
Abstract
Background
Intrauterine exposure to gestational diabetes mellitus (GDM) confers a lifelong increased risk for metabolic and other complex disorders to the offspring. GDM-induced epigenetic modifications modulating gene regulation and persisting into later life are generally assumed to mediate these elevated disease susceptibilities. To identify candidate genes for fetal programming, we compared genome-wide methylation patterns of fetal cord bloods (FCBs) from GDM and control pregnancies.
Methods and results
Using Illumina’s 450K methylation arrays and following correction for multiple testing, 65 CpG sites (52 associated with genes) displayed significant methylation differences between GDM and control samples. Four candidate genes, ATP5A1, MFAP4, PRKCH, and SLC17A4, from our methylation screen and one, HIF3A, from the literature were validated by bisulfite pyrosequencing. The effects remained significant after adjustment for the confounding factors maternal BMI, gestational week, and fetal sex in a multivariate regression model. In general, GDM effects on FCB methylation were more pronounced in women with insulin-dependent GDM who had a more severe metabolic phenotype than women with dietetically treated GDM.
Conclusions
Our study supports an association between maternal GDM and the epigenetic status of the exposed offspring. Consistent with a multifactorial disease model, the observed FCB methylation changes are of small effect size but affect multiple genes/loci. The identified genes are primary candidates for transmitting GDM effects to the next generation. They also may provide useful biomarkers for the diagnosis, prognosis, and treatment of adverse prenatal exposures.
Similar content being viewed by others


Background
The “developmental origins of health and disease (DOHAD)” or Barker hypothesis associates adverse environmental exposures in the periconceptional and/or intrauterine period with lifelong increased morbidity for metabolic, cardiovascular, and other complex diseases [1, 2]. A large number of studies provided convincing evidence that both fetomaternal under- and overnutrition negatively influence the metabolic phenotype of the exposed individuals in later life [3, 4]. The prevalence of obesity and of women developing gestational diabetes mellitus (GDM) is increasing worldwide [5, 6]. Depending on ethnicity and diagnostic criteria, GDM affects 2 to >10% of all pregnancies.
Changes in lifestyle (overnutrition and physical inactivity) and genetic risk factors [7, 8] alone cannot explain the current GDM epidemics. GDM develops during pregnancy (usually in late second trimester) when the maternal insulin production can no longer cope with increasing adiposity and insulin resistance (due to increased placental lactogen, estrogen, and prolactin) [9, 10]. It results in fetal overnutrition (with glucose, amino acids, lipids, and fatty acids) and fetal hyperinsulinism, which may cause medical problems (macrosomia, organomegaly, and neonatal hypoglycemia) in the perinatal period. In addition, the adverse intrauterine environment may lead to persistent developmental malprogramming of the metabolism [11]. The offspring of GDM mothers have increased risks of developing obesity, type 2 diabetes, and cardiovasular disease [12–15]. Moreover, GDM exposure has been associated with autism spectrum disorder and long-term neuropsychiatric morbidity [16]. Studies of Pima Indian siblings discordant for exposure to GDM indicate that in addition to shared risk alleles, the increased lifelong disease risk is at least partially mediated by the hyperglycemic intrauterine environment [17]. The most likely mechanisms for translating the effects of intrauterine GDM exposure into disease susceptibility is epigenetic dysregulation of metabolic, cardiovascular, and neuroendocrine pathways [18–20].
Epigenetic mechanisms control gene expression patterns without altering the DNA sequence. The most thoroughly studied epigenetic modification is DNA methylation, more precisely methylation of cytosine carbon 5 at cytosine phosphate guanine (CpG) dinucleotides. DNA methylation patterns are transmitted to daughter cells during somatic cell division and perhaps also from one generation to the next. Promoter methylation during development, differentiation, or disease processes leads to an inactive chromatin structure and gene silencing. In contrast, gene body methylation is usually associated with active genes [21–23]. One important hallmark of DNA methylation patterns is their enormous plasticity during development and in response to environmental factors [24, 25]. Epigenetic modifications are primary candidates for mediating the persistent effects of an adverse intrauterine environment on the metabolism of the exposed individual.
