
Abstract
Purpose
Bioremediation is widely considered the most desirable procedure for remediation of oil-contaminated soil. Few studies have focused on the relationships among microbial community, functional genes of biodegradation, and geochemical processes during field bioremediation, which provide crucial information for bioremediation.
Methods
In the current study, the microbial community and functional genes related to hydrocarbon and nitrogen metabolism, combined with the soil physico-chemical properties, were used to diagnose a set of bioremediation experiments, including bioaugmentation, biostimulation, and phytoremediation, at the field scale.
Result
The results showed that the added nutrients stimulated a variety of microorganisms, including hydrocarbon degradation bacteria and nitrogen metabolism microorganisms. The functional genes reflected the possibility of aerobic denitrification in the field, which may be helpful in biodegradation. Biostimulation was found to be the most suitable of the studied bioremediation methods in the field.
Conclusion
We offer a feasible approach to obtain useful bioremediation information and assist with the development of appropriate remediation procedures. The findings improve our knowledge of the interactions between microorganisms and edaphic parameters.
Similar content being viewed by others

Introduction
Oil contamination in water and soil is a worldwide environmental problem (Lu et al. 2014), posing a huge threat to human health and natural ecosystems (Chen et al. 2015). Compared with physical and chemical remediation, bioremediation is regarded as the optimal method for remediation of oil-contaminated soil because it is inexpensive, efficient, and applies environmentally friendly processes (Adetutu et al. 2015). The successful application of bioremediation techniques, such as bioaugmentation, biostimulation, and phytoremediation, for remediating oil spills was reported in numerous studies (Adams et al. 2015; Cai et al. 2016; Mrozik and Piotrowska-Seget 2010; Yavari et al. 2015). Field-scale bioremediation works were also conducted in some oil-contaminated fields, and the obtained results were satisfactory. Most of them were ex situ methods, such as biopiles and prepared beds (Álvarez et al. 2017; Gomez and Sartaj 2013; Gomez and Sartaj 2014; Jørgensen et al. 2000), which are always time-consuming and expensive (Farhadian et al. 2008), and therefore unsuitable for mass soil. In situ bioremediation is widely suitable and has been used for years (Han et al. 2008; Menendez-Vega et al. 2007; Mishra et al. 2001; Pizarro-Tobías et al. 2015; Zhang et al. 2017). However, few studies monitored microorganisms, so the status of degradation microorganisms in the soil could not be determined (Wu et al. 2016).
Whether bioremediation is successful mainly depends on the biodegrading microorganisms (Wu et al. 2016). These microorganisms may be affected by other microorganisms and added nutrition. An understanding of the activities of biodegrading microorganisms and the relationships between microorganisms and environmental conditions is essential for the development of appropriate remediation procedures (Boopathy 2000; Wu et al. 2017; Xue et al. 2015).
For this reason, many studies focused on the microbial community associated with oil-contaminated soil. In the early years, studies focused on the changes in microflora before and after soil contamination or during the natural attenuation process (Leys et al. 2004; Mason et al. 2014; Röling et al. 2004; Viñas et al. 2005). More recently, the microbial community and hydrocarbon-degradation-related genes have been monitored during oil bioremediation in laboratory studies (Shahi et al. 2016; Wu et al. 2016; Wu et al. 2017). On the field scale, Pizarro-Tobías et al. (2015) assessed bioremediation and rhizoremediation technologies in an oil-polluted site in Spain and monitored the variations in microbial communities and activity.
As nutrients can stimulate the metabolism of oil-degrading microorganisms, and microbial communities and activities may vary, lacking nutrients is one of restricting factors for bioremediation (Liang et al. 2011; Wu et al. 2016). However, nutrients may affect other microorganisms as well as the oil-degrading microorganisms and affect the geochemical processes. For example, ammonium as the nitrogen source may also stimulate the growth of nitrifying bacteria and enhance the nitrification process. Whether the other stimulated microorganisms can promote oil biodegradation remains unclear. Knowledge is limited about the relationships between the various geochemical processes and hydrocarbon degradation on the field scale.
To diagnose the relationship between hydrocarbon degradation and related geochemical processes on the field-scale, a set of bioremediation experiments, including bioaugmentation, biostimulation, and phytoremediation, were conducted at an oil well site. We obtained and analyzed some information about the microbial community and functional genes related to hydrocarbon and nitrogen metabolism, combined with the soil physico-chemical properties. Alfalfa was seeded at the beginning of phytoremediation, but the alfalfa did not survive, and the area was occupied by Bermuda grass that was actually applied in the phytoremediation process. Based on the field experiment, we determined (1) the relationships among hydrocarbon degradation, microbial community, functional genes, and nitrogen cycle during bioremediation, and (2) the oil degradation efficiency in the set of bioremediation experiments on the field-scale. The mechanisms of hydrocarbon biodegradation and nitrogen transformation at the field-scale were identified.
Materials and methods
Site description
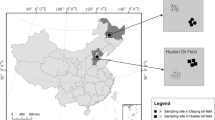
The field to be remediated was part of an abandoned oil well site located in Puyang county, Henan province, China (Fig. 1). The shallow stratum of the area was formed by the alluvial deposits of the Yellow River. The stratum is sand interbedded with clay. The shallow aquifer is mainly composed of silt and fine sand in Holocene (Q4) and Epipleistocene (Q3) strata. The upper aeration zone is mainly sandy loam soil.

Remediation site location
The oil well was abandoned in 2000 after a short period of operation due to low productivity. A blowout occurred when the well was drilled, and as a result, the dispersed oil heavily contaminated 3000 m2 of farmland soil. Surface oil deposits remain scattered around the well. An area (1 × 4 m) with similar soil concentrations (approximately 2 g kg−1 of total hydrocarbons) was designated as the study area.
Bioremediation process
The study area was divided into four blocks: blank control (Ctrl), bioaugmentation (BA), biostimulation (BS), and phytoremediation (Phyto). Each block was a soil cuboid that was 80 long, 80 cm wide, and 30 cm deep (Fig. 1). According to the dry bulk density of soil (1.56 g cm−1), there was about 300 kg dry soil in each block. The 5-cm-deep surface-layer soil covering the contaminated soil was removed. Then, the soil in each block was homogenized with plowing to even the oil concentration in the soil. Ctrl was treated as the blank to which no remediation procedure was applied. The BS block was sprinkled with nutrients dissolved in deionized water. The BA block was sprinkled with nutrients and oil-degrading microorganism, Arthrobacter, which was previously isolated from the field soil (Ning et al. 2013), and inoculated in Luria-Bertani (LB) medium at 28 °C until reaching a density of 1010 colony forming units (CFU)/mL. LB plate counts were used to estimate numbers of microorganisms (Mueller et al. 1991). The expected final density in the soil was 108 CFU/g soil. The Phyto block was treated the same as the BA block plus alfalfa seeds. The optimal ratio of carbon:nitrogen:phosphorous (C:N:P) is approximately 100:15:1 for biodegradation (Graham et al. 1999). However, as a high salt content in soil may hinder bioremediation, nutrients were added across several separate occasions when watering. In the first fertilization, ammonium sulfate ((NH4)2SO4, 0.43 g/kg soil) and monopotassium dihydrogen phosphate (KH2PO4, 0.067 g/kg soil) were added. The nutrients were added corresponding to the oil concentration of 0.67 g/kg of hydrocarbons and according to the optimal C:N:P ratio. The nutrient solution was added again in the BA, BS, and Phyto blocks after 2 weeks. Purified water was added irregularly to the BA, BS, and Phyto blocks to ensure that the water content of the soil was approximately 15% of the soil mass. The Ctrl block was not treated. The lowest and highest soil surface temperatures were monitored every day by maximum-minimum thermometers during the remediation process. When the daily lowest temperature was below 15 °C, the soil surface was covered with polyethylene ground film used for farms to promote microorganism activities. The soil in the BA and BS blocks was turned over twice after every sampling.
