
Abstract
Progressive and relatively circumscribed loss of semantic knowledge, referred to as semantic dementia (SD) which falls under the broader umbrella of frontotemporal dementia, was officially identified as a clinical syndrome less than 50 years ago. Here, we review recent neuroimaging, pathological, and genetic research in SD. From a neuroimaging perspective, SD is characterised by hallmark asymmetrical atrophy of the anterior temporal pole and anterior fusiform gyrus, which is usually left lateralised. Functional magnetic resonance imaging (fMRI) studies have revealed widespread changes in connectivity, implicating the anterior temporal regions in semantic deficits in SD. Task-related fMRI have also demonstrated the relative preservation of frontal and parietal regions alongside preserved memory performance. In addition, recent longitudinal studies have demonstrated that, with disease progression, atrophy encroaches into the contralateral temporal pole and medial prefrontal cortices, which reflects emerging changes in behaviour and social cognition. Notably, unlike other frontotemporal dementia subtypes, recent research has demonstrated strong clinicopathological concordance in SD, with TDP43 type C as the most common pathological subtype. Moreover, an underlying genetic cause appears to be relatively rare in SD, with the majority of cases having a sporadic form of the disease. The relatively clear diagnosis, clinical course, and pathological homogeneity of SD make this syndrome a promising target for novel disease-modifying interventions. The development of neuroimaging markers of disease progression at the individual level is an important area of research for future studies to address, in order to assist with this endeavour.
Similar content being viewed by others


Background
Semantic dementia (SD), a progressive neurodegenerative disorder affecting language, was empirically described only relatively recently. In the early 1970s, the conceptualisation of memory into two distinct systems, an episodic system and a semantic system by Tulving [1], coincided with the report by Warrington [2] of three individuals who presented with visual object agnosia, a profound inability to recognise or identify objects. In light of this new memory system and additional assessment, Warrington recognised that the constellation of symptoms of these patients could be conceptualised as an underlying loss of semantic memory. Since this seminal paper, the syndrome, which is characterised by circumscribed but profound loss of semantic knowledge, has been referred to as SD [3, 4] and, more recently, as semantic-variant primary progressive aphasia (PPA) [5]. Less than 50 years later, our understanding of this striking clinical syndrome has advanced. In this review, we will consider how recent studies in imaging, genetics, and pathology over the last decade have informed our knowledge of SD.
Contemporary consensus criteria for SD require individuals to first meet criteria for PPA; i.e. the most prominent clinical symptom to be in the domain of language, and evidence of subsequent impaired activities of daily living. Then, sub-classification as semantic-variant is based on impaired confrontation naming and single-word comprehension, with supportive features including impaired object knowledge, surface dyslexia or dysgraphia, spared repetition, and spared speech production. In a series of 100 cases all of whom underwent longitudinal follow-up, the mean age at presentation was 64.2 years but with a range of 40–79 years [6]. There was a 50% survival of 12.8 years indicating a slower progression than in other forms of frontotemporal dementia [6]. Studies of the prevalence and incidence of SD have been relatively limited; however, a recent epidemiology study estimated the prevalence of frontotemporal dementia at 10.8/100,000, with SD accounting for approximately one-third of these cases [7] in line with previous estimates [8]. Whether this prevalence is similar across countries, however, remains to be examined, as most existing epidemiological data hail from European studies.
Clinical presentation and cognitive profile
Clinically, patients with SD show a speech profile that is relatively fluent but empty of content, producing a pattern of so-called logorrhoea. Importantly, loss of semantic knowledge is observed irrespective of testing modality [9]. Impaired word comprehension is a mandatory feature and patients demonstrate word alienation in that they are able to repeat words such as “violin” or “caterpillar” but have no idea of their meaning. This deficit gradually progresses from low frequency and less familiar words, such as those mentioned, to more common words. Adlam et al. [10] demonstrated that SD patients are also impaired on non-verbal semantic matching tasks, tests of colour knowledge, sound knowledge, and object-use knowledge, which do not require naming or verbal comprehension even from an early stage of the disease. Such findings have provided evidence that, in SD, symptomatology reflects a profound and progressive loss of conceptual knowledge which is not limited to performance on verbal tasks [11]. There is also accompanying surface dyslexia: patients are unable to correctly pronounce irregular words such as pint which they read to rhyme with hint or flint.
In contrast, recent studies have confirmed that episodic memory is relatively preserved in SD, particularly when tasks with minimal conceptual loading are employed [12, 13]. The intact performance on traditional non-conceptually loaded episodic memory tasks converges with the performance of SD patients on autobiographical memory tasks. Patients typically show relatively preserved recollection of recent autobiographical memory in the context of poorer remote autobiographical memory (known as the reverse temporal gradient or step-function), reflecting increased semanticisation of past events (e.g. [14–16]). This is in stark contrast to the compromised ability of SD patients to project forwards in time to imagine possible future events (e.g. [17]). These deficits in future-oriented thought are attributable to semantic processing impairments, and have led to the advancement of the semantic scaffolding hypothesis which proposes that semantic knowledge is required to impart structure and meaning during the process of future simulation [18].
