
Abstract
Background
Methylation-associated SOX family genes have been proved to be involved in multiple essential processes during carcinogenesis and act as potential biomarkers for cancer diagnosis, staging, prediction of prognosis, and monitoring of response to therapy. Herein, we revealed SOX30 methylation and its clinical implication in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
Results
In the discovery stage, we identified that SOX30 methylation, a frequent event in AML, was negatively associated with SOX30 expression and correlated with overall survival (OS) and leukemia-free survival (LFS) in cytogenetically normal AML among SOX family members from The Cancer Genome Atlas (TCGA) datasets. In the validation stage, we verified that SOX30 methylation level was significantly higher in AML even in MDS-derived AML compared to controls, whereas SOX30 hypermethylation was not a frequent event in MDS. SOX30 methylation was inversely correlated with SOX30 expression in AML patients. Survival analysis showed that SOX30 hypermethylation was negatively associated with complete remission (CR), OS, and LFS in AML, where it only affected LFS in MDS. Notably, among MDS/AML paired patients, SOX30 methylation level was significantly increased in AML stage than in MDS stage. In addition, SOX30 methylation was found to be significantly decreased in AML achieved CR when compared to diagnosis time and markedly increased in relapsed AML when compared to the CR population.
Conclusions
Our findings revealed that SOX30 methylation was associated with disease progression in MDS and acted as an independent prognostic and predictive biomarker in AML.
Similar content being viewed by others


Background
Acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) are common clonal disorders in myeloid malignancies. AML is etiologically, biologically, and clinically heterogeneous disease characterized by the accumulation of excessive blasts [1], whereas MDS is characterized by ineffective hematopoiesis and has a tendency to evolve into AML [2]. Cytogenetic and molecular analyses can identify recurrent chromosomal aberrations, gene mutations, and abnormal gene expression, which are associated with MDS/AML pathogenesis, response to therapy and prognosis [3, 4]. Despite recent progresses and advancements made in the understanding of disease biology and the personalized and precision treatment regimen, clinical outcome of AML even the MDS-derived AML remains unsatisfactory [1, 2]. Thus, the identification of underlying molecular events which correlated with disease progression and prognosis could make a better understanding of cancer pathogenesis and improve treatment outcome by the use of molecular risk-adapted treatment strategies [5].
DNA methylation, a common type of epigenetic DNA modification, plays a crucial role in maintenance of genome integrity, genomic imprinting, transcriptional regulation, and developmental processes [6]. In human cancers, aberrant DNA methylation is known to contribute to various biological processes of cancer development including initiation, promotion, invasion, metastases, and chemotherapy resistance [7]. Clinically, aberrant methylation in cancer-related genes acts as potential biomarkers for diagnosis, staging, prediction of prognosis, and monitoring of response to therapy [7]. As for myeloid malignancies, studies showed that aberrant DNA methylation was a dominant mechanism in MDS progression to AML [8]. For example, our previous investigation revealed that epigenetic dysregulation of ID4, which exhibited anti-proliferation and pro-apoptosis effects in leukemia cells, and predicted disease progression and treatment outcome in myeloid malignancies [9].
SOX [sex-determining region Y (SRY) box-containing] genes encode transcription factors belonging to the HMG (High Mobility Group) superfamily [10]. There are, at least, 19 members (SOX1-15, SOX17, SOX18, SOX21, SOX30) divided into eight groups (from A to H), based on their HMG sequence identity in humans [10]. The SOX genes have emerged as modulators of canonical Wnt/β-catenin signaling and have been attributed to their properties involving in the regulation of cell differentiation, proliferation, and survival in multiple essential processes during carcinogenesis [11]. Although most SOX genes show a property of oncogenes in various cancers, different members of the SOX gene family may play distinct roles in different types of cancers including hematological malignancies: some of them show an oncogenic role contributing to cancer development, whereas others act as a tumor suppressor gene to block the growth of cancers [12]. For instance, SOX4 overexpression resulting from C/EBPα inactivation or cooperating with CREB contributed to the development of AML [13, 14]. Man et al. reported that methylation-dependent SOX7 was a novel tumor suppressor in AML via a negative modulatory effect on the Wnt/β-catenin pathway [15]. Moreover, SOX12 exhibited pro-proliferative effect involved in leukemogenesis by regulating the expression of β-catenin and then interfering with TCF/Wnt pathway [16]. Our previous study also showed that reduced SOX17 expression was associated with adverse prognosis in cytogenetically normal AML (CN-AML) [17].
In this study, we examined the methylation pattern and clinical significance of SOX30 in AML and MDS. SOX30 methylation was a novel biomarker associated with prognosis and disease recurrence in AML and correlated with disease evolution in MDS. The results might provide us with novel insights into the mechanisms of MDS/AML leukemogenesis.