Mass spectrometry revealed an increased global DNA methylation in placenta of GDM mothers [26]. Candidate gene studies have identified a number of differentially methylated genes in fetal tissues of babies from GDM mothers, including the fat-cell hormones leptin (LEP) and adiponectin (ADIPOQ), which are involved in regulation of energy metabolism and body weight [27, 28], the ATP-binding cassette transporter ABCA1, a major regulator of cellular cholesterol [29], the glucose transporters SLC2A1/GLUT1 and SLC2A3/GLUT3 [30], and the imprinted gene MEST, which plays a role in adipositas development [31]. In addition, there are already several genome-wide methylation studies in the offspring of GDM mothers [32–36], which have led to the identification of functional networks (various metabolic disease pathways and processes, cell growth and death regulation, endocytosis, inflammatory response, MAPK signaling, MODY, NOTCH signaling, type 2 diabetes) which are epigenetically programmed through GDM exposure. It is interesting to note that despite comparable sample sizes and study design, the number of identified loci with genome-wide significance ranged from none [32] to over thousand [35] and there is limited overlap between the identified genes and pathways. The GDM-susceptible genes that have been discovered so far may represent only the tip of the iceberg and also need to be replicated in independent studies. Here, we performed a 450K methylation array screen on cord bloods from GDM mothers and matched controls. To minimize the effects of confounding factors, all study subjects (the vast majority of them Caucasians) were recruited from a single obstetric clinic. Unlike other studies, diabetes during pregnancy was very well controlled. We distinguished between insulin-treated GDM (I-GDM) and dietetically treated GDM (D-GDM), assuming that I-GDM represents a more severe phenotype and more adverse fetal exposure.
Methods
Study subjects and DNA samples
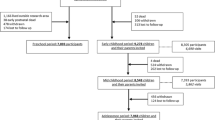
Umbilical cord bloods from newborns (singletons) of 105 mothers with I-GDM, 88 with D-GDM, and 120 controls without GDM were collected by obstetricians at the Municipal Clinics, Moenchengladbach, Germany. Blood samples were immediately frozen at −80 °C until further use. Genomic DNA was isolated with the FlexiGene DNA kit (Qiagen, Hilden, Germany) and bisulfite conversion performed with the EpiTect Fast 96 kit (Qiagen).
GDM was diagnosed between gestational weeks 24 and 27 by an elevated fasting (for 8-12 h) plasma glucose (>5.1 mmol/l) and a pathological oral glucose tolerance test (>10 mmol/l at 1 h and/or >8.5 mmol/l at 2 h after drinking a solution with 75-g glucose). Following diagnosis, women received dietary counselling by a diabetologist. According to the recommendations of the German Society of Gynecology and Obstetrics (DGGG) and the American Diabetes Association (ADA), they were put on a diet consisting of approximately 45% carbohydrate, 30–35% fat, and up to 20% protein. Protein intake was limited to approximately 0.8 g/kg body weight. The patients were not allowed to fast. If dietary treatment did not decrease glucose (<5.1 mmol/l after fasting, <7.8 mmol/l at 1 h, and <6.7 mmol/l at 2 h after meals) and HbA1C levels (<6%), patients were treated with the basis bolus insulin and rarely insulin pump therapy.
Microarray analysis
Two independent methylation array data sets (NCBI GEO accession no. GSE88929) were generated. Data set A represents 20 I-GDM and 18 control samples and data set B 24 I-GDM, 24 D-GDM, and 46 control samples (Table 1). After bisulfite conversion, the 38 samples of data set A and the 94 samples of data set B were whole-genome amplified, enzymatically fragmented, and hybridized to 4 and 8 Illumina HumanMethylation450 BeadChips, respectively, according to the manufacturer’s protocol (Illumina, San Diego, CA, USA). The arrays were scanned with an Illumina iScan. Microarray data were exported as idat files and analyzed using the statistical software package R (version 3.2.2) and the BioConductor platform (version 3.2). Preprocessing has been performed using the infrastructure implemented in the minfi [37] and watermelon [38] packages. First, sites with low signal quality (beadcount <3 and detection p value >0.05) were filtered and sites overlapping known SNPs removed. Furthermore, probes on the sex chromosomes were excluded, leaving a total number of 452,932 probes (cohort A) and 455,307 probes (cohort B), respectively, for subsequent analyses (out of >485,000 CpGs on the chip covering 99% of RefSeq genes with promoter, first exon, gene body, 5′ and 3′ UTRs and 96% of CpG islands). Intensity values were normalized using the dasen method as implemented in the watermelon package [38]. To account for potential probe-type effects, an intra-sample normalization procedure (BMIQ) has been applied which corrects for the bias of type 2 probes. Differential methylation analysis has been performed using the moderated T test model based on β values as implemented in the limma package [39]. All p values have been corrected for multiple testing using the Benjamini-Hochberg method [40].
