873. https://doi.org/10.3389/fimmu.2019.02873.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53. https://doi.org/10.1056/NEJMoa1910607.
Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136. https://doi.org/10.1186/s40425-018-0441-8.
Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33(9):1255–9. https://doi.org/10.1016/j.leukres.2008.11.024.
Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2(10):e26527. https://doi.org/10.4161/onci.26527.
Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ aggressive B-cell non-Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3(4):333–44. https://doi.org/10.1158/2326-6066.CIR-14-0114.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2020;137(5):624–36.
Shah UA, Mailankody S. CAR T and CAR NK cells in multiple myeloma: expanding the targets. Best Pract Res Clin Haematol. 2020;33(1):101141. https://doi.org/10.1016/j.beha.2020.101141.
Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JH. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020;4(9):1950–64. https://doi.org/10.1182/bloodadvances.2020001510.
Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl). 2015;3:83–92. https://doi.org/10.2147/HP.S93413.
Muz B, de la Puente P, Azab F, Luderer M, Azab AK. The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol Cancer Res. 2014;12(10):1347–54. https://doi.org/10.1158/1541-7786.MCR-14-0028.
Solocinski K, Padget MR, Fabian KP, Wolfson B, Cecchi F, Hembrough T, et al. Overcoming hypoxia-induced functional suppression of NK cells. J Immunother Cancer. 2020;8(1):e000246.
Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, Chouaib S. Hypoxia: a key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol. 2015;309(9):C569–79. https://doi.org/10.1152/ajpcell.00207.2015.
Fink T, Ebbesen P, Koppelhus U, Zachar V. Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand J Immunol. 2003;58(6):607–12. https://doi.org/10.1111/j.1365-3083.2003.01347.x.
Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. Plos One. 2013;8(5):e64835. https://doi.org/10.1371/journal.pone.0064835.
Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, Varesio L, Moretta L, Bosco MC, Vitale M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. 2013;43(10):2756–64. https://doi.org/10.1002/eji.201343448.
Viry E, Baginska J, Berchem G, Noman MZ, Medves S, Chouaib S, Janji B. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy. 2014;10(1):173–5. https://doi.org/10.4161/auto.26924.
Chambers AM, Matosevic S. Immunometabolic dysfunction of natural killer cells mediated by the hypoxia-CD73 Axis in solid tumors. Front Mol Biosci. 2019;6:60. https://doi.org/10.3389/fmolb.2019.00060.
Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, Lin G, Coudert JD, Stannard KA, Zitvogel L, Degli-Esposti MA, Vivier E, Waddell N, Linden J, Huntington ND, Souza-Fonseca-Guimaraes F, Smyth MJ. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018;78(4):1003–16. https://doi.org/10.1158/0008-5472.CAN-17-2826.
Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol. 2019;10:925. https://doi.org/10.3389/fimmu.2019.00925.
Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, Graham CH. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. 2011;71(24):7433–41. https://doi.org/10.1158/0008-5472.CAN-11-2104.
Koh YW, Lee SJ, Han JH, Haam S, Jung J, Lee HW. PD-L1 protein expression in non-small-cell lung cancer and its relationship with the hypoxia-related signaling pathways: a study based on immunohistochemistry and RNA sequencing data. Lung Cancer. 2019;129:41–7. https://doi.org/10.1016/j.lungcan.2019.01.004.
Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, Graham CH. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. 2008;68(12):4746–53. https://doi.org/10.1158/0008-5472.CAN-08-0054.
Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol. 2017;8:1300. https://doi.org/10.3389/fimmu.2017.01300.
Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Buhler L, et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1alpha unleashes NK cell activity. Immunity. 2020;52(6):1075–87 e8. https://doi.org/10.1016/j.immuni.2020.05.001.
Huang Y, Chen Z, Jang JH, Baig MS, Bertolet G, Schroeder C, Huang S, Hu Q, Zhao Y, Lewis DE, Qin L, Zhu MX, Liu D. PD-1 blocks lytic granule polarization with concomitant impairment of integrin outside-in signaling in the natural killer cell immunological synapse. J Allergy Clin Immunol. 2018;142(4):1311–21 e8. https://doi.org/10.1016/j.jaci.2018.02.050.
Lanuza PM, Pesini C, Arias MA, Calvo C, Ramirez-Labrada A, Pardo J. Recalling the biological significance of immune checkpoints on NK cells: a chance to overcome LAG3, PD1, and CTLA4 inhibitory pathways by adoptive NK cell transfer? Front Immunol. 2019;10:3010.
Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, He J, Wu G, Liu X, Zhang Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. 2015;29(2):635–41. https://doi.org/10.1016/j.intimp.2015.09.017.
da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, Osman I, Bhardwaj N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014;2(5):410–22. https://doi.org/10.1158/2326-6066.CIR-13-0171.
Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, Niiro H, Yurino A, Miyawaki K, Takenaka K, Iwasaki H, Akashi K. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17(3):341–52. https://doi.org/10.1016/j.stem.2015.07.011.
Gonçalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, Varani L, Hussain R, Siligardi G, Ceccone G, Berger SM, Ushkaryov YA, Gibbs BF, Fasler-Kan E, Sumbayev VV. The Tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine. 2017;22:44–57. https://doi.org/10.1016/j.ebiom.2017.07.018.
Gallois A, Silva I, Osman I, Bhardwaj N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Oncoimmunology. 2014;3(12):e946365. https://doi.org/10.4161/21624011.2014.946365.
Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol. 2017;139(1):335–46.e3.
Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, Cui Q, Han E, Tobin J, Bird R, Cross D, Hernandez A, Gould C, Birch S, Gandhi MK. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. 2018;131(16):1809–19. https://doi.org/10.1182/blood-2017-07-796342.
Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, Zhang J, Benson DM, He K, Caligiuri MA, Yu J. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. 2019;9(10):1422–37. https://doi.org/10.1158/2159-8290.CD-18-1259.
Ghosh A, Barba P, Perales MA. Checkpoint inhibitors in AML: are we there yet? Br J Haematol. 2020;188(1):159–67. https://doi.org/10.1111/bjh.16358.
Chen X, Liu S, Wang L, Zhang W, Ji Y, Ma X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol Ther. 2008;7(5):622–7. https://doi.org/10.4161/cbt.7.5.5689.
Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(42):17858–63. https://doi.org/10.1073/pnas.0903474106.
Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. 2018;67(3):483–94. https://doi.org/10.1007/s00262-017-2100-1.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, Sun R, Tian Z. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32. https://doi.org/10.1038/s41590-018-0132-0.
Minnie SA, Kuns RD, Gartlan KH, Zhang P, Wilkinson AN, Samson L, Guillerey C, Engwerda C, MacDonald KPA, Smyth MJ, Markey KA, Vuckovic S, Hill GR. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. 2018;132(16):1675–88. https://doi.org/10.1182/blood-2018-01-825240.
Trikha P, Lee DA. The role of AhR in transcriptional regulation of immune cell development and function. Biochim Biophys Acta Rev Cancer. 1873;2020(1):188335.
Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. 2011;17(22):6985–91. https://doi.org/10.1158/1078-0432.CCR-11-1331.
Arandi N, Ramzi M, Safaei F, Monabati A. Overexpression of indoleamine 2,3-dioxygenase correlates with regulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res. 2018;53(4):294–8. https://doi.org/10.5045/br.2018.53.4.294.
Curti A, Aluigi M, Pandolfi S, Ferri E, Isidori A, Salvestrini V, Durelli I, Horenstein AL, Fiore F, Massaia M, Piccioli M, Pileri SA, Zavatto E, D'Addio A, Baccarani M, Lemoli RM. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia. 2007;21(2):353–5. https://doi.org/10.1038/sj.leu.2404485.
Kim MS, Park TI, Son SA, Lee HW. Immunohistochemical features of indoleamine 2,3-dioxygenase (IDO) in various types of lymphoma: a single center experience. Diagnostics (Basel). 2020;10(5):275.
Ninomiya S, Hara T, Tsurumi H, Hoshi M, Kanemura N, Goto N, Kasahara S, Shimizu M, Ito H, Saito K, Hirose Y, Yamada T, Takahashi T, Seishima M, Takami T, Moriwaki H. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse large B-cell lymphoma treated with R-CHOP. Ann Hematol. 2011;90(4):409–16. https://doi.org/10.1007/s00277-010-1093-z.
Chen X, Zang Y, Li D, Guo J, Wang Y, Lin Y, Wei Z. IDO, TDO, and AHR overexpression is associated with poor outcome in diffuse large B-cell lymphoma patients in the rituximab era. Medicine (Baltimore). 2020;99(21):e19883. https://doi.org/10.1097/MD.0000000000019883.
Choe JY, Yun JY, Jeon YK, Kim SH, Park G, Huh JR, Oh S, Kim JE. Indoleamine 2,3-dioxygenase (IDO) is frequently expressed in stromal cells of Hodgkin lymphoma and is associated with adverse clinical features: a retrospective cohort study. BMC Cancer. 2014;14(1):335. https://doi.org/10.1186/1471-2407-14-335.