Changes in behaviour and social cognition are increasingly recognised in SD [19]. Clinically, SD patients often show mental rigidity and inflexible behaviour. For example, patients may become obsessive in tasks they engage in (e.g. we have noticed patients spending hours completing jigsaw puzzles), food preferences (usually restricted to specific foods), or daily routines (e.g. clockwatching). In addition, SD patients may have increased apathy and changes in eating behaviour, as well as loss of empathy, impaired emotion perception and emotional memories, and reduced theory of mind capacity [20–24]. Over time, many patients become essentially mute with only a limited repertoire of stereotypic phrases and a complete loss of word comprehension. Changes in emotional capacity as well as increased rigid behaviours are associated with higher carer burden (e.g. [25]), and progressive behavioural changes and/or increasing disability leads to residential care in most cases [6] (Table 1).
Imaging
Structural imaging
An extensive body of brain imaging studies have investigated structural and functional brain abnormalities in patients with SD. At presentation, visual inspection of magnetic resonance imaging (MRI) typically reveals hallmark bilateral, but asymmetric atrophy of the anterior temporal lobes, which is usually left lateralised (Fig. 1). With the development of neuroimaging techniques to statistically measure this degeneration, whole-brain structural MRI studies using voxel-based morphometry (VBM) have confirmed grey matter loss, which is relatively localised to the temporal lobe (left-predominant), with some involvement of frontal and limbic regions (e.g. [26]). Specifically, these regions include asymmetric but bilateral involvement of the temporal pole, the fusiform gyrus, middle and inferior temporal gyrus, ventromedial prefrontal cortex, amygdala, hippocampus, and the insula, which have been confirmed by a recent meta-analysis [27]. Importantly, the asymmetric hippocampal involvement is also considered one of the hallmark features of SD. In addition, surface-based imaging studies have demonstrated predominantly left anterior temporal cortical thinning on both the lateral and ventral surfaces of the temporal lobe (e.g. [28, 29]). Debate continues concerning the most critical region, but it appears that bilateral atrophy of the anterior fusiform region is required to generate the syndrome of SD.

Axial MRI scans showing typical anterior and middle temporal structural abnormalities in early left and right lateralised SD. L left, R right, SD semantic dementia
More recently, white matter changes in SD have also been mapped using diffusion tensor imaging (DTI) and tractrography analyses. These studies have demonstrated that patients with SD also have reduced white matter integrity in the left temporal lobe, periventricular white matter, corpus callosum, and in white matter tract areas of the fornix, inferior longitudinal fasciculus, and the uncinate fasciculus [30]. Studies using DTI have also shown reduced structural connectivity in frontotemporal pathways in SD, particularly in the uncinate, arcuate, and inferior longitudinal fasciculi [31, 32]. Although a range of DTI metrics have been applied across studies, the areas of abnormality show spatial overlap and are mostly adjacent to regions showing grey matter atrophy [33, 34]. Recently, a tractrography study has uncovered the role of the frontal aslant tract in verbal fluency whilst degeneration of the uncinate fasciculus is uniquely correlated with semantic deficits [35].
Despite the hallmark pattern of atrophy at presentation in this syndrome, how atrophy progresses over time has been relatively poorly understood, in part due to the methodological difficulties in acquiring and analysing longitudinal neuroimaging data. In recent years, however, rapid progress has been made in this area, with an emergence of longitudinal neuroimaging studies tracking progression. These studies have revealed that, in SD, atrophy extends from the anterior temporal lobe into the posterior temporal and/or the inferior frontal lobes with disease progression [36]. Some studies have suggested left greater than right hemisphere atrophy with disease progression [37]. A recent longitudinal imaging study in a larger cohort, however, demonstrated progressive right hemisphere cortical atrophy in SD, despite patients showing left-dominant atrophy at presentation [38]. Longitudinal studies of white matter changes have also revealed involvement of the right hemisphere with disease progression, with focal left lateralized degeneration involving the uncinate and anterior inferior longitudinal fasciculi at baseline, which spreads to the right hemisphere with disease progression [39, 40]. In summary, although the cortical atrophy is relatively localised in the left hemisphere early in the disease, progressive grey and white matter involvement of the frontal and contralateral temporal lobe is observed as the disease progresses (Fig. 2).