Methods
Patients and samples
A total of 196 AML patients (184 de novo AML and 12 MDS-derived AML), 104 MDS patients, and 28 healthy donors were enrolled in the present study approved by the Institutional Ethics Committee of the Affiliated People’s Hospital of Jiangsu University. The diagnosis and classification of MDS and AML patients were based on the 2016 World Health Organization (WHO) criteria [18]. The main clinical and laboratory features of AML and MDS patients were presented in Tables 1 and 2. After signing the written informed consents, bone marrow (BM) was collected from all participants at diagnosed time. Moreover, BM from 49 AML patients who achieved complete remission (CR) after induction therapy and 27 relapsed AML patients were also included. BM mononuclear cells (BMMNCs) were separated by density-gradient centrifugation using Lymphocyte Separation Medium (Absin, Shanghai, China) [9].
Treatment regimen
The treatment for MDS patients with lower IPSS scores (Low/Int-1) was symptomatic and supportive treatment with/without thalidomide/lenalidomide or EPO or cyclosporine together with ATG, whereas patients with higher IPSS scores (Int-2/High) received symptomatic and supportive treatment with/without chemotherapy included decitabine or HAG protocol (cytarabine, homoharringtonine, and granulocyte colony stimulating factor) or CAG protocol (cytarabine, aclacinomycin, and granulocyte colony stimulating factor) [19]. AML patients received chemotherapy including induction therapy and subsequent consolidation treatment. For non-M3 patients, induction therapy was daunorubicin/homoharringtonine/mitoxantrone combined with cytarabine. Subsequent consolidation treatment included high-dose cytarabine, mitoxantrone with cytarabine, and homoharringtonine combined with cytarabine. Meanwhile, for M3 patients, induction therapy was oral all-trans retinoic acid (ATRA) together with daunorubicin in combination with cytarabine. Maintenance therapy was oral mercaptopurine, oral methotrexate, and oral ATRA over 2 years [9, 20].
Cytogenetic analysis and gene mutation detection
BM cells were harvested after 1 to 3 days of unstimulated culture in RPMI 1640 medium (BOSTER, Wuhan, China) containing 20% fetal calf serum (ExCell Bio, Shanghai, China). The metaphase cells were banded by trypsin-Giemsa technique and karyotyped according to the recommendations of the International System for Human Cytogenetic Nomenclature (ISCN). Cytogenetics for AML and MDS patients were analyzed at the new diagnosis time by conventional R-banding method and karyotype risk was classified according to what was reported previously [21]. Mutations in NPM1, C-KIT, DNMT3A, N/K-RAS, U2AF1, and SRSF2 were detected by high-resolution melting analysis (HRMA) as reported previously [22,23,24,25,26], whereas mutations in FLT3-ITD and CEBPA were detected by DNA sequencing as reported previously [27, 28].
RNA isolation, reverse transcription, and RQ-PCR
Total RNA was isolated by using Trizol reagent and was synthesized into cDNA through reverse transcription as reported previously [9]. The primers used for SOX30 transcript detection were 5′-TGTCACACTTTTCCAGCCCA-3′ (forward) and 5′-TGAAATCCTGTTGGCGCTCT-3′ (reverse). Real-time quantitative PCR (RQ-PCR) was performed to detect SOX30 transcript using SYBR Premix Ex Taq II (TaKaRa, Tokyo, Japan). RQ-PCR conditions for SOX30 transcript level detection were 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, 66 °C for 30 s, 72 °C for 30 s, 88 °C for 30 s (collect fluorescence), and finally followed by the melting program at 95 °C for 15 s, 60 °C for 60 s, 95 °C for 15 s, and 60 °C for 15 s. The housekeeping gene ABL detected by RQ-PCR using 2× SYBR Green PCR Mix (Multisciences, Hangzhou, China) was used to calculate the abundance of SOX30 transcript. Relative SOX30 transcript level was calculated by 2−∆∆CT methods.
DNA isolation, bisulfite modification, and RQ-MSP
Genomic DNA isolation and modification were performed as reported previously [9]. Real-time quantitative methylation-specific PCR (RQ-MSP) was applied to examine SOX30 methylation level using AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Piscataway, NJ, USA) with primers reported previously [29]. RQ-MSP conditions for SOX30 methylation level detection were 95 °C for 5 min, 40 cycles for 10 s at 95 °C, 1 min at 68 °C, 1 min at 72 °C, 80 °C for 30 s (collect fluorescence), and finally followed by the melting program at 95 °C for 15 s, 60 °C for 60 s, 95 °C for 15 s, and 60 °C for 15 s. The gene ALU was used to calculate the abundance of SOX30 methylation level. Relative SOX30 methylation level was calculated by 2−∆∆CT methods.
BSP
The primers used for SOX30 methylation density detection were 5′-TTTTTGGGTAGTAGTTATGGAG-3′ (forward) and 5′-AACTTAACCACCCTAAAAACTC-3′ (reverse). Bisulfite sequencing PCR (BSP) was conducted using TaKaRa Taq™ Hot Start Version kit (Tokyo, Japan). BSP conditions for SOX30 methylation density detection were 10 s at 98 °C, 40 cycles for 10 s at 98 °C, 30 s at 58 °C, 30 s at 72 °C, and followed by a final 7 min at 72 °C. Clone sequencing of BSP products was performed as described previously [9], and five/six independent clones were sequenced (BGI Tech Solutions Co., Shanghai, China).