Karihtala K, Leivonen SK, Bruck O, Karjalainen-Lindsberg ML, Mustjoki S, Pellinen T, et al. Prognostic impact of tumor-associated macrophages on survival is checkpoint dependent in classical hodgkin lymphoma. Cancers (Basel). 2020;12(4):877.
Platten M, von Knebel DN, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. 2014;5:673.
Scoville SD, Nalin AP, Chen L, Chen L, Zhang MH, McConnell K, Beceiro Casas S, Ernst G, Traboulsi AAR, Hashi N, Williams M, Zhang X, Hughes T, Mishra A, Benson DM, Saultz JN, Yu J, Freud AG, Caligiuri MA, Mundy-Bosse BL. Human AML activates the aryl hydrocarbon receptor pathway to impair NK cell development and function. Blood. 2018;132(17):1792–804. https://doi.org/10.1182/blood-2018-03-838474.
David CJ, Massagué J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat Rev Mol Cell Biol. 2018;19(7):419–35. https://doi.org/10.1038/s41580-018-0007-0.
Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. 2008;181(6):3784–92. https://doi.org/10.4049/jimmunol.181.6.3784.
Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. 2004;172(12):7335–40. https://doi.org/10.4049/jimmunol.172.12.7335.
Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, Haura EB, Schabath MB, Coppola D, Wei S, Djeu JY. TGF-β-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci U S A. 2014;111(11):4203–8. https://doi.org/10.1073/pnas.1319269111.
Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-beta 1. J Immunol. 1995;155(3):1066–73.
Keskin DB, Allan DS, Rybalov B, Andzelm MM, Stern JN, Kopcow HD, et al. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc Natl Acad Sci U S A. 2007;104(9):3378–83. https://doi.org/10.1073/pnas.0611098104.
Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, Krzewski K, Coligan JE. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the γc cytokines and TGF-β1. Blood. 2011;118(11):3019–27. https://doi.org/10.1182/blood-2011-04-346825.
Tanyildiz HG, Kaygusuz G, Unal E, Tacyildiz N, Dincaslan H, Yavuz G. The prognostic importance of TGF-β, TGF-β receptor, and fascin in childhood solid tumors. Pediatr Hematol Oncol. 2017;34(4):238–53. https://doi.org/10.1080/08880018.2017.1363838.
Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol Med. 2019;25(11):1010–23. https://doi.org/10.1016/j.molmed.2019.06.007.
Achyut BR, Yang L. Transforming growth factor-β in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology. 2011;141(4):1167–78. https://doi.org/10.1053/j.gastro.2011.07.048.
Roy LO, Poirier MB, Fortin D. Transforming growth factor-beta and its implication in the malignancy of gliomas. Target Oncol. 2015;10(1):1–14. https://doi.org/10.1007/s11523-014-0308-y.
Roy LO, Poirier MB, Fortin D. Differential expression and clinical significance of transforming growth factor-beta isoforms in GBM tumors. Int J Mol Sci. 2018;19(4):1113.
Close HJ, Stead LF, Nsengimana J, Reilly KA, Droop A, Wurdak H, Mathew RK, Corns R, Newton-Bishop J, Melcher AA, Short SC, Cook GP, Wilson EB. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin Exp Immunol. 2020;200(1):33–44. https://doi.org/10.1111/cei.13403.
Pelizzo G, Veschi V, Mantelli M, Croce S, Di Benedetto V, D'Angelo P, et al. Microenvironment in neuroblastoma: isolation and characterization of tumor-derived mesenchymal stromal cells. BMC Cancer. 2018;18(1):1176. https://doi.org/10.1186/s12885-018-5082-2.
Krneta T, Gillgrass A, Chew M, Ashkar AA. The breast tumor microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol. 2016;13(5):628–39. https://doi.org/10.1038/cmi.2015.42.
Zhao Y, Ma J, Fan Y, Wang Z, Tian R, Ji W, Zhang F, Niu R. TGF-β transactivates EGFR and facilitates breast cancer migration and invasion through canonical Smad3 and ERK/Sp1 signaling pathways. Mol Oncol. 2018;12(3):305–21. https://doi.org/10.1002/1878-0261.12162.
Bewersdorf JP, Zeidan AM. Transforming growth factor (TGF)-β pathway as a therapeutic target in lower risk myelodysplastic syndromes. Leukemia. 2019;33(6):1303–12. https://doi.org/10.1038/s41375-019-0448-2.
Bataller A, Montalban-Bravo G, Soltysiak KA, Garcia-Manero G. The role of TGFβ in hematopoiesis and myeloid disorders. Leukemia. 2019;33(5):1076–89. https://doi.org/10.1038/s41375-019-0420-1.