Brain imaging findings in SD at presentation and with disease progression. a Cross-sectional multimodal imaging findings in 10 SD patients versus 21 healthy controls: reduced regional grey matter density (top row), reduced FDG-PET (second row), increased radial diffusivity (third row), and composite of multimodal findings (fourth row). From Acosta-Carbonero et al. [31] with permission. b Baseline and longitudinal changes in cortical thickness in 22 left SD vs 9 right SD patients. From Kumfor et al. [14, 38] with permission. c Longitudinal white matter changes from baseline in 11 SD patients. From Lam et al. [40] with permission. FA fractional anisotropy, FDG-PET fluorodeoxyglucose positron emission tomography, FDR, L left, MD mean diffusivity, R right, RadialD radial diffusivity, SD semantic dementia, TBSS tract-based spatial statistics, VBM voxel-based morphometry, FDR false discovery rate
Molecular imaging
In addition to MRI, 2-(Fluorine-18)fluoro-2-deoxy-d-glucose (18F-FDG) positron emission tomography (PET) (FDG-PET) [41] has been used to visualize cerebral glucose metabolism, a measure which increases with regional synaptic activity and decreases with synaptic dysfunction or neural degeneration. As such, FDG-PET is a functional imaging marker useful for early diagnosis of dementia, although evidence regarding its utility for differential diagnosis in PPA syndromes is limited [42]. In SD, unsurprisingly, left dominant cerebral glucose metabolism is reduced in the temporal lobes [3], especially the left temporal pole [43] and the hippocampus [36]. These metabolic reductions correspond to observed regional grey matter atrophy patterns [44]. Longitudinal FDG-PET studies have also reported right lateralised reduction in glucose metabolism in the temporal lobes with disease progression, which is associated with impaired cognitive performance [45].
Functional imaging
Recent studies have also begun to investigate functional brain changes in SD. Functional MRI (fMRI) measures brain activity by detecting changes associated with the blood flow and the blood oxygen level-dependent (BOLD) response. fMRI can be performed either at rest (i.e. resting-state fMRI) or during performance of specific tasks to examine baseline brain activity or activation concomitant with cognitive performance [46]. Whole-brain and regional functional connectivity can be further derived from resting-state fMRI by measuring the correlation of the time series of BOLD signals across brain regions [47, 48]. Resting-state fMRI studies in SD have generally found reduced functional connectivity in the executive [30] and language [32] networks. Seed-based analyses in SD have described extensive disruptions in functional connectivity between the anterior temporal lobe and a broad range of brain regions across the temporal, frontal, parietal, and occipital lobes [49]. Independent component analysis has indicated that SD is also associated with changes in functional connectivity in the prefrontal cortex bilaterally, the anterior cingulate, and in different components within the default mode, salience, and emotion networks [50]. In summary, these results show that SD patients manifest extensive functional connectivity alterations beyond the core atrophic regions in the anterior temporal lobes and related language networks. It should be noted that while these studies have revealed extensive changes in functional connectivity associated with SD, most have not examined how these changes relate to hallmark cognitive and behavioural symptoms in these patients. As such, future studies addressing the behavioural relevance of the observed brain connectivity changes are warranted.
Imaging studies have recently begun to establish the neural correlates of the clinical and cognitive changes associated with SD. In SD, bilateral (yet asymmetrical left > right) neurodegeneration of the anterior temporal lobes is associated with profound semantic deficits, yet syntax, phonology, and fluency are strikingly spared. A set of recent fMRI studies sought to differentiate components of language processing in SD. These studies have described both task-positive and task-negative changes in the language network in SD patients compared to controls showing: i) decreased activation in the fusiform and superior temporal gyrus [51, 52]; ii) increased activation in the intraparietal sulcus [51], the inferior frontal gyrus [52], and the left superior temporal gyrus [53]; and iii) failure of deactivation in the anterior temporal lobe [54]. These results further emphasise the suggested role of the anterior temporal lobes in the combination of both low-level perceptual processing and higher-level integration of semantic processing. Taken together, these findings suggest that spared syntactic processing in SD depends on preserved functionality of left frontal, parietal and, to lesser degree, posterior temporal regions. The relative integrity of regions beyond the medial temporal cortices, such as the posterior cingulate and frontal cortices, also seems to sub-serve other cognitive functions such as the preserved episodic memory performance in SD patients [13, 55].
Right lateralised semantic dementia
A proportion of patients present with right greater than left lateralised atrophy, referred to as right SD, or right temporal variant frontotemporal dementia [56]. These patients often present with profound behavioural changes, which can make distinction from the behavioural variant of frontotemporal dementia challenging [57]. Importantly, increasing evidence has revealed that the extent of behavioural and social cognition changes is related to integrity of the right temporal pole in this syndrome [21, 38]. Patients with right lateralised atrophy also tend to show greater social cognition deficits than patients with left lateralised SD, while a subset present with prosopagnosia as the primary clinical feature [20, 38]. Improved diagnosis of right SD, together with better understanding of features which give rise to the manifestation of this syndrome, will be important for future studies to address.
Pathology
Volumetric analyses in autopsy tissue have shown that, in addition to the frontal and anterior temporal cortices, significant degeneration in the cingulate cortices, anterior thalamus, and hippocampal head is apparent by the end of the disease course [58]. The regions within the limbic memory circuit (also known as the Papez circuit) that remain intact include the mammillary bodies, hippocampal body and tail, and memory relays between these key regions, a finding that almost certainly accounts for the relative preservation of episodic memory in patients with SD [15, 58]. Interestingly, a selective loss of von Economo neurons has been identified in the anterior cingulate cortices in SD [58, 59] and may account for some of the behavioural deficits that emerge with disease progression [19].