TCGA databases
SOX gene family methylation (HM450) and mRNA expression (RNA Seq V2 RSEM) data in a cohort of 200 AML patients (NEJM 2013) from The Cancer Genome Atlas (TCGA) [30] were downloaded via cBioPortal (http://www.cbioportal.org) [31, 32].
Bioinformatics analyses
The human disease methylation database DiseaseMeth version 2.0 (http://www.bio-bigdata.com/diseasemeth/analyze.html) was used for differential methylation analysis. The Genomicscape Survival Analysis (http://genomicscape.com/microarray/survival.php) was applied to determine the impact of SOX30 expression on survival of CN-AML patients.
Statistical analyses
Statistical analyses were conducted using SPSS software version 20.0 and GraphPad Prism 5.0. Mann-Whitney U test was carried to compare the difference of continuous variables between two groups, whereas Pearson chi-square analysis/Fisher exact test was applied to compare the difference of categorical variables between two groups. Correlation analysis was performed by Spearman test. Receiver operating characteristic (ROC) curve and area under the ROC curve (AUC) were carried out to test the performance of SOX30 methylation level in distinguishing AML patients from controls. CR was obtained after one or two courses of chemotherapy. Overall survival (OS) was measured from diagnosis to last follow-up or death from any cause. Leukemia-free survival (LFS) for MDS was calculated from diagnosis to progression to acute leukemia or the end of follow-up, whereas for AML was calculated from the day that CR was established until either relapse or death without relapse. The prognostic value of SOX30 methylation for survival (OS and LFS) was analyzed by Kaplan-Meier analysis and Cox regression analyses (univariate and multivariate analyses). All tests were two sided, and P < 0.05 was defined as statistically significant.
Results
Identification of methylation-dependent SOX gene associated with prognosis in AML
For initial selection of prognostic relevant methylation of SOX genes, we analyzed 19 members of SOX gene family by utilizing TCGA data. Among all SOX genes, methylation data was available for SOX5, SOX7, SOX8, SOX10, SOX12, SOX15, SOX18, and SOX30. To investigate their prognostic value in AML, we divided the patients into two groups by the median methylation level of each gene respectively. In whole-cohort AML patients, we did not observe the prognostic impact of SOX genes methylation on OS and LFS besides SOX30 showed a trend (Additional file 1: Figure S1). However, among CN-AML, OS and LFS were adversely affected by methylation in SOX10 and SOX30, but not in SOX5, SOX7, SOX8, SOX12, SOX15, and SOX18 (Fig. 1a).

Identification of methylation-dependent SOX genes associated with prognosis in AML. a The prognostic value of SOX gene methylation for OS and LFS among CN-AML patients from TCGA databases. SOX gene methylation (HM450) data was downloaded via cBioPortal (http://www.cbioportal.org). AML patients were divided into two groups by the median methylation level of each gene respectively. b Correlation between SOX genes expression and methylation among AML patients from TCGA databases. SOX gene methylation (HM450) and mRNA expression (RNA Seq V2 RSEM) data was downloaded via cBioPortal (http://www.cbioportal.org). The correlation analysis was conducted by Spearman test. c SOX30 methylation level in AML patients and controls obtained by bioinformatics analysis. SOX30 promoter (CpG island) methylation level was obtained through the human disease methylation database DiseaseMeth version 2.0 (http://www.bio-bigdata.com/diseasemeth/analyze.html). d The prognostic value of SOX30 expression for OS among CN-AML patients obtained by bioinformatics analysis. The effect of SOX30 expression on prognosis was determined by the Genomicscape Survival Analysis (http://genomicscape.com/microarray/survival.php)
As is well known, DNA methylation is a major mechanism that regulates gene expression. We next analyzed the association between SOX gene methylation and expression in AML patients from TCGA data. Significant negative association was shown in SOX8, SOX18, and SOX30, but not in SOX5, SOX7, SOX10, SOX12, and SOX15 (Fig. 1b). These data suggested methylation in SOX8, SOX18, and SOX30 may play a major role in regulating gene expression.
Finally, we used DiseaseMeth version 2.0 (based on TCGA and Gene Expression Omnibus) to identify whether SOX30 promoter (CpG island) was differentially methylated in AML. According to the analyses, the methylation level of SOX30 in AML was significantly higher than normal controls (P < 0.001, Fig. 1c). In addition, a recent investigation reported SOX gene family expression in leukemia and found SOX30 expression was significantly downregulated in AML [15]. By GenomicScape, patients with lower SOX30 mRNA level tended to have a shorter OS time than those with higher SOX30 mRNA level (P = 0.078, Fig. 1d). Taken together, we deduced that methylation-dependent SOX30 played a crucial role in leukemogenesis.