Bisulfite pyrosequencing
The PyroMark Assay Design 2.0 software (Biotage, Uppsala, Sweden) was used for design of PCR and sequencing primers (Additional file 1: Table S1). Assays were established using the EpiTect PCR Control DNA set (Qiagen) with 0, 25, 50, 75, and 100% methylation. PCR reactions were performed in a total volume of 25 μl using the FastStart Taq DNA polymerase system (Roche Diagnostics, Mannheim, Germany). The 25 μl reaction consisted of 2.5 μl 10× PCR buffer, 20 mM MgCl2, 1.0 μl dNTP (10 mM) mix, 10 pmol of forward and reverse primer, 1 IU of FastStart polymerase, 1 μl (approximately 100 ng) bisulfite converted template DNA, and 18.3 μl PCR-grade water. For SLC17A4, 2.0 μl template DNA and 17.3 μl water were used.
To reduce technical noise (batch effects), bisulfite conversion and PCR (of D-GDM, I-GDM, and control samples) were performed simultaneously in 96-well microtiter plates. Pyrosequencing was performed on a PyroMark Q96 MD system (Qiagen) using the PyroMark Gold Q96 CDT reagent kit (Qiagen), 10 pmol of sequencing primer, and Pyro Q-CpG software (Qiagen). In our experience, the average methylation difference between technical replicates (including bisulfite conversion, PCR, and pyrosequencing) is approximately 1–2 percentage points. Artificially methylated and unmethylated DNA standards (Qiagen) were included as controls in each pyrosequencing run.
Statistical testing
Statistical analyses were performed with the statistical software package R (version 3.2.2) and IBM SPSS Statistics 23. The DNA methylation levels at each individual CpG site and the mean of all CpGs for the targeted region were compared between groups using the Mann-Whitney U test. To adjust for potential confounding factors, multivariate linear regression models have been used for the analysis of the pyrosequencing data. Potential confounders have been selected based on known and observed factors potentially influencing DNA methylation. The regression coefficients of the final model were adjusted for maternal BMI, gestational age, and fetal sex.
Results
Methylation array screens
Our genome-wide study of DNA methylation patterns was based on fetal cord bloods (FCBs) from pregnancies with D-GDM, I-GDM, and without GDM. Clinical parameters of the different cohorts and subgroups are presented in Table 1. The vast majority (>90%) of study subjects were of middle European descent with the remaining few percent from South-Eastern Europe and Turkey. Array cohort A consisted of 20 FCB samples from mothers with I-GDM and 18 controls. Samples were carefully matched for gestational week, fetal sex, maternal BMI, and age. The independent array cohort B consisted of 24 samples from mothers with D-GDM, 24 with I-GDM, and 48 controls. Due to the larger sample size, it was not possible to match for all relevant clinical parameters. Maternal BMI and gestational age differed between groups. In general, women with GDM were managed very well during pregnancy, displaying average HbA1c levels <6%. Only a few women in each group presented with comorbidities such as hypertension, preeclampsia, or thyroid dysfunction. Ten to 17% of women with I-GDM but none with D-GDM group suffered from type 1 or 2 diabetes before pregnancy (Table 1). There were only few preterm births (before 37th week of gestation) and small or large for gestational age (SGA, LGA) babies, respectively. Since white blood counts were not available, the relative proportion of different cell types in the FCBs was estimated from genome-wide methylation profiles using statistical methods [41]. None of the two analyzed cohorts showed a significant difference in cell composition between GDM and control samples (Additional file 2: Figure S1).