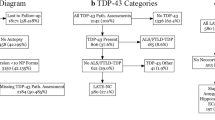
At a microscopic level, the main class of pathology identified in patients with SD is frontotemporal lobar degeneration with TAR DNA-binding protein 43 (FTLD-TDP) [6, 60, 61]. Based on the morphology and cortical distribution of the TDP43 lesions, four classes of FTLD-TDP (subtypes A–D) are recognised [62, 63]. In contrast to TDP subtypes A, B, and D, where TDP-43 manifests as neuronal cytoplasmic inclusions with and without short dystrophic neurites and/or intranuclear inclusions, TDP type C is characterised by long dystrophic neurites [62, 63] (see Fig. 3). Notably, clinicopathological studies have consistently found TDP type C to account for the majority of patients with SD. Neuroimaging analyses of TDP subtypes have found TDP type C to be associated with asymmetric anterior temporal lobe atrophy, but this likely reflects the high representation of SD in patients with this pathological subtype [64]. Prominent left anterior temporal thinning is also seen in patients with SD who have FTLD-ubiquitin pathology at autopsy [65].

llustration of the FTLD-TDP subtypes. From Tan et al. [63] with permission
Nevertheless, FTLD-TDP types A and B, Tau-positive frontotemporal lobar degeneration (FTLD-tau) and Alzheimer’s disease (AD) have been reported in 17% to 32% of patients with SD [6, 64, 66, 67]. Based on the morphology and distribution of the predominant species of tau deposited, FTLD-tau cases are subclassified into 3R (i.e. Pick’s disease) and 4R tauopathies (i.e. corticobasal degeneration, progressive supranuclear palsy and, more recently, globular glial tauopathy) [68, 69]. Almost all patients with SD and underlying FTLD-tau pathology demonstrate the 3R tauopathy Pick’s disease [6, 61, 67], with only one recent report of a patient in which the 4R globular glial tauopathy was identified [70].
Finally, recent research efforts have attempted to identify FTLD pathological subtypes via cerebrospinal fluid (CSF) biomarkers. Increased neurofilament light chain (NfL) levels in the CSF are associated with neuronal and axonal degeneration, and have been reported in patients with neurodegenerative diseases, particularly in patients with probable TDP43 pathology [71–74]. Specifically, increased NfL levels in the CSF have been reported in patients with SD [71, 72]. Although preliminary, these studies indicate that increased NfL levels in the CSF may represent a promising marker of underlying TDP pathology. In summary, emerging clinicopathological research demonstrates that, in the vast majority of cases, TDP43 type C is the most common pathological subtype, rendering SD one of the most pathologically homogenous frontotemporal syndromes.
Genetics
Unlike other forms of frontotemporal dementia, SD is typically sporadic, and a suggestive family history is identified in around 5% of patients only [6, 75]. In the patients that do have a highly positive family history of dementia, mutation in the progranulin (GRN) or expansion of the chromosome 9 open reading frame 72 (C9ORF72) gene have been described [76, 77]. Notably, however, these mutations are very rarely found in sporadic SD [78] and the C9ORF72 expansion has only been described in two patients, both of whom demonstrated co-existing behavioural changes [79, 80].
Interestingly, although a diagnosis of SD in association with a frontotemporal dementia genetic abnormality is rare, semantic impairment appears to develop quite commonly in individuals carrying a mutation in the GRN or microtubule associated protein tau (MAPT) genes [75, 81]. Given that SD is consistently associated with FTLD-TDP type C pathology, the rarity of mutations identified in this syndrome is consistent with other recognized geno-pathological associations within the frontotemporal dementia spectrum (e.g. GRN with FTLD-TDP subtype A; C9ORF72 with FTLD-TDP subtype B; and MAPT with FTLD-tau) [62]. With this in mind, in individuals clinically presenting with SD and with an absence of a strong family history, clinicians can be confident that an underlying genetic abnormality is unlikely.
Conclusions
As this review reveals, despite a relatively short history much knowledge has been gained about the SD syndrome, particularly over the last decade. Indeed, SD appears to be one of the more straightforward frontotemporal dementia subtypes. It has a clear clinical course, which begins with language features and, with progression, affects behaviour and social cognition; this reflects early and relatively circumscribed neurodegeneration of the anterior temporal pole, which encroaches into medial prefrontal and posterior temporal regions as well as into the contralateral hemisphere with disease progression. Pathologically, it is most commonly associated with TDP43 type C and genetic causes are rare.