The alfalfa in the Phyto block died 2 days after sprouting. The alfalfa was then replanted. Shading and moisture-retention measurements were taken to promote alfalfa growth in this block. However, no alfalfa plants survived for more than a few days. Instead, the block was occupied by the surrounding weed, Bermuda grass, after 2 weeks. Bermuda grass may be helpful for the remediation of oil-contaminated soil (Razmjoo and Adavi 2012). Therefore, it was allowed to follow its own course in this block.
Sampling and physico-chemical analyses
A five-point sampling, coning, and quartering method was used to collect samples; thus, a mixed sample was collected in each block at each time. About 50 g soil for each sample was collected from 15 cm below the surface using a sterile knife and placed in aluminum boxes previously sterilized at high temperature. The collected samples were immediately stored in a cooler with an ice pack and then transferred to the laboratory and stored at − 70 °C until analysis. These samples were used for the extraction of genomic DNA for the microbiological and functional gene analysis. Another soil subsample was stored in automatically sealing plastic bags for analysis of physical and chemical characteristics. Samples were collected on days 0, 3, 7, 11, 16, 22, 28, 34, 47, 62, 74, and 125 of the remediation process in Ctrl, BA, and BS blocks. Samples were only collected on days 0, 3, 7, 11, 62, 74, and 125 in the Phyto block because there was grass growing after the 11th day, and the sampling would have destroyed the grass. The samples were also collected on the site and its neighboring farmland on day 0 and 1 year prior to the start of the experiment. The oil content in the soil samples was determined using supersonic extraction and ultraviolet-visible spectrophotometric methods. Trichloromethane was used as the oil extraction solvent, and oil was extracted supersonically for 15 min twice. Then, the extraction solvent was evaporated at 65 °C, and the remnant was dissolved by petroleum ether. An ultraviolet-visible spectrophotometer was used to determine oil content at a 225-nm wavelength (Li 1999). The water content, pH, electrical conductivity, and nitrate, nitrite, ammonium, and chloridion contents in the soil samples were measured as previously described (Lu 2000). Each parameter was measured twice. The samples collected on days 0, 3, 11, 47, and 125 of the experimental blocks and on farmland neighboring the contaminated field this year (F_ty) and 1 year ago (F_oya), and in the contaminated field 1 year ago (Ctrl_oya) were chosen to analyze the DNA information, including the functional genes and microbial communities.
DNA extraction and gene quantification
DNA was extracted from the soils using the E.Z.N.ATM Mag-Bind Soil DNA Kit (Omega Bio-tek, Inc., Norcross, GA, USA) in accordance with the manufacturer’s protocol.
To quantify the distribution of anammox bacteria (AB), ammonia-oxidizing archaea (AOA), ammonia-oxidizing bacteria (AOB), denitrifying bacteria, dissimilatory nitrite-reducing bacteria, and hydrocarbon-degrading bacteria, the corresponding functional genes, including amx (corresponds to AB), amoA (corresponds to AOA), amoA (corresponds to AOB), nosz (corresponds to denitrifying bacteria), nap (corresponds to dissimilatory nitrite-reducing bacteria), alkB (corresponds to alkane-degrading bacteria), and alcvx (corresponds to a hydrocarbon-degrading bacteria genus Alcanivorax) were quantified with a StepOne™ Real-Time PCR System (Applied Biosystems™, Foster City, CA, USA) based on SYBR® Premix Ex Taq™ (Tli RNase H Plus) (TaKaRa, Dalian, China). SYBR Green and absolute quantification methods were used to quantify the target genes. The acceptable R2 values of standard curves of each gene were greater than 0.99. The primer sequences, annealing temperatures, amplified fragment size, and targets are listed in Table 1, and standard plasmids were prepared according to previous studies (Kostka et al. 2011; Shu et al. 2016; Sun et al. 2012). To validate the reproducibility, each reaction was performed in duplicate.
Microbial community sequencing
The DNA was PCR amplified using 25 cycles with the Bakt_341F (CCTACGGGNGGCWGCAG) and Bakt_805R (GACTACHVGGGTATCTAATCC) primers (Herlemann et al. 2011). The PCR products were purified and then adjusted to 10–20 ng of DNA. Metagenomic sequencing was conducted on the MiSeq sequencing platform at Sangon Biotech Co., Ltd. (Shanghai, China) (Hatta et al. 2016). The sequence reads were trimmed, optimized, subsampled, aligned, and clustered into operational taxonomic units (OTUs) (Liu et al. 2017). The representative sequences were annotated with the RDP-classifier 2.2 (QIIME) (Lan et al. 2012). The species diversity index was calculated according to the α diversity analysis to evaluate the species diversity in the soil samples (Korenblum et al. 2012). The community composition of each sample was determined to the genus level.
Statistical analysis
The relationships between the samples and the microbial community at the gene level were revealed by principal component analysis (PCA), which was used to cluster the distribution of microbial community composition (Tong et al. 2019). The abundances of functional genes and physicochemical parameters were set as environmental factors (Liu et al. 2016). The relationships between functional genes and environmental factors, including the ammonium, nitrite, nitrate, and oil contents were determined by redundancy analysis (RDA). Both PCA and RDA were conducted using the CANOCO4.5 software (Microcomputer Power, Ithaca, NY, USA) (Chen et al. 2013). Cluster analysis was conducted based on the weighted UniFrac distance to compare the microbial communities in the different soil samples (Lozupone and Knight 2005).
Results and discussion
Bioremediation and nitrogen transformation characteristics
The variations in the oil contents of the soil are shown in Fig. 2a. Although the curves varied under the heterogeneity of the oil distribution, after 125 days of remediation, the oil content in the BA and BS blocks decreased from 2000 to approximately 500 mg kg−1, which is a level similar to that in the soil sampled in the nearby farmland (F_oya and F_ty). The oil contents in the Phyto and Ctrl blocks also decreased but with a lower decrease rate. The oil-removal efficiencies were approximately 78%, 77%, 60%, and 46% in the BA, BS, Phyto, and Ctrl blocks, respectively. The BA and BS treatments yielded the same degradation efficiency. This suggests that the added microorganism, Arthrobacter, played a negligible role, which is consistent with some studies (Wu et al. 2016; Yu et al. 2005). However, some other studies found that bioaugmentation with exogenous bacteria may enhance the degradation (Bento et al. 2005; Bidja Abena et al. 2019; Roy et al. 2018). The contradictory results may be caused by the different soil characteristics, different strains and their concentrations, and different experimental conditions. The saline-alkaline soil and changing temperature and moisture of the field experiment may have been unsuitable for the survival of our added microorganism.