Samples of cohort A were hybridized to 4 and cohort B to 8 Illumina HumanMethylation450 BeadChips. We did not find significant differences in global (array CpG) methylation between control, D-GDM, and I-GDM samples in cohort A (p = 0.87) and B (p = 0.94), respectively. Since methylation levels differ markedly between CpG island (CGI)-related sites and the remaining genome, we performed separate analyses for the array GpG subsets in CGIs, north/south shelfs and shores, and open sea (Additional file 3: Table S2). Although there were no significant between-group differences, it is noteworthy that in cohort B, mean methylation of all targeted CpG subsets was 0.3–1.1 percentage points lower in both the D-GDM and I-GDM groups, compared to controls.
In cohort A, none of the analyzed 452,932 CpGs showed a significant between-group methylation difference after correction for multiple testing. However, the p value distribution (histogram) displayed an accumulation of p values in the low significance range, indicating the presence of a weak signal in the data set. In cohort B, 11,195 of 455,307 analyzed CpGs exhibited a significant (FDR-adjusted p < 0.05) methylation difference between I-GDM and controls and none between D-GDM and controls. Comparative analysis of data sets A and B revealed high concordance (R = 0.999, p < 2.2E−16) of single CpG methylation values. Both data sets showed a significant correlation of methylation differences (R = 0.126; p < 2.2E−16) and T values (R = 0.078; p < 2.2E−16) between I-GDM and control samples, consistent with the presence of a shared signal. To extract robust signals, the p values of both analyses were combined using order statistics of two uniformly distributed random variables. The first-order statistics revealed 1564 and the more robust second-order statistics 65 significant CpG sites, 52 of which are associated with genes (Table 2). The lack of significant signals in the D-GDM samples may be due to the lower sample size or the less severe metabolic phenotype.
Since earlier studies reported a correlation between placental ADIPOQ methylation and maternal blood glucose concentration [28], we analyzed the association of HbA1c levels with array CpG methylation in both GDM subgroups (Hb1Ac values were not available for controls). However, neither cohort A nor B displayed any significant sites after multiple testing correction. In addition, we tested the anthropometric surrogate parameters birth weight and gestational age for their association with DNA methylation. In cohort A, none of the analyzed array CpG sites reached genome-wide significance. In cohort B, there were no significant CpG sites for birth weight after multiple testing correction. A small number (823 of 455,307, 0.2%) of CpGs showed a significant association between gestational age and DNA methylation.
Validation of candidate genes by bisulfite pyrosequencing
Four candidate genes from our methylation array screen, ATP5A1, MFAP4, PRKCH, and SLC17A4, were analyzed by bisulfite pyrosequencing in 61 I-GDM, 64 D-GDM, and 56 control samples, including some (mainly control) samples that had been on the array. These genes were selected, because they exhibited between-group methylation differences >2% in both array data sets and were associated with common complications of diabetes in the literature [42–48]. It is noteworthy that maternal BMI, weight before pregnancy and birth, respectively, were significantly higher and the gestational age significantly lower in the I-GDM group, compared to D-GDM and controls (Table 1). HbAC1 levels were also significantly higher in I-GDM than in D-GDM women, but the majority of samples were still in the normal range. Eight of 61 (13%) women with I-GDM had diabetes before pregnancy. Thus, metabolic disturbances appear to be more pronounced in women requiring insulin treatment.