Wolfraim LA, Fernandez TM, Mamura M, Fuller WL, Kumar R, Cole DE, Byfield S, Felici A, Flanders KC, Walz TM, Roberts AB, Aplan PD, Balis FM, Letterio JJ. Loss of Smad3 in acute T-cell lymphoblastic leukemia. N Engl J Med. 2004;351(6):552–9. https://doi.org/10.1056/NEJMoa031197.
Huang CH, Liao YJ, Chiou TJ, Huang HT, Lin YH, Twu YC. TGF-beta regulated leukemia cell susceptibility against NK targeting through the down-regulation of the CD48 expression. Immunobiology. 2019;224(5):649–58. https://doi.org/10.1016/j.imbio.2019.07.002.
Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, Barese C, Murali V, Wu MF, Liu H, Shpall EJ, Bollard CM, Rabin KR, Rezvani K. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia. 2016;30(4):800–11. https://doi.org/10.1038/leu.2015.327.
Han X, Ruan J, Zhang W, Zhou D, Xu D, Pei Q, Ouyang M, Zuo M. Prognostic implication of leucocyte subpopulations in diffuse large B-cell lymphoma. Oncotarget. 2017;8(29):47790–800. https://doi.org/10.18632/oncotarget.17830.
Hsu SM, Lin J, Xie SS, Hsu PL, Rich S. Abundant expression of transforming growth factor-beta 1 and -beta 2 by Hodgkin’s reed-Sternberg cells and by reactive T lymphocytes in Hodgkin’s disease. Hum Pathol. 1993;24(3):249–55. https://doi.org/10.1016/0046-8177(93)90034-E.
Ma X, Zha J, He J, Chen L, Huang J, Wu W, Tian P, Qian BH, Yu L, Jiang Y, Xu B. T follicular helper cell-mediated IL-21 production suppresses FOXP3 expression of T follicular regulatory-like cells in diffuse large B cell lymphoma patients. Hum Immunol. 2020;81(8):452–9. https://doi.org/10.1016/j.humimm.2020.05.008.
Akhurst RJ. Targeting TGF-beta signaling for therapeutic gain. Cold Spring Harb Perspect Biol. 2017;9(10):a022301.
Tran BH, Yu Y, Chang L, Tan B, Jia W, Xiong Y, Dai T, Zhong R, Zhang W, le VM, Rose P, Wang Z, Mao Y, Zhu YZ. A novel liposomal S-propargyl-cysteine: a sustained release of hydrogen sulfide reducing myocardial fibrosis via TGF-beta1/Smad pathway. Int J Nanomedicine. 2019;14:10061–77. https://doi.org/10.2147/IJN.S216667.
Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain SA, Elmas E, Foltz JA, et al. Generation of knock-out primary and expanded human NK cells using Cas9 ribonucleoproteins. J Vis Exp. 2018;(136):58237.
Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz CRY, Bollard CM. Engineering the TGFbeta receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin Cancer Res. 2019;25(14):4400–12. https://doi.org/10.1158/1078-0432.CCR-18-3183.
Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, Nguyen T, Abdel-Baki MS, Bollard CM. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19(3):408–18. https://doi.org/10.1016/j.jcyt.2016.12.005.
Foltz JA, Moseman JE, Thakkar A, Chakravarti N, Lee DA. TGFβ imprinting during activation promotes natural killer cell cytokine hypersecretion. Cancers (Basel). 2018;10(11):423.
Schmidt S, Tramsen L, Rais B, Ullrich E, Lehrnbecher T. Natural killer cells as a therapeutic tool for infectious diseases - current status and future perspectives. Oncotarget. 2018;9(29):20891–907. https://doi.org/10.18632/oncotarget.25058.
Acknowledgments
Images created with BioRender.com
Funding
Not applicable—review manuscript.
Author information
Authors and Affiliations
Contributions
M.G.L: writing—original draft preparation. H.G.R: writing—original draft preparation. B.T: visualization D.A.L: conceptualization, writing—reviewing and editing. The authors critically reviewed and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable—review manuscript.
Consent for publication
All authors have reviewed the manuscript and consented to publication.
Competing interests
D.A.L is on the scientific advisory board of the Caribou Biosciences and Courier Therapeutics. He is also on the scientific advisory board, provides consulting, owns stock, and has IP licensed to Kiadis Pharma.
B.P.T has IP licensed to Kiadis Pharma.
M.G.L and H.G.R have no conflicts of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lamb, M.G., Rangarajan, H.G., Tullius, B.P. et al. Natural killer cell therapy for hematologic malignancies: successes, challenges, and the future. Stem Cell Res Ther 12, 211 (2021). https://doi.org/10.1186/s13287-021-02277-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-021-02277-x