Validation of SOX30 hypermethylation was a frequent event and correlated with prognosis in AML
We designed RQ-MSP and BSP primer sets and assays at the CpG island of SOX30 gene promoter (Fig. 2a) to validate SOX30 methylation in AML patients and analyzed its clinical significance. Firstly, SOX30 methylation was examined by RQ-MSP, and AML patients had a significantly higher SOX30 methylation level than controls (Fig. 2b). Among the tested AML patients with available RNA samples, SOX30 expression, detected by RQ-PCR, was inversely correlated with SOX30 methylation (R = − 0.302, P = 0.001, n = 125, Fig. 2c). ROC curve analysis showed that SOX30 methylation may be acted as a potential biomarker for differentiating AML from controls with an AUC of 0.685 (95% CI 0.614–0.756, P = 0.002) (Additional file 1: Figure S2). We classified AML patients into two groups (hypermethylated and non-hypermethylated) based on the methylation level (1.024) at the cutoff point by ROC curve analysis (sensitivity = 51%, specificity = 100%, positive predictive value = 100%, negative predictive value = 23%). SOX30 hypermethylated patients had significantly lower SOX30 expression level than SOX30 non-hypermethylated patients (P = 0.027, Fig. 2d). Secondly, we performed BSP in eight representative patients (two controls selected randomly, two AML patients with lowest SOX30 methylation level, and four AML patients with highest SOX30 methylation level) to validate the RQ-MSP results. As a result, SOX30 methylation density was heavily correlated with SOX30 methylation level, and the results of SOX30 methylation density in representative AML patients were shown in Fig. 2e.

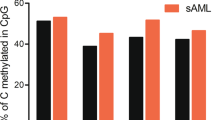
Validation of SOX30 methylation in MDS/AML patients. a The genomic coordinates (GC) of SOX30 promoter region CpG island and primer locations. The panel plots the GC content as a percentage of the total. Each vertical bar in the bottom panel represents the presence of a CpG dinucleotide. Black horizontal lines indicate regions amplified by RQ-MSP primer pairs and BSP primer pairs. CpGplot (http://emboss.bioinformatics.nl/cgi-bin/emboss/cpgplot) and Methyl Primer Express v1.0 software were used for creating the figure. TSS: transcription start site; RQ-MSP: real-time quantitative methylation-specific PCR; BSP: bisulfite sequencing PCR. b SOX30 methylation level in controls and MDS/AML patients. SOX30 methylation level was examined by RQ-MSP. Low/Int and High indicated MDS subtypes based on the classification of IPSS risks. AML included de novo AML and sAML which indicated MDS-derived AML. Each was compared to controls. NS: no significance; *: P < 0.05; **: P < 0.01; ***: P < 0.001. c Correlation between SOX30 methylation level and expression level in AML patients. SOX30 methylation level and expression level were examined by RQ-MSP and RQ-PCR, respectively. The correlation analysis was conducted by Spearman test. d SOX30 expression level in SOX30 hypermethylated and non-hypermethylated AML patients. SOX30 methylation level and expression level were examined by RQ-MSP and RQ-PCR, respectively. e SOX30 methylation density in controls and representative AML patients. SOX30 methylation density was determined by BSP. P1-P2 indicated two controls selected randomly. P3-P4 represented two AML patients with lower SOX30 methylation level. P5-P8 showed four AML patients with highest SOX30 methylation level
In order to analyze the clinical significance of SOX30 methylation in AML, we compared the clinical and laboratory features between SOX30 hypermethylated and SOX30 non-hypermethylated groups, and results were presented in Table 1. There were no significant differences between two groups among sex, white blood cells, hemoglobin, platelets, and BM blasts. However, SOX30 hypermethylation was correlated with higher age. Moreover, significant differences were observed between two groups in the distribution of FAB classifications and karyotype. SOX30 hypermethylation was less frequently occurred in M3/t(15;17) subtypes. Moreover, among gene mutations, there was no significant association of SOX30 hypermethylation with gene mutations besides IDH1/2 mutations with a trend.
We next determined the prognostic impact of SOX30 methylation in AML patients. Follow-up data was available in 175 patients with a survival time ranged from 0.5 to 136 months (median 8 months). Firstly, we observed the association of SOX30 hypermethylation with CR rate in AML patients. Among whole-cohort AML, SOX30 hypermethylated patients showed significantly lower CR rate than SOX30 non-hypermethylated patients (Table 1). In non-M3 AML and CN-AML, patients with SOX30 hypermethylation tended to have lower CR rate than those with SOX30 non-hypermethylation [44% (30/68) vs 31% (23/74), P = 0.121 and 54% (20/37) vs 33% (15/45), P = 0.074]. Secondly, by Kaplan-Meier analysis, SOX30 hypermethylated patients had significantly shorter OS and LFS than SOX30 non-hypermethylated patients (Fig. 3a, b). Significant difference was also observed among non-M3 and CN-AML patients (Fig. 3c–f). In addition, by Cox regression analysis, SOX30 hypermethylation was an independently adverse prognostic biomarker for OS among whole-cohort AML, non-M3 AML, and CN-AML patients (P = 0.014, 0.012, and 0.054, Additional file 1: Table S1-S3).