The pyrosequencing assay for ATP5A1 targeted two CpG sites in the promoter region. The array CpG (CpG2 of the pyrosequencing assay) displayed a significant methylation difference (β = −2%; p = 0.014) between GDM and control samples. When comparing I-GDM versus controls, CpG1 and the mean of both CpGs were significantly (p = 0.001 and 0.007) different between groups. The comparison of D-GDM versus controls did not reveal significant results. Similarly, four CpGs were analyzed in the MFAP4 promoter-flanking region; however, neither individual CpG nor mean methylation differed between GDM and control group. The only significant difference was observed for CpG4 between I-GDM and control samples (β = -0.4%; p = 0.048). The PRKCH assay targeted three CpGs in an enhancer region. Each individual CpG (CpG3 being the array CpG) and their mean methylation were significantly hypomethylated (β = −1.1 to −1.9%; p < 0.005) in GDM samples, compared with controls. The same was true (p < 0.001) when comparing I-GDM samples versus controls. A weaker effect was seen for CpG1 (p = 0.034), CpG2 (p = 0.003), and mean methylation of all CpGs (p = 0.015) in D-GDM versus controls. Three CpGs were analyzed in SLC17A4. Consistent with the methylation screen, the array CpG (CpG3 of the pyrosequencing assay) was hypomethylated (β =-0.8% to -2.0%) in GDM, I-GDM, and D-GDM samples, but the results were not significant. Surprisingly, CpG2, which is 141 bp upstream of CpG3, was significantly (p < 0.001) hypermethylated (β = 4.4–5.2%) in GDM, I-GDM, and D-GDM, compared with controls (Fig. 1). Thus, the methylation difference between CpG2 and CpG3 was 4.5–6.5 percentage points (p < 0.001) higher in the GDM, I-GDM, and D-GDM groups than in controls.

Methylation difference in fetal cord blood of GDM versus non-GDM pregnancies. Differences in methylation percentages between GDM and control FCBs are shown per CpG site for each gene studied (ATP5A1, blue bars; MFAP4, red bars; PRKCH, green bars; SLC17A, mauve bars; HIF3A, orange bars), adjusted for maternal BMI, gestational week, and fetal sex. Significant sites are indicated by star symbols (*p < 0.05; **p < 0.005)
Previously, HIF3A methylation in adult blood was positively correlated with BMI [49] and adipose tissue dysfunction [50]. Although it was not among the top candidate genes in our methylation screen, HIF3A was also analyzed by bisulfite pyrosequencing, targeting 11 CpGs in the HIF3A promoter (array CpGs 6, 8, and 11). Mean methylation of all 11 CpGs was significantly higher between GDM and controls (β = 1.3%; p = 0.001), I-GDM and controls (β = 1.5%; p = 0.005), and D-GDM and controls (β = 1.2%; p = 0.002). At the individual CpG level, nine of 11 CpGs were significantly hypermethylated in GDM and six of 11 in I-GDM and D-GDM samples, respectively. CpGs 3, 4, 5, 10, and 11 were significant in all three between-group comparisons, CpG 2 and 8 in none of the comparisons.
After adjusting for the potential confounding factors maternal BMI, gestational week, and fetal sex in our multivariate regression analysis (Additional file 4: Table S3), GDM was associated with significant FCB methylation changes of CpG2 (β = −2.2%; p = 0.02) in ATP5A1, of CpG4 (β = −0.4%; p = 0.04) in MFAP4, of CpG1 (β = −1.1%; p = 0.03), CpG2 (β = −1.9%; p = 0.003), and CpG3 (β = −1.2%; p = 0.05) in PRKCH, of CpG2 (β = 5.3%; p < 0.001) in SLC17A4, and of CpG5 (β = 1.7%; p = 0.03), CpG6 (β = 3.1%; p = 0.03), CpG10 (β = 3.7%; p = 0.01), and CpG11 (β = 3.7%; p = 0.03) in HIF3A. Mean methylation of all CpGs in the target region was significant for PRCHK (β = −1.4%; p = 0.008), SLC17A (β = 1.5%; p = 0.03), and HIF3A (β = 2.3%; p = 0.05).
Discussion
The prevalence of GDM and maternal obesity is constantly increasing worldwide and gives rise to a vicious cycle in which babies exposed to GDM in utero are more likely to develop metabolic (and other) disorders later in life [12–16]. The mechanisms increasing the risk for long-term morbidity in the offspring are still poorly understood, but epigenetics is thought to be a key player in this process [18–20]. A growing number of studies in human postpartum tissues [26–36] have demonstrated GDM-related changes in the offspring’s DNA methylation patterns. In the mouse model, there is evidence that epigenetic changes in the germ cells of offspring from diabetic/obese mothers may contribute to transgenerational inheritance of a metabolic phenotype [51, 52].