Variations in a oil content, b–d nitrogen concentration, and e–h other physical and chemical parameters. A five-point sampling, coning, and quartering method was used to collect samples, thus a mixed sample was collected in each block at each time. Error bars present the standard deviation (SD) of test results (n = 2)
The differences between the oil-removal in BA (or BS) and Ctrl blocks were approximately equal to the calculated oil consumed by the added nutrient stimulated microorganisms according to the optimal C:N:P ratio (100:15:1). These findings suggest that the added C:N:P ratio was suitable for the field. Another factor, aeration by plowing, may have increased the difference in oil-removal efficiencies between BA (BS) blocks and the control block, as there was no additional plowing other than the blending at the beginning. The Phyto block was also not turned over for the growth of grass and had the lowest oil-removal efficiency.
The oil content in the Ctrl block dramatically fluctuated, which may have been due to the heterogeneity of the oil content in the field and the soil not being turned over as much as in the BA and BS blocks. The decreasing oil content in the block may have been caused by natural attenuation, which was mainly due to microorganisms (Agnello et al. 2016). The initial turning over the soil may supply oxygen.
The forms of nitrogen dramatically changed once ammonium were added to the oil-contaminated soil. The ammonium (Fig. 2b) concentration decreased, while that of nitrate (Fig. 2d) increased from about 50 to 300 mg/L during the first 10 days. The nitrate concentrations peaked (about 600 mg/L) on days 16 and 22 in the BS and BA blocks, respectively, and peaked again (about 800 mg/L) on day 47 in both blocks. After day 47, the nitrate concentrations sharply decreased and reached a low point (about 100 mg/L) on day 62, gradually increasing thereafter. Nitrite (Fig. 2c) is formed during the process, and its concentration peaked on day 16 in both the BS and BA blocks. Ammonium and nitrite were gradually eliminated in the final days of the experiment. The nitrogen concentration, including that in ammonium, nitrate, and nitrite, in the BA block was greater than in the BS block for the culture medium containing nitrogen. The Phyto block was similar to the BA block. Figure 2 also suggests that some nitrogen was imported into this block through nitrogen fixation by microorganisms and other processes, such as soil organic matter mineralization (Peoples et al. 1995). This fixed nitrogen can be used as a nitrogen source for hydrocarbon biodegradation. This result may also be explained by the volatilization of the hydrocarbons and photolysis (Fine et al. 1997), which were promoted on summer days by the high temperature and strong light intensity.
The moisture content (Fig. 2e) in the BA and BS blocks was maintained at approximately 15% after day 28, while the moisture content in the Ctrl block was below 15% during that period. The pH (Fig. 2f) dramatically decreased in the BA and BS blocks due to the acidity of the nutrient solution, whereas that of the Ctrl block remained the same. The amplitudes of the variations in the electrical conductivity (Fig. 2g) and chloride ion concentration (Fig. 2h) were small in all the blocks, except in the initial samples.
Functional genes diagnosis
The 16S rRNA concentration and the ratio of functional genes to 16S rRNA are shown in Fig. 3. This figure shows that in most cases, nitrogen cycle-related genes, amoA (AOA), amoA (AOB), and nosZ were more abundant in the BA and BS blocks than in the Ctrl block. However, the hydrocarbon-degrading bacterial genes were not all the same. The variations in the alcvx gene in the Ctrl, BA, and BS blocks were almost parallel. AlkB was more abundant in BA and BS than that in Ctrl during the first 11 days, but in the following days, alkB in Ctrl was more abundant than in BA or BS. The 16S rRNA gene concentrations in the BA and BS blocks both decreased during the first 11 days but increased after nutrients were added. The concentrations then either decreased or remained the same concentrations. The 16S rRNA gene concentrations in the Ctrl block exhibited a decreasing trend.

Variations in a 16S rRNA gene concentrations and b–h the concentration ratios (R) of functional genes to 16S rRNA genes with remediation time in the Ctrl, BS, and BA blocks. Error bars present the standard deviation (SD) of test results (n = 2)
The relationship between the functional genes and environmental factors is shown in the RDA plot (Fig. 4). All the genes except alcvx had close relationships with the ammonium and oil contents.

RDA biplot analysis of the functional genes and environmental variables
The nitrogen concentration variation and formation, as well as the functional genes, indicate that microorganisms related to nitrogen transformation remained active in the oil-contaminated soil. This finding is consistent with that of previous studies (Xu et al. 1995). Figure 4 shows that nitrogen transformation bacteria had a close relationship with hydrocarbon degradation.
Chloride ions (Cl−) cannot be metabolized by microorganisms or plants, adsorbed by soil particles or easily formed by precipitation, and are often used as a tracer of soil water movement (White and Broadley 2001). Except for the initial raining days, the chloride ion concentrations (Fig. 2h) in the BA and BS blocks were almost constant throughout the remediation process. This finding indicated that the added nitrogen had not seeped into the lower soil. Therefore, the decrease in inorganic nitrogen, including that in ammonium, nitrite, and nitrate, was mainly caused by the volatilization of ammonium and microbial mediation. Nitrification caused a dramatic decrease in ammonium and an increase in nitrate after the nutrients were added (Wde and Kowalchuk 2001). The nitrate concentrations dramatically decreased during the days after nutrients were added, which may have been due to the denitrification and assimilation of microorganisms (Cabrera et al. 2016). The variations in related functional genes were consistent with the nitrogen variations.
Most microorganisms, including hydrocarbon-degrading bacteria, consistently use ammonium as their optimal nitrogen source (Hasinger et al. 2012; Kern et al. 2017). Figure 4 shows that all the genes except the alcvx gene had close relationships with ammonium. Nitrification is a natural process that has been considered for bioremediation for many years (Sayavedra-Soto et al. 2010). The nitrification gene amoA is positively correlated with oil content, and alkB is crucial in hydrocarbon degradation (Viggor et al. 2015). Nitrate can be used as an electron acceptor, and hydrocarbons can be used as electron donors during denitrification (Lueders 2017). Therefore, the process of denitrification can be regarded as the hydrocarbon degradation process in oil-contaminated soil. The genes involved in denitrification, including nap and nosz, had positive correlations with oil content and alkB gene expression. Consequently, these processes of nitrogen transformation are conducive to oil degradation. The qPCR of the alcvx gene targeted the genus Alcanivorax, which contains alkane-degrading species (Barbato et al. 2015). Alcanivorax always correlates with a high nitrate level (Mulla et al. 2017), which is consistent with our findings.
Microbial community diagnosis
The coverage of this sequencing was greater than 0.97, which suggests that the results of the sequencing were reliable. The abundance distributions of microorganisms are shown in Fig. 5.