Prognostic and predictive value of SOX30 methylation in MDS/AML patients. a–f The prognostic value of SOX30 methylation for OS and LFS among AML patients. The survival analysis was performed among different subtypes of AML patients included whole-cohort AML, non-M3-AML, and CN-AML patients. g Dynamic changes of SOX30 methylation level in paired MDS/sAML patients. SOX30 methylation level was examined by RQ-MSP. sAML indicated MDS-derived AML patients. h, i The prognostic value of SOX30 methylation for OS and LFS among MDS patients
SOX30 hypermethylation increased the risk of leukemia transformation in MDS
Notably, for AML, SOX30 methylation level in MDS-derived AML was significantly higher than de novo AML patients (Fig. 2a). Next, we further determined SOX30 methylation in 12 paired patients during progression from MDS to AML. Expectedly, SOX30 methylation level was significantly increased in AML stage than in MDS stage among all the paired patients (Fig. 3g).
SOX30 hypermethylation was associated with higher IPSS risks and leukemia-free survival in MDS
We further investigated SOX30 methylation in a large cohort of MDS patients. SOX30 methylation level in MDS patients was found similar to controls (Fig. 2a). Nevertheless, SOX30 methylation in the MDS patients with high IPSS risks were significantly higher than in controls (P = 0.035), and also higher than in MDS patients with low/Int IPSS risks (P = 0.068) (Fig. 2a). We also used the same cutoff value to define SOX30 hypermethylation and non-hypermethylation in MDS patients. The comparison of clinical and laboratory features between hypermethylated and non-hypermethylated MDS patients was shown in Table 2. There were no significant differences between two groups among sex, age, white blood cells, and platelets. However, SOX30 hypermethylation was associated with lower hemoglobin and higher BM blasts. Moreover, significant differences were observed between two groups in the distribution of WHO classifications and IPSS scores. SOX30 hypermethylation was associated with MDS higher IPSS risks (Int-2/High) and WHO classifications (MDS-EB2). In addition, among gene mutations, we observed the association of SOX30 hypermethylation with U2AF1 mutation.
Prognostic impact of SOX30 methylation in MDS patients was performed in 96 patients with available follow-up data (range 1–113 months, median 19 months). Kaplan-Meier analysis showed that SOX30 hypermethylated patients had a tendency of shorter OS (Fig. 3h) and significantly shorter LFS (Fig. 3i) than SOX30 non-hypermethylated patients. However, Cox regression analysis showed that SOX30 hypermethylation may act as an independently adverse prognostic biomarker for LFS in MDS patients (P = 0.102, Additional file 1: Table S4).
SOX30 methylation was a predictive biomarker in monitoring disease recurrence in AML
To investigate whether SOX30 methylation was a potential biomarker in the surveillance of AML, we examined SOX30 methylation level in different clinical stages of AML patients (49 patients achieved CR and 27 relapsed patients). Significantly, SOX30 methylation level in CR stage was significantly decreased than in diagnosis time and was markedly increased in relapsed population when compared to CR stage (Fig. 4a). Moreover, the dynamic changes of SOX30 methylation level in 13 paired patients were also shown in Fig. 4b.

SOX30 methylation as a predictive biomarker in monitoring disease recurrence in AML. a SOX30 methylation level in AML patients of different clinical stages. SOX30 methylation level was examined by RQ-MSP. **: P < 0.01; ***: P < 0.001. b Dynamic changes of SOX30 methylation level in paired patients of different clinical stages when compared individually
Discussion
SOX30, a member of the SOX family, encodes a sequence-specific transcription factor that plays a vital role in gonadal differentiation and development. In species of mouse and human, SOX30 is considered to be closely related to spermatogonial differentiation and spermatogenesis [33]. SOX30 was reported to be highly expressed in male germ cells, human oocytes, or more differentiated cells [34]. Recently, SOX30 has been validated to be a diagnostic, prognostic, and functional factor in several solid cancers. Han et al. demonstrated that SOX30 was epigenetically downregulated by promoter methylation and functioned as a novel tumor suppressor partly by transcriptional activating p53 in lung cancer [29]. SOX30 could also inhibited tumor metastasis through attenuating Wnt signaling via the regulation of β-Catenin in a transcriptional and posttranslational manner in lung cancer [35]. Furthermore, the expression of SOX30 was verified to be closely associated with clinical outcomes in lung cancer patients [36]. Also, Guo et al. showed the anti-proliferation effect of SOX30 overexpression in colon cancer [37]. These results indicated a non-negligible role of SOX30 that played in the development of cancer.