The observed GDM-associated epigenetic changes in cord blood and/or placenta are small (in the order of a few percentage points) at the single-gene level but appear to be widespread. Nevertheless and similar to the hits of genome-wide association studies (GWAS), despite small effect size, the identified differentially methylated loci may uncover genes that are essential for fetal programming of a metabolic phenotype in GDM offspring. Considering the enormous variation of DNA methylation patterns among non-exposed neonates/infants, the measured methylation values in GDM offspring are still in the normal range and, thus, their diagnostic or prognostic value is currently too low for clinical implementation. Again similar to GWAS, the development of polygenic risk scores may allow better predictions of the outcome of adverse intrauterine exposures. When interpreting the functional relevance of epigenetic markers, it is important to emphasize that the epigenomes differ between cell types and tissues. Alterations in cord blood DNA methylation cells do not necessarily reflect alterations in the organs (pancreatic islets, fat, liver, skeletal muscle, and hypothalamus) that play a role in the pathogenesis of GDM. Due to ethical and legal restrictions, the target tissues for fetal programming of metabolic disease in GDM-exposed offspring are not accessible.
Moreover, there are numerous confounding factors on the maternal and offspring’s side. Differences in ethnicity (genetic background), comorbidities, diagnostic criteria for GDM, and treatment during pregnancy may explain the huge discrepancies in the number of genome-wide significant hits in conceptually very similar 450K methylation array studies [32, 35]. To the extent possible, we tried to minimize the effects of ethnicity and comorbitidies. More than 90% of our study participants from a single big obstetric clinic were Caucasians. Only a few women in each cohort/subgroup suffered from hypertension, preeclamsia, thyroid dysfunction, or other medical problems. In addition, there were only few preterm births and babies with SGA or LGA. Typical for the situation in Germany, diabetes in our GDM cohorts was very well controlled. Most GDM mothers exhibited HbA1c values in the normal range (5.5 ± 0.3% in D-GDM and 5.8 ± 0.4% in I-GDM), which may explain the relatively low number of differentially methylated CpGs in exposed offspring, compared to a recent study on South Asian pregnant women [35]. It seems plausible to assume that an early diagnosis and optimum treatment of GDM reduces epigenetic effects due to adverse intrauterine exposure. In general, we observed more significant effects in I-GDM than in D-GDM. This may be due to epigenetic effects of insulin itself or, more likely, to a more severe phenotype in women requiring insulin treatment. Ten to 17% of women in the analyzed I-GDM subgroups (but none with D-GDM) have been diagnosed with diabetes before pregnancy, consistent with an adverse environmental exposure of the embryo/fetus during early development. Maternal BMI and HbA1c levels were significantly higher in pregnant women with I-GDM, compared to D-GDM, whereas gestational age at birth was lower. In addition to maternal BMI and gestational week, fetal sex-dependent endocrine effects may play an important role in the pathogenesis of GDM [53]. However, following adjustment for the maternal BMI, gestational week, and fetal sex in a multivariate regression model, the GDM effect on the methylation patterns of the four analyzed candidate genes remained significant. This argues in favor of the robustness of our approach and the quality of our array data. In addition, there were no detectable differences in cell composition of FCB and control bloods, which could explain the observed effects.
Although the number of GDM and control samples analyzed here meets current standards for genome-wide methylation studies, the sample size is still two orders of magnitude lower than that of recent GWAS for complex phenotypes. Therefore, existing methylation array data sets are likely still polluted with false positives and false negatives. Overall, we identified 65 GDM-associated CpG methylation changes. The 55 associated genes are mainly novel and reliable candidates for fetal programming by GDM. In addition, one candidate gene, HIF3A, from the literature [49, 50] was validated.