In spite of this progress, a number of outstanding questions remain which we hope research over the next decade will address. Clinical phenotype is clearly influenced by the laterality of pathology and associated atrophy in this syndrome, with left lateralised atrophy initially manifesting as loss of semantic knowledge (i.e. anomia) and right lateralised atrophy initially manifesting as loss of person knowledge (i.e. prosopagnosia, knowledge of social norms), giving important insights into the representation of conceptual knowledge across hemispheres [36, 82]. Currently, it remains unclear why only a subset of cases (~30% [56]) present with right lateralised atrophy, with the majority of patients presenting with left lateralised neurodegeneration. Future studies that consider pre-clinical variables (e.g. handedness, occupational history, learning disabilities) with the potential to influence vulnerability of brain hemispheres to disease, may shed light on this issue (e.g. [83]). Indeed, it has been suggested that patients with PPA have a higher rate of pre-existing language disorders than would be expected in the general population, but this has not been investigated specifically in the SD clinical phenotype [83]. In addition, from a management perspective, changes in behaviour, capacity to engage in social situations, and reduced empathy lead to increased burden and stress in carers, which is often greater than in carers of patients with the behavioural variant of frontotemporal dementia [14]. This may reflect inadequate psychoeducation of carers regarding the manifestation of behavioural change in SD patients which is important for clinicians to consider when interacting with family members and carers of SD patients.
Clinically, one of the key issues on the horizon is the development of drugs that target the deposition or clearance of pathology. As these are likely to be specific to pathological subtypes, SD appears to be a promising syndrome for drug developers to target, given the striking clinicopathological concordance. Development of such agents, however, has been hindered by the lack of suitable animal models showing pathological changes that mirror those seen in SD. Knowledge is also lacking about the basic biological processes that underlie the deposition of TDP43 type C in SD. Recent advances in CSF studies may represent a promising avenue to identify FTLD pathological subtypes in vivo. The development of biomarkers such as these (e.g. CSF NfL, neuroimaging, blood biomarkers) are essential for discrimination, prognosis, and prediction of disease progression in SD. Improved understanding of the pathophysiological mechanisms which give rise to SD will also be essential for the development of novel drug interventions. From an imaging perspective, new techniques to measure change in brain integrity and function with disease progression have already started to make some headway. Applications of these techniques at the individual level are likely to be key to track disease progression and potentially measure the efficacy of interventions as these become available.
Abbreviations
- 18F-FDG:
-
2-(Fluorine-18)fluoro-2-deoxy-d-glucose
- AD:
-
Alzheimer’s disease
- BOLD:
-
Blood oxygen level-dependent
- CSF:
-
Cerebrospinal fluid
- DTI:
-
Diffusion tensor imaging
- fMRI:
-
Functional magnetic resonance imaging
- FTLD:
-
Frontotemporal lobar degeneration
- MRI:
-
Magnetic resonance imaging
- NfL:
-
Neurofilament light chain
- PET:
-
Positron emission tomography
- PPA:
-
Primary progressive aphasia
- SD:
-
Semantic dementia
- TDP:
-
TAR DNA-binding protein 43
- VBM:
-
Voxel-based morphometry
References
Tulving E. Episodic and semantic memory. New York and London: Academic Press; 1972.
Warrington EK. The selective impairment of semantic memory. Q J Exp Psychol. 1975;27(4):635–57.
Hodges JR, Patterson K, Oxbury S, Funnell E. Semantic dementia: progressive fluent aphasia with temporal lobe atrophy. Brain. 1992;115(6):1783–806.
Snowden JS, Goulding P, Neary D. Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol. 1989;2(3):167–182.
Gorno-Tempini ML, Hillis A, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14.
Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, Nestor PJ, Patterson K. Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain. 2010;133(Pt 1):300–6.
Coyle-Gilchrist IT, Dick KM, Patterson K, Rodríquez PV, Wehmann E, Wilcox A, Lansdall CJ, Dawson KE, Wiggins J, Mead S. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736–43.
Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58(11):1615–21.
Savage S, Hsieh S, Leslie F, Foxe D, Piguet O, Hodges JR. Distinguishing subtypes in primary progressive aphasia: application of the Sydney Language Battery. Dement Geriatr Cogn Disord. 2013;35(3–4):208–18.
Adlam A-L, Patterson K, Rogers T, Nestor P, Salmond C, Acosta-Cabronero J, Hodges J. Semantic dementia and fluent primary progressive aphasia: two sides of the same coin? Brain. 2006;129(11):3066–80.
Patterson K, Nestor PJ, Rogers TT. Where do you know what you know? The representation of semantic knowledge in the human brain. Nat Rev Neurosci. 2007;8(12):976–87.
Adlam A-LR, Patterson K, Hodges JR. “I remember it as if it were yesterday”: memory for recent events in patients with semantic dementia. Neuropsychologia. 2009;47:1344–51.
Irish M, Bunk S, Tu S, Kamminga J, Hodges JR, Hornberger M, Piguet O. Preservation of episodic memory in semantic dementia: the importance of regions beyond the medial temporal lobes. Neuropsychologia. 2016;81:50–60.
Kumfor F, Teo D, Miller L, Lah S, Mioshi E, Hodges JR, Piguet O, Irish M. Examining the relationship between autobiographical memory impairment and carer burden in dementia syndromes. J Alzheimers Dis. 2016;51(1):237–48.