In this study, we first identified SOX30 methylation in AML from TCGA datasets and further confirmed that SOX30 methylation were a frequent event in AML patients. In clinics of AML, SOX30 methylation was found to be associated with older age and less frequently in FAB-M3/t(15;17), which may be caused by disease entity (less blasts in BM). Notably, SOX30 methylation seemed to be associated with IDH1/2 mutations despite that the P did not attach statistical significance. As is well known, cancer-associated IDH mutations are characterized by neomorphic enzyme activity and resultant 2-hydroxyglutarate production [38] and also contributed to 5-hydroxymethylcytosine depletion in cancer cells [39, 40]. Mutational and epigenetic profiling of a large cohort of AML patients revealed that IDH1/2-mutant AMLs displayed global DNA hypermethylation and a specific hypermethylation signature [38]. These indicated that SOX30 methylation during leukemogenesis may be caused by IDH1/2 mutations. For MDS, we did not observe the significant association of SOX30 methylation with IDH1/2 mutations and older age, which may be caused by limited cases in MDS. However, we found the association of SOX30 methylation with U2AF1 mutations. U2AF1 mutations altered sequence specificity of pre-mRNA binding and splicing and was an important feature of the pathogenesis of MDS and related myeloid neoplasms [41]. Studies showed that U2AF1 mutations caused differential splicing of hundreds of genes, affecting biological pathways such as DNA methylation (DNMT3B) [42]. However, the potential molecular mechanism between U2AF1 mutation and SOX30 methylation needs further studies. Importantly, SOX30 methylation was associated with higher blasts, high-risk MDS, and shorter LFS time. Moreover, SOX30 methylation showed a higher methylation level in MDS-derived AML compared to de novo AML, and the detection of SOX30 methylation in 12 paired MDS/sAML patients showed that SOX30 methylation level was significantly increased in AML stage than in MDS stage. These results suggested SOX30 methylation might play a crucial role in MDS progression. By gene array technologies, Jiang et al. demonstrated that aberrant DNA methylation, more frequently than chromosome aberrations, was the dominant mechanism for tumor suppressor gene silencing and clonal variation in MDS evolution to AML [8]. However, it was the first time to report SOX30 methylation in myeloid malignancies, whether SOX30 functioned as a progression-related driver in MDS needed further studies.
Epigenetic modifications not only played crucial roles in cancer biology but also acted as biomarkers for cancer diagnosis and prognosis especially in blood cancer. Our previous study showed that the long non-coding RNA H19 overexpression promoted leukemogenesis and predicted unfavorable prognosis in AML [43]. Moreover, besides mutation, dysregulation of CEBPA caused by its methylation was also regarded as a prognostic biomarker to guide treatment plan for AML patients [44]. SOX30 as a prognostic biomarker has been reported in lung cancer [36]. From our study, although SOX30 methylation was not an independent indicator in MDS, we revealed that SOX30 methylation could act as a promising biomarker in AML. Firstly, SOX30 hypermethylation was associated with lower CR rate, which indicated SOX30 methylation was associated with chemotherapy response in AML. Secondly, SOX30 methylation was associated with shorter LFS/OS and acted as independent prognostic factor in AML. Lastly, the dynamic changes of SOX30 methylation in the surveillance of AML showed it could act as a predictor in monitoring disease recurrence. All these results suggested that determination of the SOX30 methylation may be useful to predict long-term survival and to guide post-remission therapy in MDS and AML. Interestingly, Božić et al. also used DNA methylation profiles of AML patients from TCGA and identified a CpG site in complement component 1 subcomponent R (C1R) as best suited biomarker to further complement risk assessment in AML [45]. Obviously, prospective studies and integrative analysis are needed before we can routinely use the promising biomarkers for risk stratification and planning therapy in MDS and AML.
Conclusions
Taken together, SOX30 methylation was associated with disease progression in MDS and acted as an independent prognostic and predictive biomarker in AML.
Abbreviations
- AML:
-
Acute myeloid leukemia
- ATRA:
-
All-trans retinoic acid
- AUC:
-
Area under the ROC curve
- BM:
-
Bone marrow
- BMMNCs:
-
BM mononuclear cells
- BSP:
-
Bisulfite sequencing PCR
- CN-AML:
-
Cytogenetically normal AML
- CR:
-
Complete remission
- GEO:
-
Gene Expression Omnibus
- HRMA:
-
High-resolution melting analysis
- ISCN:
-
International System for Human Cytogenetic Nomenclature
- LFS:
-
Leukemia-free survival
- MDS:
-
Myelodysplastic syndromes
- OS:
-
Overall survival
- ROC:
-
Receiver operating characteristic
- RQ-MSP:
-
Real-time quantitative methylation-specific PCR
- RQ-PCR:
-
Real-time quantitative PCR
- TCGA:
-
The Cancer Genome Atlas
References
Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–907.
Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. 2014;383:2239–52.
Rowley JD. Chromosomal translocations: revisited yet again. Blood. 2008;112:2183–9.
Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86.
Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23–36.
Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20.
Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60:376–92.
Jiang Y, Dunbar A, Gondek LP, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113:1315–25.