ATP5A1 encodes a subunit of mitochondrial ATP synthetase, which prevents oxidative damage by mitochondrial superoxide generation. ATP synthetase disruption by high glucose levels promotes diabetic cardiomyopathy in mouse models [47]. Genetic mutations in mitochondrial ATP synthetase cause very severe metabolic disorders, presenting as early-onset encephalo-cardiomyopathies [54]. The microfibrillar-associated protein 4 (MFAP4) is involved in cell adhesion and intercellular interactions and is highly expressed in blood vessels. Plasma MFAP4 levels have been associated with various cardiovascular complications [44] and diabetic neuropathy [55]. PRKCH, which is hypomethylated in GDM offspring, belongs to the protein kinase C family that is involved in diverse cellular signaling pathways. It can promote cellular senescence through transcriptional upregulation of cell cycle inhibitors p21 and p27 [56]. PRKCH variants have been associated with early-onset obesity [48] and increased stroke risk [45, 46]. The epigenetic regulation of the intestinal sodium/phosphate cotransporter SLC17A4 by GDM appears to be complex. The methylation difference between two neighboring CpGs was increased, one being hypermethylated and one being hypomethylated in GDM samples. Previously, we have shown that the methylation difference between neighboring CpGs not only is due to stochastic fluctuations but also may reflect epigenetic signatures of tissue, environment, etc. [57]. Transcription factor binding site searches [58] revealed that the hypomethylated C is important to create a p53 binding site. It is tempting to speculate that the regional DNA methylation profile modulates access of transcription factors to their binding sites. A common variant near the SLC17A4 gene has been associated with measures of atherosclerotic disease [42]. Hypoxia inducible factors (HIFs) are heterodimeric transcription factors that mediate hypoxia response in various tissues [59]. HIF3A is one of the several isoforms of the α subunit that can form dimers with the β subunit (ARNT). HIF3A plays a role in glucose and amino acid metabolism and adipocyte differentiation [60]. The increased FCB methylation in GDM offspring is consistent with an increased risk for adipositas development [49, 50].
Conclusions
Accumulating evidence suggests that GDM leads to changes in the epigenome(s) of the exposed offspring. Since DNA methylation plays a key role in the control of gene regulation [21–23], it is plausible to assume a causal relationship between GDM-related methylation changes at births and increased disease risks in later life. Longitudinal studies on well-characterized mother-infant pairs and larger sample sizes are needed to demonstrate persistence of the epigenetic alterations into adulthood and the effect of possible (nutritional, pharmacological, and behavioral) interventions during pregnancy and postnatal period (lactation and weaning). On the long term, only meta-analyses combining genome-wide data sets generated in different laboratories with different GDM cohorts will reveal a more complete picture.
Abbreviations
- BMI:
-
Body mass index
- CGI:
-
CpG island
- CpG:
-
Cytosine phosphate guanine
- D-GDM:
-
Dietetically treated GDM
- GDM:
-
Gestational diabetes mellitus
- GWAS:
-
Genome-wide association study
- HIF:
-
Hypoxia inducible factor
- I-GDM:
-
Insulin-treated GDM
- LGA:
-
Large for gestational age
- SGA:
-
Small for gestational age
References
Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–7.
Gillman MW. Developmental origins of health and disease. N Engl J Med. 2005;353:1848–50.
Roseboom T, De Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev. 2006;82:485–91.
El Hajj N, Schneider E, Lehnen H, Haaf T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction. 2014;148:R111–20.
Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond). 2008;32:1431–7.
Ben-Haroush A, Yogev Y, Hod M. Epidemiology of gestational diabetes mellitus and its association with type 2 diabetes. Diabet Med. 2004;21:103–13.
Robitaille J, Grant AM. The genetics of gestational diabetes mellitus: evidence for relationship with type 2 diabetes mellitus. Genet Med. 2008;10:240–50.
Kwak SH, Kim SH, Cho YM, Go MJ, Cho YS, Choi SH, Moon MK, Jung HS, Shin HD, Kang HM, et al. A genome-wide association study of gestational diabetes mellitus in Korean women. Diabetes. 2012;61:531–41.
Di Cianni G, Miccoli R, Volpe L, Lencioni C, Del Prato S. Intermediate metabolism in normal pregnancy and in gestational diabetes. Diabetes Metab Res Rev. 2003;19:259–70.
Buchanan TA, Xiang AH. Gestational diabetes mellitus. J Clin Invest. 2005;115:485–91.
Plagemann A. Maternal diabetes and perinatal programming. Early Hum Dev. 2011;87:743–7.
Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers. Relationship to fetal hyperinsulinism. Diabetes Care. 1995;18:611–7.