Irish M, Hornberger M, Lah S, Miller L, Pengas G, Nestor PJ, Hodges JR, Piguet O. Profiles of recent autobiographical memory retrieval in semantic dementia, behavioural-variant frontotemporal dementia, and Alzheimer’s disease. Neuropsychologia. 2011;49(9):2694–702.
Viard A, Desgranges B, Matuszewski V, Lebreton K, Belliard S, De La Sayette V, Eustache F, Piolino P. Autobiographical memory in semantic dementia: new insights from two patients using fMRI. Neuropsychologia. 2013;51(13):2620–32.
Irish M, Addis DR, Hodges JR, Piguet O. Considering the role of semantic memory in episodic future thinking: evidence from semantic dementia. Brain. 2012;135(7):2178–91.
Irish M, Piguet O. The pivotal role of semantic memory in remembering the past and imagining the future. Front Behav Neurosci. 2013;7:27.
Kumfor F, Piguet O. Disturbance of emotion processing in frontotemporal dementia: a synthesis of cognitive and neuroimaging findings. Neuropsychol Rev. 2012;22(3):280–97.
Irish M, Kumfor F, Hodges JR, Piguet O. A tale of two hemispheres: contrasting patterns of socioemotional dysfunction in left- versus right-lateralised semantic dementia. Dement Neuropsychologia. 2013;7(1):88–95.
Irish M, Hodges JR, Piguet O. Right anterior temporal lobe dysfunction underlies theory of mind impairments in semantic dementia. Brain. 2014;137(4):1241–53.
Van Langenhove T, Leyton CE, Piguet O, Hodges JR: Comparing longitudinal behavior changes in the primary progressive aphasias. J Alzheimers Dis. 2016; 53(3):1033–42.
Kumfor F, Irish M, Hodges JR, Piguet O. The orbitofrontal cortex is involved in emotional enhancement of memory: evidence from the dementias. Brain. 2013;136:2992–3003.
Kumfor F, Miller L, Lah S, Hsieh S, Savage S, Hodges JR, Piguet O. Are you really angry? The effect of intensity on emotion recognition in frontotemporal dementia. Soc Neurosci. 2011;6(5-6):502–14.
Hsieh S, Irish M, Daveson N, Hodges JR, Piguet O. When one loses empathy: its effect on carers of patients with dementia. J Geriatr Psychiatry Neurol. 2013;26(3):174–84.
Mummery CJ, Patterson K, Price CJ, Ashburner J, Frackowiak RS, Hodges JR. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol. 2000;47(1):36–45.
Yang J, Pan P, Song W, Shang HF. Quantitative meta-analysis of gray matter abnormalities in semantic dementia. J Alzheimers Dis. 2012;31(4):827–33.
Rohrer JD, Warren JD, Modat M, Ridgway GR, Douiri A, Rossor MN, Ourselin S, Fox NC. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. 2009;72(18):1562–9.
Rohrer JD, Ridgway GR, Crutch SJ, Hailstone J, Goll JC, Clarkson MJ, Mead S, Beck J, Mummery C, Ourselin S, et al. Progressive logopenic/phonological aphasia: erosion of the language network. Neuroimage. 2010;49(1):984–93.
Yang Q, Guo QH, Bi YC. The brain connectivity basis of semantic dementia: a selective review. CNS Neurosci Ther. 2015;21(10):784–92.
Acosta-Cabronero J, Patterson K, Fryer TD, Hodges JR, Pengas G, Williams GB, Nestor PJ. Atrophy, hypometabolism and white matter abnormalities in semantic dementia tell a coherent story. Brain. 2011;134(Pt 7):2025–35.
Pievani M, Filippini N, van den Heuvel MP, Cappa SF, Frisoni GB. Brain connectivity in neurodegenerative diseases—from phenotype to proteinopathy. Nat Rev Neurol. 2014;10(11):620–33.
La Joie R, Landeau B, Perrotin A, Bejanin A, Egret S, Pelerin A, Mezenge F, Belliard S, de La Sayette V, Eustache F, et al. Intrinsic connectivity identifies the hippocampus as a main crossroad between Alzheimer’s and semantic dementia-targeted networks. Neuron. 2014;81(6):1417–28.
Mahoney CJ, Malone IB, Ridgway GR, Buckley AH, Downey LE, Golden HL, Ryan NS, Ourselin S, Schott JM, Rossor MN, et al. White matter tract signatures of the progressive aphasias. Neurobiol Aging. 2013;34(6):1687–99.
Catani M, Mesulam MM, Jakobsen E, Malik F, Martersteck A, Wieneke C, Thompson CK, Thiebaut de Schotten M, Dell’Acqua F, Weintraub S, et al. A novel frontal pathway underlies verbal fluency in primary progressive aphasia. Brain. 2013;136(Pt 8):2619–28.
Hodges JR, Patterson K. Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol. 2007;6(11):1004–14.
Rogalski E, Cobia D, Martersteck A, Rademaker A, Wieneke C, Weintraub S, Mesulam MM. Asymmetry of cortical decline in subtypes of primary progressive aphasia. Neurology. 2014;83(13):1184–91.