Zhou JD, Zhang TJ, Li XX, Ma JC, Guo H, Wen XM, Zhang W, Yang L, Yan Y, Lin J, Qian J. Epigenetic dysregulation of ID4 predicts disease progression and treatment outcome in myeloid malignancies. J Cell Mol Med. 2017;21:1468–81.
Bowles J, Schepers G, Koopman P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol. 2000;227:239–55.
Kormish JD, Sinner D, Zorn AM. Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev Dyn. 2010;239:56–68.
Dong C, Wilhelm D, Koopman P. Sox genes and cancer. Cytogenet Genome Res. 2004;105:442–7.
Sandoval S, Kraus C, Cho EC, Cho M, Bies J, Manara E, Accordi B, Landaw EM, Wolff L, Pigazzi M, Sakamoto KM. Sox4 cooperates with CREB in myeloid transformation. Blood. 2012;120:155–65.
Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber PB, Di Ruscio A, Welner RS, Ebralidze A, Zhang J, Levantini E, Lefebvre V, Valk PJ, Delwel R, Hoogenkamp M, Nerlov C, Cammenga J, Saez B, Scadden DT, Bonifer C, Ye M, Tenen DG. Sox4 is a key oncogenic target in C/EBPα mutant acute myeloid leukemia. Cancer Cell. 2013;24:575–88.
Man CH, Fung TK, Wan H, Cher CY, Fan A, Ng N, Ho C, Wan TS, Tanaka T, So CW, Kwong YL, Leung AY. Suppression of SOX7 by DNA methylation and its tumor suppressor function in acute myeloid leukemia. Blood. 2015;125:3928–36.
Wan H, Cai J, Chen F, Zhu J, Zhong J, Zhong H. SOX12: a novel potential target for acute myeloid leukaemia. Br J Haematol. 2017;176:421–30.
Tang CY, Lin J, Qian W, Yang J, Ma JC, Deng ZQ, Yang L, An C, Wen XM, Zhang YY, Qian J. Low SOX17 expression: prognostic significance in de novo acute myeloid leukemia with normal cytogenetics. Clin Chem Lab Med. 2014;52:1843–50.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Zhou JD, Lin J, Zhang TJ, Ma JC, Yang L, Wen XM, Guo H, Yang J, Deng ZQ, Qian J. GPX3 methylation in bone marrow predicts adverse prognosis and leukemia transformation in myelodysplastic syndrome. Cancer Med. 2017;6:267–74.
Li Y, Lin J, Yang J, Qian J, Qian W, Yao DM, Deng ZQ, Liu Q, Chen XX, Xie D, An C, Tang CY. Overexpressed let-7a-3 is associated with poor outcome in acute myeloid leukemia. Leuk Res. 2013;37:1642–7.
Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK. National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–65.
Lin J, Yao DM, Qian J, Chen Q, Qian W, Li Y, Yang J, Wang CZ, Chai HY, Qian Z, Xiao GF, Xu WR. Recurrent DNMT3A R882 muta-tions in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS One. 2011;6:e26906.
Qian J, Yao DM, Lin J, Qian W, Wang CZ, Chai HY, Yang J, Li Y, Deng ZQ, Ma JC, Chen XX. U2AF1 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS One. 2012;7:e45760.
Lin J, Yang J, Wen XM, Yang L, Deng ZQ, Qian Z, Ma JC, Guo H, Zhang YY, Qian W, Qian J. Detection of SRSF2-P95 mutation by high-resolution melting curve analysis and its effect on prognosis in myelodysplastic syndrome. PLoS One. 2014;9:e115693.
Yang J, Yao DM, Ma JC, Yang L, Guo H, Wen XM, Xiao GF, Qian Z, Lin J, Qian J. The prognostic implication of SRSF2 mutations in Chinese patients with acute myeloid leukemia. Tumour Biol. 2016;37:10107–14.
Lin J, Yao DM, Qian J, Chen Q, Qian W, Li Y, Yang J, Wang CZ, Chai HY, Qian Z, Xiao GF, Xu WR. IDH1 and IDH2 mutation analysis in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. Ann Hematol. 2012;91:519–25.
Wen XM, Lin J, Yang J, Yao DM, Deng ZQ, Tang CY, Xiao GF, Yang L, Ma JC, Hu JB, Qian W, Qian J. Double CEBPA mutations are prognostically favorable in non-APL acute myeloid leukemia patients with wild-type NPM1 and FLT3-ITD. Int J Clin Exp Pathol. 2014;7:6832–40.
Wen XM, Hu JB, Yang J, Qian W, Yao DM, Deng ZQ, Zhang YY, Zhu XW, Guo H, Lin J, Qian J. CEBPA methylation and mutation in myelodysplastic syndrome. Med Oncol. 2015;32:192.
Han F, Liu W, Jiang X, Shi X, Yin L, Ao L, Cui Z, Li Y, Huang C, Cao J, Liu J. SOX30, a novel epigenetic silenced tumor suppressor, promotes tumor cell apoptosis by transcriptional activating p53 in lung cancer. Oncogene. 2015;34:4391–402.