Distributions of bacteria in the 13 samples at the genus level. Taxa were represented at > 0.5% frequency in at least one sample. “Others” refers to the taxa with a maximum abundance of < 0.5% in any sample. Unclassified refers to the sequences that could not be assigned to known bacterial genus. F_ty stands for the sample collected from a farmland neighbouring the contaminated field this year. BA_3d, BA_11d, BA_47d, and BA_125d represent the samples collected after 3, 11, 47, and 125 days, respectively, from the beginning of the remediation process in the BA block. BS_11d, BS_47d, and BS_125d represent the samples collected after 11, 47, and 125 days, respectively, from the beginning of the remediation process in the BS block. Phyto_125d represents the sample collected 125 days after the beginning of the remediation process in the Phyto block. Ctrl_oya is a sample that was collected in the contaminated site 1 year ago. F_oya is the sample collected from a farmland neighbouring the contaminated field 1 year ago
The sum of Pseudomonas, Acinetobacter, and unclassified bacteria accounted for more than half of the total bacteria in all the samples. The abundance of Pseudomonas was greater than that of any other bacteria. The added bacteria Arthrobacter, isolated from the field soil and used for bioaugmentation, was only present in the F_ty, BA_11d, and BS_47d samples, and the relative abundances were only 0.03%, 0.01%, and 0.01%, respectively. The percentages of unclassified bacteria in the contaminated soils were greater than those in the uncontaminated soils, whereas Pseudomonas exhibited the opposite trend. Samples F_oya and F_ty, which were collected from uncontaminated farmland in different years, had similar abundance distributions. In contrast, the abundance distributions in Ctrl_oya and Ctrl_0 samples, which were collected from the same contaminated site but in different years, were less similar.
The cluster analysis (Fig. 6) also shows similar results. The early remediation stage samples (days 3 and 11), untreated contaminated soils (Ctrl _0 and Ctrl _oya), Ctrl _125d, and farmland samples were grouped together. Except for Ctrl _125d, the later remediation stage samples (days 47 and 125) were grouped together. The samples collected at the same time from the BS and BA blocks had similar microflora.

Cluster analysis of the samples based on the weighted UniFrac distance and 16S rRNA genes
The microbial community structure of Ctrl _125d was similar to that of the uncontaminated soils. This finding suggests that this sample experienced similar environmental conditions to those in the uncontaminated soils. The sample was collected at the surface of this block, which was constantly exposed to the air and sun and received no nutrient additions, which was similar to the uncontaminated soils. Nutrient and other environmental factors, such as moisture and aeration, strongly affect microorganism communities (Jungmann et al. 2015).
During the remediation processes, the variation characteristics of microbial communities, as well as the amoA (AOA), amoA (AOB), and nosZ genes in the BA and BS blocks were similar. In addition, except for the various nitrogen concentrations in the early remediation stage, the soils collected in the BA and BS blocks at the same time had similar nitrogen concentrations, but different physical and chemical parameters. This result suggests that the added strain played a negligible role in oil degradation and the transformation of nitrogen, which is consistent with previous research (Wu et al. 2016). Although the added, Arthrobacter strain was initially isolated from this contaminated field, they could barely survive alongside the existing indigenous bacteria. Therefore, regardless of the strain added, there were no differences between the BA and BS blocks in terms of treatment effects; both had similar parameters and microbial communities.
The Phyto block, whose soil was turned over only once and had the same added nutrients as the BA and BS blocks, demonstrated characteristics similar to those in the BA and BS blocks rather than those in the Ctrl block according to samples collected on the last day. We speculate that the nutrients affected the microbial activity more than the tillage. We observed some differences between the Phyto and BA (or BS) blocks due to the undisturbed soil and the growth of Bermuda grass in the Phyto block, and plant growth in the plot may also play on a crucial role in microbial community evolution.
The PCA of the samples and microorganisms (at the genus level) is presented in Fig. 7. The sample points can be divided into the following groups: the pre-remediation-stage group (group 1), the earlier-stage group (group 2), middle- and later stage group (group 3), and uncontaminated plus Ctrl_125d sample group (group 4). The Meniscus, Ignavibacterium, Thiobacillus, Gillisia, Sphingopyxis, and Parvibaculum bacteria were included in the first group samples. The third group corresponded to the Ohtaekwangia, Salinimicrobium, TM7, Sphingomonas, Nitrosomonas, Flavisolibacter, Luteimonas, Pedobacter, Chitinophaga, and Spartobacteria bacteria. The samples in the last group had close relationships with the Lactococcus, Pseudomonas, Acinetobacter, Psychrobacter, Chryseobacterium, and Enhydrobacter bacteria.

PCA biplot analysis of samples and microorganisms (at the genus level). The green points represent the samples. The black rays with solid arrows represent the microorganism species. The red rays with open arrows represent the environmental factors, including the nitrogen and functional genes
Those in the first group of samples (Fig. 7) were related to variations in hydrocarbon degradation and the oxidization of reduced sulfur compounds, which are present in crude oil. Thiobacillus can oxidize the reduced sulfur compounds to sulfate (Pronk et al. 1990). The abundance of Thiobacillus was reduced in the later samples because the added nutrients contained sulfur, which affects the oxidization of sulfur compounds. Meniscus and Gillisia are associated with hydrocarbon degradation (Guibert et al. 2012). Ignavibacterium, a strictly anaerobic chemoheterotroph with versatile metabolic characteristics (Sun et al. 2015), can use aniline (Sun et al. 2015) and hydrocarbons (Salam et al. 2017). Sphingopyxis, a crude-oil-degrading marine bacterium (Kim et al. 2014), can degrade aromatic hydrocarbons (Rodriguezr et al. 2015) associated with ammonia assimilation (Williams et al. 2009). Parvibaculum can oxidize alkane (Rosario-Passapera et al. 2012), linear alkylbenzene sulfonate (Schleheck et al. 2011), and polycyclic aromatic hydrocarbons (PAHs) (Lai et al. 2011). This capability is in line with the functional genes (alkB and amoA (AOB)) and environmental factors (oil and ammonium contents) shown in Fig. 7.
The second group was a transient group. The samples collected in the early days were distributed in this region. These days, adequate ammonium, moisture, and nitrite were transformed from ammonium. Except for sedimen, there was no specific bacterium in the group.
The third group (Fig. 7) corresponded to bacteria associated with nitrogen transformation and easily degradable hydrocarbon compounds as well as some bacteria that are sensitive to oil. Ohtaekwangia, a nitrifying organism, can oxidize ammonium to nitrate (Rodríguez-Caballero et al. 2017) and can metabolize easily degradable organic matter (Li et al. 2014). Salinimicrobium could barely survive in the highly contaminated soil because it is sensitive to petroleum hydrocarbons. As a result, these bacteria can be used as oil contamination indicators (Wang 2011). The BA125, Phyto125, and BS125 samples, which were collected in the final sampling, were teeming with these types of bacteria, which indicate that the oil content had sufficiently decreased in these treated blocks. TM7 and Spartobacteria genera incertae sedis are associated with hydrocarbon degradation (Huang and Li 2014; Salam et al. 2017; Song et al. 2015). Sphingomonas strains are capable of metabolizing polycyclic aromatic hydrocarbons under low nutrient conditions (Ye et al. 2006; Zylstra and Kim 1997). Nitrosomonas, a nitrifier organism, can oxidize ammonium to nitrate (Suzuki et al. 1974) and can nitrify and denitrify at the same time when grown under oxygen limitation (Bock et al. 1995). The Flavisolibacter level has significant positive relationships with a number of denitrification genes (Huang et al. 2016). Luteimonas has the ability to reduce nitrates to nitrogen (Young et al. 2007), Pedobacter has the ability to degrade oil hydrocarbons (Margesin et al. 2003; Zhang et al. 2010), and Chitinophaga has the ability to reduce nitrate (Kim and Jung 2007). These processes were also consistent with the functional genes and environmental factors present.