Kumfor F, Landin-Romero R, Devenney E, Hutchings R, Grasso R, Hodges JR, Piguet O. On the right side? A longitudinal study of left- versus right-lateralized semantic dementia. Brain. 2016;139(Pt 3):986–98.
Tu S, Leyton CE, Hodges JR, Piguet O, Hornberger M. Divergent longitudinal propagation of white matter degradation in logopenic and semantic variants of primary progressive aphasia. J Alzheimers Dis. 2015;49(3):853–61.
Lam BY, Halliday GM, Irish M, Hodges JR, Piguet O. Longitudinal white matter changes in frontotemporal dementia subtypes. Hum Brain Mapp. 2014;35(7):3547–57.
Phelps ME, Huang SC, Hoffman EJ, Selin C, Sokoloff L, Kuhl DE. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Ann Neurol. 1979;6(5):371–88.
Kato T, Inui Y, Nakamura A, Ito K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res Rev. 2016;30:73–84.
Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, O’Neil JP, Lal RA, Dronkers NF, Miller BL, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64(4):388–401.
Diehl J, Grimmer T, Drzezga A, Riemenschneider M, Forstl H, Kurz A. Cerebral metabolic patterns at early stages of frontotemporal dementia and semantic dementia. A PET study. Neurobiol Aging. 2004;25(8):1051–6.
Diehl-Schmid J, Grimmer T, Drzezga A, Bornschein S, Perneczky R, Forstl H, Schwaiger M, Kurz A. Longitudinal changes of cerebral glucose metabolism in semantic dementia. Dement Geriatr Cogn Disord. 2006;22(4):346–51.
Buchbinder BR. Functional magnetic resonance imaging. Handb Clin Neurol. 2016;135:61–92.
Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8(9):700–11.
Smith SM, Vidaurre D, Beckmann CF, Glasser MF, Jenkinson M, Miller KL, Nichols TE, Robinson EC, Salimi-Khorshidi G, Woolrich MW, et al. Functional connectomics from resting-state fMRI. Trends Cogn Sci. 2013;17(12):666–82.
Guo CC, Gorno-Tempini ML, Gesierich B, Henry M, Trujillo A, Shany-Ur T, Jovicich J, Robinson SD, Kramer JH, Rankin KP, et al. Anterior temporal lobe degeneration produces widespread network-driven dysfunction. Brain. 2013;136(Pt 10):2979–91.
Farb NA, Grady CL, Strother S, Tang-Wai DF, Masellis M, Black S, Freedman M, Pollock BG, Campbell KL, Hasher L, et al. Abnormal network connectivity in frontotemporal dementia: evidence for prefrontal isolation. Cortex. 2013;49(7):1856–73.
Wilson SM, Brambati SM, Henry RG, Handwerker DA, Agosta F, Miller BL, Wilkins DP, Ogar JM, Gorno-Tempini ML. The neural basis of surface dyslexia in semantic dementia. Brain. 2009;132(Pt 1):71–86.
Agosta F, Henry RG, Migliaccio R, Neuhaus J, Miller BL, Dronkers NF, Brambati SM, Filippi M, Ogar JM, Wilson SM, et al. Language networks in semantic dementia. Brain. 2010;133(Pt 1):286–99.
Goll JC, Ridgway GR, Crutch SJ, Theunissen FE, Warren JD. Nonverbal sound processing in semantic dementia: a functional MRI study. Neuroimage. 2012;61(1):170–80.
Wilson SM, DeMarco AT, Henry ML, Gesierich B, Babiak M, Mandelli ML, Miller BL, Gorno-Tempini ML. What role does the anterior temporal lobe play in sentence-level processing? Neural correlates of syntactic processing in semantic variant primary progressive aphasia. J Cogn Neurosci. 2014;26(5):970–85.
Nestor PJ, Fryer TD, Hodges JR. Declarative memory impairments in Alzheimer’s disease and semantic dementia. Neuroimage. 2006;30(3):1010–20.
Chan D, Anderson V, Pijenburg Y, Whitwell JL, Barnes J, Scahill R, Stevens JM, Barkhof F, Scheltens P, Rossor MN, et al. The clinical profile of right temporal lobe atrophy. Brain. 2009;132:1287–98.
Kamminga J, Kumfor F, Burrell JR, Piguet O, Hodges JR, Irish M. Differentiating between right-lateralised semantic dementia and behavioural-variant frontotemporal dementia: an examination of clinical characteristics and emotion processing. J Neurol Neurosurg Psychiatry. 2015;86:1082–8.
Tan RH, Wong S, Kril JJ, Piguet O, Hornberger M, Hodges JR, Halliday GM. Beyond the temporal pole: limbic memory circuit in the semantic variant of primary progressive aphasia. Brain. 2014;137(Pt 7):2065–76.
Chow TW, Links KA, Masterman DL, Mendez MF, Vinters HV. A case of semantic variant primary progressive aphasia with severe insular atrophy. Neurocase. 2012;18(6):450–6.
Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, Dickson DW. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol (Berl). 2011;122(2):137–53.
Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM, Beck J, Hardy J, de Silva R, Warrington E, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011;134:2565–81.
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol (Berl). 2011;122(1):111–3.
Tan RH, Shepherd CE, Kril JJ, McCann H, McGeachie A, McGinley C, Affleck A, Halliday GM. Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol Commun. 2013;1:33.
Rohrer JD, Geser F, Zhou J, Gennatas ED, Sidhu M, Trojanowski JQ, Dearmond SJ, Miller BL, Seeley WW. TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology. 2010;75(24):2204–11.
Grossman M, Libon DJ, Forman MS, Massimo L, Wood E, Moore P, Anderson C, Farmer J, Chatterjee A, Clark CM, et al. Distinct antemortem profiles in patients with pathologically defined frontotemporal dementia. Arch Neurol. 2007;64(11):1601–9.
Chare L, Hodges JR, Leyton CE, McGinley C, Tan RH, Kril JJ, Halliday GM. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry. 2014;85(8):865–70.
Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, Dickson DW. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122(2):137–53.
Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, Giaccone G, Hatanpaa KJ, Holton JL, Josephs KA, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. 2013;126(4):537–44.
Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, White CL, Schneider JA, Grinberg LT, Halliday G, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22.
Graff-Radford J, Josephs KA, Parisi JE, Dickson DW, Giannini C, Boeve BF. Globular glial tauopathy presenting as semantic variant primary progressive aphasia. JAMA Neurol. 2016;73(1):123–5.
Landqvist Waldo M, Frizell Santillo A, Passant U, Zetterberg H, Rosengren L, Nilsson C, Englund E. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol. 2013;13:54.
Pijnenburg YA, Verwey NA, van der Flier WM, Scheltens P, Teunissen CE. Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimers Dement (Amst). 2015;1(4):505–12.
Tortelli R, Ruggieri M, Cortese R, D’Errico E, Capozzo R, Leo A, Mastrapasqua M, Zoccolella S, Leante R, Livrea P, et al. Elevated cerebrospinal fluid neurofilament light levels in patients with amyotrophic lateral sclerosis: a possible marker of disease severity and progression. Eur J Neurol. 2012;19(12):1561–7.
Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, Kramer JH, Rabinovici G, Ahlijanian M, Miller BL, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75(1):116–26.
Pickering-Brown SM, Richardson AM, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu WK, Yen SH, Hardy J, et al. Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain. 2002;125(Pt 4):732–51.
Cerami C, Marcone A, Galimberti D, Villa C, Fenoglio C, Scarpini E, Cappa SF. Novel missense progranulin gene mutation associated with the semantic variant of primary progressive aphasia. J Alzheimers Dis. 2013;36(3):415–20.
Le Ber I, Camuzat A, Guillot-Noel L, Hannequin D, Lacomblez L, Golfier V, Puel M, Martinaud O, Deramecourt V, Rivaud-Pechoux S, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis. 2013;34(2):485–99.
Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, Galloway NM, Feldman H, Woltjer R, Miller CA, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161–70.
Abbate C, Arosio B, Galimberti D, Nicolini P, Chiara LR, Rossi PD, Ferri E, Gussago C, Deriz M, Fenoglio C, et al. Phenotypic variability associated with the C9ORF72 hexanucleotide repeat expansion: a sporadic case of frontotemporal lobar degeneration with prodromal hyposmia and predominant semantic deficits. J Alzheimers Dis. 2014;40(4):849–55.
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135(Pt 3):693–708.
Rohrer JD, Crutch SJ, Warrington EK, Warren JD. Progranulin-associated primary progressive aphasia: a distinct phenotype? Neuropsychologia. 2010;48(1):288–97.
Olson IR, Plotzker A, Ezzyat Y. The Enigmatic temporal pole: a review of findings on social and emotional processing. Brain. 2007;130(Pt 7):1718–31.
Rogalski E, Johnson N, Weintraub S, Mesulam M. Increased frequency of learning disability in patients with primary progressive aphasia and their first-degree relatives. Arch Neurol. 2008;65(2):244–8.
Funding
RLR is supported by the ARC Centre of Excellence in Cognition and its Disorders Memory Node (CE11000102). RT is supported by a National Health and Medical Research Council (NHMRC)–Australian Research Council (ARC) Dementia Research Development Fellowship (APP1110369). FK is supported by an NHMRC-ARC Dementia Research Development Fellowship (APP1097026). This work was also supported by funding to Forefront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neurone disease, from NHMRC of Australia program grant (#1037746) and the ARC Centre of Excellence in Cognition and its Disorders Memory Node (#CE110001021).
Authors’ contributions
RLR, RT, JRH, and FK reviewed the literature and were involved in manuscript preparation and revision. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Landin-Romero, R., Tan, R., Hodges, J.R. et al. An update on semantic dementia: genetics, imaging, and pathology. Alz Res Therapy 8, 52 (2016). https://doi.org/10.1186/s13195-016-0219-5
Published:
DOI: https://doi.org/10.1186/s13195-016-0219-5