Network CGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Osaki E, Nishina Y, Inazawa J, Copeland NG, Gilbert DJ, Jenkins NA, et al. Identification of a novel Sry-related gene and its germ cell-specific expression. Nucleic Acids Res. 1999;27:2503–10.
Han F, Wang Z, Wu F, Liu Z, Huang B, Wang D. Characterization, phylogeny, alternative splicing and expression of Sox30 gene. BMC Mol Biol. 2010;11:98.
Han F, Liu WB, Shi XY, Yang JT, Zhang X, Li ZM, Jiang X, Yin L, Li JJ, Huang CS, Cao J, Liu JY. SOX30 inhibits tumor metastasis through attenuating Wnt-signaling via transcriptional and posttranslational regulation of β-catenin in lung cancer. EBioMedicine. 2018; https://doi.org/10.1016/j.ebiom.2018.04.026.
Han F, Liu W, Xiao H, Dong Y, Sun L, Mao C, Yin L, Jiang X, Ao L, Cui Z, Cao J, Liu J. High expression of SOX30 is associated with favorable survival in human lung adenocarcinoma. Sci Rep. 2015;5:13630.
Guo ST, Guo XY, Wang J, Wang CY, Yang RH, Wang FH, Li XY, Hondermarck H, Thorne RF, Wang YF, Jin L, Zhang XD, Jiang CC. MicroRNA-645 is an oncogenic regulator in colon cancer. Oncogene. 2017;6:e335.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Kroeze LI, Aslanyan MG, van Rooij A, Koorenhof-Scheele TN, Massop M, Carell T, Boezeman JB, Marie JP, Halkes CJ, de Witte T, Huls G, Suciu S, Wevers RA, van der Reijden BA, Jansen JH. EORTC leukemia group and GIMEMA. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood. 2014;124:1110–8.
Chou NH, Tsai CY, Tu YT, Wang KC, Kang CH, Chang PM, Li GC, Lam HC, Liu SI, Tsai KW. Isocitrate dehydrogenase 2 dysfunction contributes to 5-hydroxymethylcytosine depletion in gastric cancer cells. Anticancer Res. 2016;36:3983–90.
Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, Kielkopf CL, Ley TJ, Walter MJ, Graubert TA. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015;29:909–17.
Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14–26.
Zhang TJ, Zhou JD, Zhang W, Lin J, Ma JC, Wen XM, Yuan Q, Li XX, Xu ZJ, Qian J. H19 overexpression promotes leukemogenesis and predicts unfavorable prognosis in acute myeloid leukemia. Clin Epigenetics. 2018;10:47.
Lin TC, Hou HA, Chou WC, Ou DL, Yu SL, Tien HF, Lin LI. CEBPA methylation as a prognostic biomarker in patients with de novo acute myeloid leukemia. Leukemia. 2011;25:32–40.
Božić T, Lin Q, Frobel J, Wilop S, Hoffmann M, Müller-Tidow C, Brümmendorf TH, Jost E, Wagner W. DNA-methylation in C1R is a prognostic biomarker for acute myeloid leukemia. Clin Epigenetics. 2015;7:116.
Funding
This work was supported by National Natural Science Foundation of China (81270630), Medical Innovation Team of Jiangsu Province (CXTDB2017002), 333 Project of Jiangsu Province (BRA2016131), Six Talent Peaks Project in Jiangsu Province (2015-WSN-115), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX17_1821, KYCX18_2281), Key Medical Talent Program of Zhenjiang City.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
JQ, JL and J-d Z conceived and designed the experiments; J-d Z and Y-x W performed the experiments; J-dZ and T-j Z analyzed the data; X-x L, YG and WZ collected the clinical data; J-c M offered technique support; J-d Z wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The present study approved by the Ethics Committee and Institutional Review Board of the Affiliated People’s Hospital of Jiangsu University.
Consent for publication
Written informed consents were obtained from all enrolled individuals prior to their participation.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. The prognostic value of SOX genes methylation for OS and LFS among whole-cohort AML patients from TCGA databases. SOX genes methylation (HM450) data was downloaded via cBioPortal (http://www.cbioportal.org). AML patients were divided into two groups by the median methylation level of each gene respectively. Figure S2. ROC curve analysis of SOX30 methylation for discriminating AML patients form controls. Table S1. Univariate and multivariate analyses of prognostic factors for overall survival in AML patients. Table S2. Univariate and multivariate analyses of prognostic factors for overall survival in non-M3 AML patients. Table S3. Univariate and multivariate analyses of prognostic factors for overall survival in CN-AML patients. Table S4. Univariate and multivariate analyses of prognostic factors for overall survival and leukemia free survival in MDS patients. (DOCX 660 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Jd., Wang, Yx., Zhang, Tj. et al. Identification and validation of SRY-box containing gene family member SOX30 methylation as a prognostic and predictive biomarker in myeloid malignancies. Clin Epigenet 10, 92 (2018). https://doi.org/10.1186/s13148-018-0523-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0523-y