The samples in the last group had close relationships with bacteria that had few or negative correlations with oil and nitrogen. Pseudomonas and Acinetobacter occupied larger proportions than any other bacteria in all the samples, especially in the fourth group. Pseudomonas is a genus of aerobic bacteria that contains 191 validly described species. This genus includes human pathogens, plant pathogens, soil bacteria, and plant growth-promoting bacteria (EUZéBY 1997) that can also degrade hydrocarbons (O’Mahony et al. 2006). The high abundance of Pseudomonas in uncontaminated soil may have been caused by the crops and the lack of influence of oil. Acinetobacter (Doughari et al. 2011) and Chryseobacterium (Bernardet et al. 2015) are widely distributed in nature and commonly occur in soil and water, but oil and anaerobic environments may harm them. Lactococcus produces a single product, lactic acid, as the major or only product of glucose fermentation (Crisan and Jay 2005), and glucose may exist in crops not contaminated by crude oil. Psychrobacter is oxidase-positive, with a strictly oxidative metabolism, and they are moderately halotolerant (Bozal et al. 2003). In consequence, the presence of salt in the contaminated soil may have affected their survival.
Relationships between oil biodegradation and nitrogen transformation
The above discussion suggests that the process of oil biodegradation is always accompanied by nitrogen transformations, such as denitrification or nitrification. Denitrification is one of the important hydrocarbon biodegradation processes under anaerobic conditions (Hutchins et al. 1991). The study field was under aerobic conditions because there was adequate available oxygen from the frequently turned over soil, and the ammonium was transformed into nitrite and nitrate rapidly through nitrification. Therefore, the denitrification in the soil could be identified as aerobic denitrification. Previous studies showed aerobic denitrifying bacteria degrade hydrocarbons (Kwapisz et al. 2008) but they lacked field data. This study may supply the data to support this finding.
Studies showed that the petroleum hydrocarbon may slow down nitrification (Chang and Weaver 1997; Deni and Penninckx 1999), but no report has shown that oil biodegradation is hindered by the presence of ammonium, even if both of the processes consume oxygen and may compete for the limited oxygen. Some studies showed that a few pure cultured nitrifying bacteria can oxidize a wide variety of hydrocarbon substrates through the action of ammonia monooxygenase, the key enzyme for nitrification (Chang et al. 2002; Deni and Penninckx 1999). Our results show that nitrification has no negative effect on oil biodegradation. Although alkB and amoA had a positive relationship, abundances of both in the oil biodegradation and nitrification microorganisms were promoted by ammonium. Thus, future research should consider whether nitrification has a positive effect on oil biodegradation in the field.
Our findings showed that the added ammonium promoted the oil biodegradation. There are two potential reasons for this finding. Firstly, ammonium supplied the nitrogen source for the biodegradation microorganisms. Secondly, ammonium may transform into nitrate by nitrification, and the nitrate may serve as the electron acceptor for biodegradation through denitrification.
Practical application suggestion
The current study and many previous studies revealed that contaminated oil exploit sites are always accompanied by saline and alkaline stress (Liu et al. 2019; Liu et al. 2014; Wang et al. 2012). Therefore, the added alien creatures, including microorganisms and plants, struggle to survive in the saline-alkaline environment. Cultivating biodegradation microorganisms and plants is laborious and time-consuming. Therefore, bioaugmentation and phytoremediation methods are not recommended. From this study, biostimulation with ammonium and phosphate could be a good choice for site remediation in practical applications.
Conclusions
The nutrients added to oil-contaminated soil stimulated a variety of microorganisms, including hydrocarbon-degrading bacteria and nitrogen metabolizing microorganisms. The functional genes in nitrogen metabolism, such as amx, amoA, nap, and nos, showed a positive correlation with the alkane monooxygenase gene alkB. Thus, aerobic denitrification may occur in the field, which could be helpful for oil biodegradation. Phytoremediation and bioaugmentation nearly failed in the field, potentially because the environment was unsuitable for the survival of added bacterial strains, Arthrobacter, and alfalfa. Biostimulation was found to be the most suitable bioremediation method in the field and could have wide suitability because no new organisms are introduced to the contaminated soil. Our findings improve our knowledge about the interactions between microorganisms and edaphic parameters and offer a feasible approach to obtain useful bioremediation information and assist in the development of appropriate remediation procedures.
References
Adams GO, Fufeyin PT, Okoro SE, Ehinomen I (2015) Bioremediation, biostimulation and bioaugmention: a review. Int J Environ Bioremediation Biodegradation 3:28–39
Adetutu EM et al (2015) Exploiting the intrinsic hydrocarbon-degrading microbial capacities in oil tank bottom sludge and waste soil for sludge bioremediation. Int J Environ Sci Technol 12:1427–1436
Agnello AC, Bagard M, van Hullebusch ED, Esposito G, Huguenot D (2016) Comparative bioremediation of heavy metals and petroleum hydrocarbons co-contaminated soil by natural attenuation, phytoremediation, bioaugmentation and bioaugmentation-assisted phytoremediation. Sci Total Environ 563-564:693–703. https://doi.org/10.1016/j.scitotenv.2015.10.061
Álvarez LM, Ruberto LAM, Balbo AL, Mac Cormack WP (2017) Bioremediation of hydrocarbon-contaminated soils in cold regions: development of a pre-optimized biostimulation biopile-scale field assay in Antarctica. Sci Total Environ 590:194–203
Barbato M, Mapelli F, Chouaia B, Crotti E, Daffonchio D, Borin S (2015) Draft genome sequence of the hydrocarbon-degrading bacterium alcanivorax dieselolei KS-293 isolated from surface seawater in the Eastern Mediterranean Sea. Genome Announc 3. https://doi.org/10.1128/genomeA.01424-15
Bento FM, Camargo FAO, Okeke BC, Frankenberger WT (2005) Comparative bioremediation of soils contaminated with diesel oil by natural attenuation, biostimulation and bioaugmentation. Bioresour Technol 96:1049–1055. https://doi.org/10.1016/j.biortech.2004.09.008
Bernardet J-F, Hugo CJ, Bruun B (2015) Chryseobacterium. In: Bergey’s Manual of Systematics of Archaea and Bacteria. Wiley. https://doi.org/10.1002/9781118960608.gbm00301
Bidja Abena MT, Li T, Shah MN, Zhong W (2019) Biodegradation of total petroleum hydrocarbons (TPH) in highly contaminated soils by natural attenuation and bioaugmentation. Chemosphere 234:864–874. https://doi.org/10.1016/j.chemosphere.2019.06.111
Bock E, Schmidt I, Stüven R, Zart D (1995) Nitrogen loss caused by denitrifying Nitrosomonas cells using ammonium or hydrogen as electron donors and nitrite as electron acceptor. Arch Microbiol 163:16–20. https://doi.org/10.1007/bf00262198
Boopathy R (2000) Factors limiting bioremediation technologies. Bioresour Technol 74:63–67
Bozal N, Montes MJ, Tudela E, Guinea J (2003) Characterization of several Psychrobacter strains isolated from Antarctic environments and description of Psychrobacter luti sp. nov. and Psychrobacter fozii sp. nov. Int J Syst Evol Microbiol 53:1093
Cabrera JJ, Salas A, Torres MJ, Bedmar EJ, Richardson DJ, Gates AJ, Delgado MJ (2016) An integrated biochemical system for nitrate assimilation and nitric oxide detoxification in Bradyrhizobium japonicum. Biochem J 473:297–309
Cai B, Ma J, Yan G, Dai X, Li M, Guo S (2016) Comparison of phytoremediation, bioaugmentation and natural attenuation for remediating saline soil contaminated by heavy crude oil. Biochem Eng J 112:170–177
Chang SW, Hyman MR, Williamson KJ (2002) Cooxidation of naphthalene and other polycyclic aromatic hydrocarbons by the nitrifying bacterium, Nitrosomonas europaea. Biodegradation 13:373–381
Chang Z, Weaver R (1997) Nitrification and utilization of ammonium and nitrate during oil bioremediation at different soil water potentials. Soil Sediment Contam 6:149–160
Chen L, Wang L-Y, Liu S-J, Hu J-Y, He Y, Zhou H-W, Zhang X-H (2013) Profiling of microbial community during in situ remediation of volatile sulfide compounds in river sediment with nitrate by high throughput sequencing. Int Biodeterior Biodegradation 85:429–437
Chen M, Xu P, Zeng G, Yang C, Huang D, Zhang J (2015) Bioremediation of soils contaminated with polycyclic aromatic hydrocarbons, petroleum, pesticides, chlorophenols and heavy metals by composting: applications, microbes and future research needs. Biotechnol Adv 33:745–755
Crisan EV, Jay JM (2005) Modern Food Microbiology. Bioscience 101:167
Deni J, Penninckx MJ (1999) Nitrification and autotrophic nitrifying bacteria in a hydrocarbon-polluted soil. Appl Environ Microbiol 65:4008–4013
Doughari HJ, Ndakidemi PA, Human IS, Benade S (2011) The ecology, biology and pathogenesis of Acinetobacter spp.: an overview. Microbes Environ 26:101–112
Euzéby JP (1997) List of bacterial names with standing in nomenclature: a folder available on the Internet. Int J Syst Bacteriol 47:590
Farhadian M, Vachelard C, Duchez D, Larroche C (2008) In situ bioremediation of monoaromatic pollutants in groundwater: a review. Bioresour Technol 99:5296–5308
Fine P, Graber ER, Yaron B (1997) Soil interactions with petroleum hydrocarbons: abiotic processes. Soil Technol 10:133–153
Gomez F, Sartaj M (2013) Field scale ex-situ bioremediation of petroleum contaminated soil under cold climate conditions. Int Biodeterior Biodegradation 85:375–382
Gomez F, Sartaj M (2014) Optimization of field scale biopiles for bioremediation of petroleum hydrocarbon contaminated soil at low temperature conditions by response surface methodology (RSM). Int Biodeterior Biodegradation 89:103–109
Graham DW, Smith VH, Cleland DL, Law KP (1999) Effects of nitrogen and phosphorus supply on hexadecane biodegradation in soil systems. Water Air Soil Pollut 111:1–18
Guibert LM, Loviso CL, Marcos MS, Commendatore MG, Dionisi HM, Lozada M (2012) Alkane biodegradation genes from chronically polluted subantarctic coastal sediments and their shifts in response to oil exposure. Microb Ecol 64:605–616
Han H-L, Chen Z, Yang J-M, Miao C-C, Zhang K, Jin W-B, Liu Z (2008) Field scale demonstration of fungi-bacteria augmented remediation of petroleum-contaminated soil. Huanjing Kexue 29:454–461
Hasinger M, Scherr KE, Lundaa T, Bräuer L, Zach C, Loibner AP (2012) Changes in iso- and n-alkane distribution during biodegradation of crude oil under nitrate and sulphate reducing conditions. J Biotechnol 157:490–498. https://doi.org/10.1016/j.jbiotec.2011.09.027
Hatta Y et al (2016) Detection of <i > Campylobacter jejuni</i > in rectal swab samples from <i > Rousettus amplexicaudatus</i > in the Philippines. J Vet Med Sci 78:1347–1350. https://doi.org/10.1292/jvms.15-0621
Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF (2011) Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. https://doi.org/10.1038/ismej.2011.41
Huang X, Liu L, Teng W, Zhang J, Wang F, Cai Z (2016) Changes in the soil microbial community after reductive soil disinfestation and cucumber seedling cultivation. Appl Microbiol Biotechnol 100(12):1–13.
Huang Y, Li L (2014) Biodegradation characteristics of naphthalene and benzene, toluene, ethyl benzene, and xylene (BTEX) by bacteria enriched from activated sludge water. Environ Res 86:277–284
Hutchins SR, Sewell GW, Kovacs DA, Smith GA (1991) Biodegradation of aromatic hydrocarbons by aquifer microorganisms under denitrifying conditions. Environ Sci Technol 25:68–76
Jørgensen K, Puustinen J, Suortti A-M (2000) Bioremediation of petroleum hydrocarbon-contaminated soil by composting in biopiles. Environ Pollut 107:245–254
Jungmann D, Ludwichowski KU, Faltin V, Benndorf J (2015) A field study to investigate environmental factors that could effect microcystin synthesis of a microcystis population in the Bautzen reservoir. Int Rev Hydrobiol 81:493–501
Kern M, Watzinger A, Scherr KE (2017) 15 N-nitrate and 34S-sulfate isotopic fractionation reflects electron acceptor ‘recycling’ during hydrocarbon biodegradation. New Biotechnol 38:91–100. https://doi.org/10.1016/j.nbt.2016.03.004
Kim J et al (2014) Draft genome sequence of Sphingopyxis sp. strain MWB1, a crude-oil-degrading marine bacterium. Genome Announc 2:e01256–e01214
Kim MK, Jung H-Y (2007) Chitinophaga terrae sp. nov., isolated from soil. Int J Syst Evol Microbiol 57:1721–1724. https://doi.org/10.1099/ijs.0.64964-0
Korenblum E, Souza DB, Penna M, Seldin L (2012) Molecular analysis of the bacterial communities in crude oil samples from two Brazilian offshore petroleum platforms. Int J Microbiol 2012:156537
Kostka JE et al (2011) Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the deepwater horizon oil spill. Appl Environ Microbiol 77:7962–7974. https://doi.org/10.1128/aem.05402-11
Kwapisz E, Wszelaka J, Marchut O, Bielecki S (2008) The effect of nitrate and ammonium ions on kinetics of diesel oil degradation by Gordonia alkanivorans S7. Int Biodeterior Biodegradation 61:214–222. https://doi.org/10.1016/j.ibiod.2007.08.002
Lai Q, Wang L, Liu Y, Yuan J, Sun F, Shao Z (2011) Parvibaculum indicum sp. nov., isolated from deep-sea water. Int J Syst Evol Microbiol 61:271–274
Lan Y, Wang Q, Cole JR, Rosen GL (2012) Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS One 7:e32491
Leys NM, Ryngaert A, Bastiaens L, Verstraete W, Top EM, Springael D (2004) Occurrence and phylogenetic diversity of Sphingomonas strains in soils contaminated with polycyclic aromatic hydrocarbons. Appl Environ Microbiol 70:1944–1955
Li J (1999) Determination of oil content in soil by ultravioletvisible spectrophotometric metod and super sonic extraction. J Univ Petroleum China
Li X, Rui J, Mao Y, Yannarell A, Mackie R (2014) Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol Biochem 68:392–401
Liang Y et al (2011) Functional gene diversity of soil microbial communities from five oil-contaminated fields in China. ISME J 5:403
Liu Q, Tang J, Liu X, Song B, Zhen M, Ashbolt NJ (2019) Vertical response of microbial community and degrading genes to petroleum hydrocarbon contamination in saline alkaline soil. J Environ Sci 81:80–92. https://doi.org/10.1016/j.jes.2019.02.001
Liu W, Hou J, Wang Q, Ding L, Luo Y (2014) Isolation and characterization of plant growth-promoting rhizobacteria and their effects on phytoremediation of petroleum-contaminated saline-alkali soil. Chemosphere 117:303–308. https://doi.org/10.1016/j.chemosphere.2014.07.026
Liu W, Yin D, Li N, Hou X, Wang D, Li D, Liu J (2016) Influence of environmental factors on the active substance production and antioxidant activity in Potentilla fruticosa L. and its quality assessment. Sci Rep 6:28591
Liu Y, Zhang Z, Li Y, Wen Y, Fei Y (2017) Response of soil microbial communities to roxarsone pollution along a concentration gradient. J Environ Sci Health Part A 52:819–827. https://doi.org/10.1080/10934529.2017.1281687
Lozupone C, Knight R (2005) UniFrac: a newphylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228
Lu L, Tyler H, Song J, Yi Z, Zhiyong Jason R (2014) Microbial metabolism and community structure in response to bioelectrochemically enhanced remediation of petroleum hydrocarbon-contaminated soil. Environ Sci Technol 48:4021
Lu R (2000) Analysis methods of soil agricultural chemistry. China Agricultural Science and Technology Publishing House (In Chinese). China Agricultural Science and Technology Press, Beijing
Lueders T (2017) The ecology of anaerobic degraders of BTEX hydrocarbons in aquifers. FEMS Microbiol Ecol 93:fiw220. https://doi.org/10.1093/femsec/fiw220
Margesin R, Spröer C, Schumann P, Schinner F (2003) Pedobacter cryoconitis sp. nov., a facultative psychrophile from alpine glacier cryoconite. Int J Syst Evol Microbiol 53:1291–1296. https://doi.org/10.1099/ijs.0.02436-0
Mason OU et al (2014) Metagenomics reveals sediment microbial community response to Deepwater Horizon oil spill. ISME J
Menendez-Vega D, Gallego JLR, Pelaez AI, Fernandez de Cordoba G, Moreno J, Muñoz D, Sanchez J (2007) Engineered in situ bioremediation of soil and groundwater polluted with weathered hydrocarbons. Eur J Soil Biol 43:310–321. https://doi.org/10.1016/j.ejsobi.2007.03.005
Mishra S, Jyot J, Kuhad RC, Lal B (2001) Evaluation of inoculum addition to stimulate in situ bioremediation of oily-sludge-contaminated soil. Appl Environ Microbiol 67:1675–1681
Mrozik A, Piotrowska-Seget Z (2010) Bioaugmentation as a strategy for cleaning up of soils contaminated with aromatic compounds. Microbiol Res 165:363–375. https://doi.org/10.1016/j.micres.2009.08.001
Mueller JG, Lantz SE, Blattmann BO, Chapman PJ (1991) Bench-scale evaluation of alternative biological treatment processes for the remediation of pentachlorophenol-and creosote-contaminated materials. Solid Phase Bioremediation Environ Sci Technol 25:1045–1055
Mulla A, Fernandes G, Menezes L, Meena RM, Naik H, Gauns M, Damare S (2017) Diversity of culturable nitrate-reducing bacteria from the Arabian Sea oxygen minimum zone. Deep Sea Research Part II: Topical Studies in Oceanography. https://doi.org/10.1016/j.dsr2.2017.12.014
Ning Z, Liu YC, Zhang S, Zhang CY, Ze HE, Yin MY (2013) Study of the isolation and screening of oil degradation microorganism and the construction and optimization of the mixed strains Sichuan environment
O’Mahony MM, Dobson AD, Barnes JD, Singleton I (2006) The use of ozone in the remediation of polycyclic aromatic hydrocarbon contaminated soil. Chemosphere 63:307–314
Peoples MB, Herridge DF, Ladha JK (1995) Biological nitrogen fixation: an efficient source of nitrogen for sustainable agricultural production? Plant Soil 174:3–28
Pizarro-Tobías P et al (2015) Field trial on removal of petroleum-hydrocarbon pollutants using a microbial consortium for bioremediation and rhizoremediation. Environ Microbiol Rep 7:85–94. https://doi.org/10.1111/1758-2229.12174
Pronk J, Meulenberg R, Hazeu W, Bos P, Kuenen J (1990) Oxidation of reduced inorganic sulphur compounds by acidophilic thiobacilli. FEMS Microbiol Rev 75:293–306
Razmjoo K, Adavi Z (2012) Assessment of bermudagrass cultivars for phytoremediation of petroleum contaminated soils. Int J Phytoremediation 14:14
Rodríguez-Caballero G, Caravaca F, Alguacil MM, Fernández-López M, Fernández-González AJ, Roldán A (2017) Striking alterations in the soil bacterial community structure and functioning of the biological N cycle induced by Pennisetum setaceum invasion in a semiarid environment. Soil Biol Biochem 109:176–187. https://doi.org/10.1016/j.soilbio.2017.02.012
Rodriguezr LM, Overholt WA, Hagan C, Huettel M, Kostka JE, Konstantinidis KT (2015) Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J 9:1928
Röling WF, Couto de Brito IR, Swannell RP, Head IM (2004) Response of archaeal communities in beach sediments to spilled oil and bioremediation. Appl Environ Microbiol 70:2614–2620
Rosario-Passapera R, Keddis R, Wong R, Lutz RA, Starovoytov V, Vetriani C (2012) Parvibaculum hydrocarboniclasticum sp. nov., a mesophilic, alkane-oxidizing alphaproteobacterium isolated from a deep-sea hydrothermal vent on the East Pacific Rise. Int J Syst Evol Microbiol 62:2921–2926. https://doi.org/10.1099/ijs.0.039594-0
Roy A et al (2018) Biostimulation and bioaugmentation of native microbial community accelerated bioremediation of oil refinery sludge. Bioresour Technol 253:22–32. https://doi.org/10.1016/j.biortech.2018.01.004
Salam LB, Ilori MO, Amund OO, LiiMien Y, Nojiri H (2017) Characterization of bacterial community structure in a hydrocarbon-contaminated tropical African soil Environmental Technology, pp 1–16. https://doi.org/10.1080/09593330.2017.1317838
Sayavedra-Soto LA, Gvakharia B, Bottomley PJ, Arp DJ, Dolan ME (2010) Nitrification and degradation of halogenated hydrocarbons—a tenuous balance for ammonia-oxidizing bacteria. Appl Microbiol Biotechnol 86:435–444. https://doi.org/10.1007/s00253-010-2454-1
Schleheck D et al (2011) Complete genome sequence of Parvibaculum lavamentivorans type strain (DS-1 T). Stand Genomic Sci 5:298–310. https://doi.org/10.4056/sigs.2215005
Shahi A, Aydin S, Ince B, Ince O (2016) Evaluation of microbial population and functional genes during the bioremediation of petroleum-contaminated soil as an effective monitoring approach. Ecotoxicol Environ Saf 125:153–160. https://doi.org/10.1016/j.ecoenv.2015.11.029
Shu D, He Y, Yue H, Wang Q (2016) Metagenomic and quantitative insights into microbial communities and functional genes of nitrogen and iron cycling in twelve wastewater treatment systems. Chem Eng J 290:21–30. https://doi.org/10.1016/j.cej.2016.01.024
Song M, Luo C, Jiang L, Zhang D, Wang Y, Zhang G (2015) Identification of Benzo[a]pyrene-Metabolizing Bacteria in Forest Soils by Using DNA-Based Stable-Isotope Probing. Appl Environ Microbiol 81:7368–7376. https://doi.org/10.1128/aem.01983-15
Sun J-Q, Xu L, Tang Y-Q, Chen F-M, Wu X-L (2012) Simultaneous degradation of phenol and n-hexadecane by Acinetobacter strains. Bioresour Technol 123:664–668. https://doi.org/10.1016/j.biortech.2012.06.072
Sun W et al (2015) Identification of anaerobic aniline-degrading bacteria at a contaminated industrial site. Environ Sci Technol 49:11079–11088
Suzuki I, Dular U, Kwok SC (1974) Ammonia or ammonium ion as substrate for oxidation by nitrosomonas europaea cells and extracts. J Bacteriol 120:556–558
Tong J et al (2019) Microbial community evolution and fate of antibiotic resistance genes along six different full-scale municipal wastewater treatment processes. Bioresour Technol 272:489–500. https://doi.org/10.1016/j.biortech.2018.10.079
Viggor S, Jõesaar M, Vedler E, Kiiker R, Pärnpuu L, Heinaru A (2015) Occurrence of diverse alkane hydroxylase alkB genes in indigenous oil-degrading bacteria of Baltic Sea surface water. Mar Pollut Bull 101:507–516. https://doi.org/10.1016/j.marpolbul.2015.10.064
Viñas M, Sabaté J, Espuny MJ, Solanas AM (2005) Bacterial community dynamics and polycyclic aromatic hydrocarbon degradation during bioremediation of heavily creosote-contaminated soil. Appl Environ Microbiol 71:7008–7018
Wang S et al (2012) Degradation of pyrene by immobilized microorganisms in saline-alkaline soil. J Environ Sci 24:1662–1669. https://doi.org/10.1016/S1001-0742(11)60963-7
Wang X (2011) Microbial diversity and bioremediation of petroleum-contaminated saline-alkali soil. University of Chinese Academy of Sciences
Wde B, Kowalchuk GA (2001) Nitrification in acid soils: micro-organisms and mechanisms. Soil Biol Biochem 33:853–866
White PJ, Broadley MR (2001) Chloride in soils and its uptake and movement within the Plant: a review. Ann Bot 88:967–988. https://doi.org/10.1006/anbo.2001.1540
Williams TJ, Ertan H, Ting L, Cavicchioli R (2009) Carbon and nitrogen substrate utilization in the marine bacterium Sphingopyxis alaskensis strain RB2256. ISME J 3:1036
Wu M, Li W, Dick WA, Ye X, Chen K, Kost D, Chen L (2017) Bioremediation of hydrocarbon degradation in a petroleum-contaminated soil and microbial population and activity determination. Chemosphere 169:124–130
Wu M et al (2016) Bioaugmentation and biostimulation of hydrocarbon degradation and the microbial community in a petroleum-contaminated soil. Int Biodeterior Biodegradation 107:158–164. https://doi.org/10.1016/j.ibiod.2015.11.019
Xu JG, Johnson RL, Yeung PY, Wang Y (1995) Nitrogen transformations in oil-contaminated, bioremediated, solvent-extracted and uncontaminated soils. Toxicol Environ Chem 47:109–118. https://doi.org/10.1080/02772249509358133
Xue J, Yu Y, Bai Y, Wang L, Wu Y (2015) Marine oil-degrading microorganisms and biodegradation process of petroleum hydrocarbon in marine environments: a review. Curr Microbiol 71:220–228
Yavari S, Malakahmad A, Sapari NB (2015) A review on phytoremediation of crude oil spills. Water Air Soil Pollut 226:279
Ye D, Siddiqi MA, Maccubbin AE, Subodh Kumar A, Sikka HC (2006) Degradation of polynuclear aromatic hydrocarbons by Sphingomonas paucimobilis. Environ Sci Technol 30:136–142
Young C-C et al (2007) Luteimonas composti sp. nov., a moderately thermophilic bacterium isolated from food waste. Int J Syst Evol Microbiol 57:741–744. https://doi.org/10.1099/ijs.0.64701-0
Yu KSH, Wong AHY, Yau KWY, Wong YS, Tam NFY (2005) Natural attenuation, biostimulation and bioaugmentation on biodegradation of polycyclic aromatic hydrocarbons (PAHs) in mangrove sediments. Mar Pollut Bull 51:1071–1077. https://doi.org/10.1016/j.marpolbul.2005.06.006
Zhang D-C, Schinner F, Margesin R (2010) Pedobacter bauzanensis sp. nov., isolated from soil. Int J Syst Evol Microbiol 60:2592–2595. https://doi.org/10.1099/ijs.0.018903-0
Zhang S et al (2017) Application of bioremediation in oil contaminated soil. J Groundwater Sci Engin 5:116–123
Zylstra GJ, Kim E (1997) Aromatic hydrocarbon degradation by Sphingomonas yanoikuyae B1. J Ind Microbiol Biotechnol 19:408–414
Acknowledgements
This research was funded by the National Key R&D Program of China (2018YFC1803301), The National Natural Science Foundation of China (No.41402233), and The Institute of Hydrogeology and Environmental Geology, CAGS Research Fund] grant number (SK201702 and SK201214). We thank Alex Boon, Ph.D., from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Ethics approval and consent to participate
Not applicable.
Funding
This research was funded by the National Key R&D Program of China (2018YFC1803301), The National Natural Science Foundation of China (No.41402233), and The Institute of Hydrogeology and Environmental Geology, CAGS Research Fund] grant number (SK201702 and SK201214).
Author information
Authors and Affiliations
Contributions
Pingping Cai carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. Zhuo Ning designed the study and carried out the experiments. Yaci Liu and Ze He participated in the experiments and data analyses. Manlan Niu and Jiansheng Shi participated in its design and coordination. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, P., Ning, Z., Liu, Y. et al. Diagnosing bioremediation of crude oil-contaminated soil and related geochemical processes at the field scale through microbial community and functional genes. Ann Microbiol 70, 36 (2020). https://doi.org/10.1186/s13213-020-01580-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-020-01580-x