Franks PW, Looker HC, Kobes S, Touger L, Tataranni PA, Hanson RL, Knowler WC. Gestational glucose tolerance and risk of type 2 diabetes in young Pima Indian offspring. Diabetes. 2006;55:460–5.
Moore TR. Fetal exposure to gestational diabetes contributes to subsequent adult metabolic syndrome. Am J Obstet Gynecol. 2010;202:643–9.
West NA, Crume TL, Maligie MA, Dabelea D. Cardiovascular risk factors in children exposed to maternal diabetes in utero. Diabetologia. 2011;54:504–7.
Nahum Sacks K, Friger M, Shoham-Vardi I, Abokaf H, Spiegel E, Sergienko R, Landau D, Sheiner E. Prenatal exposure to gestational diabetes mellitus as an independent risk factor for long-term neuropsychiatric morbidity of the offspring. Am J Obstet Gynecol. 2016;215:380. e1-7.
Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH, Knowler WC. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49:2208–11.
Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol. 2009;5:401–8.
Lehnen H, Zechner U, Haaf T. Epigenetics of gestational diabetes mellitus and offspring health: the time for action is in early stages of life. Mol Hum Reprod. 2013;19:415–22.
Holbrook JD. Catching diabetes. Epigenomics. 2016;8:1173–7.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–54.
Vaissière T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–8.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Haaf T. Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol. 2006;310:13–22.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
Reichetzeder C, Dwi Putra SE, Pfab T, Slowinski T, Neuber C, Kleuser B, Hocher B. Increased global placental DNA methylation levels are associated with gestational diabetes. Clin Epigenetics. 2016;8:82.
Bouchard L, Thibault S, Guay SP, Santure M, Monpetit A, St-Pierre J, Perron P, Brisson D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care. 2010;33:2436–41.
Bouchard L, Hivert MF, Guay SP, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes. 2012;61:1272–80.
Houde AA, Guay SP, Desgagné V, Hivert MF, Baillargeon JP, St-Pierre J, Perron P, Gaudet D, Brisson D, Bouchard L. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics. 2013;8:1289–302.
Novakovic B, Gordon L, Robinson WP, Desoye G, Saffery R. Glucose as a fetal nutrient: dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J Nutr Biochem. 2013;24:282–8.
El Hajj N, Pliushch G, Schneider E, Dittrich M, Müller T, Korenkov M, Aretz M, Zechner U, Lehnen H, Haaf T. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes. 2013;62:1320–8.
Ruchat SM, Houde AA, Voisin G, St-Pierre J, Perron P, Baillargeon JP, Gaudet D, Hivert MF, Brisson D, Bouchard L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics. 2013;8:935–43.
Del Rosario MC, Ossowski V, Knowler WC, Bogardus C, Baier LJ, Hanson RL. Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation. Metabolism. 2014;6:654–60.
Quilter CR, Cooper WN, Cliffe KM, Skinner BM, Prentice PM, Nelson L, Bauer J, Ong KK, Constância M, Lowe WL, et al. Impact on offspring methylation patterns of maternal gestational diabetes mellitus and intrauterine growth restraint suggest common genes and pathways linked to subsequent type 2 diabetes risk. FASEB J. 2014;28:4868–79.
Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, Sinha A, Williams D, Rakyan VK, Hitman GA. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. 2015;24:3021–9.
Rong C, Cui X, Chen J, Qian Y, Jia R, Hu Y. DNA methylation profiles in placenta and its association with gestational diabetes mellitus. Exp Clin Endocrinol Diabetes. 2015;123:282–8.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. JR Statist Soc B. 1995;57:289–300.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31.
Bis JC, Kavousi M, Franceschini N, Isaacs A, Abecasis GR, Schminke U, Post WS, Smith AV, Cupples LA, Markus HS, et al. Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. 2011;43:940–7.
Schwenk RW, Vogel H, Schürmann A. Genetic and epigenetic control of metabolic health. Mol Metab. 2013;2:337–47.
Wulf-Johansson H, Lock Johansson S, Schlosser A, Trommelholt Holm A, Rasmussen LM, Mickley H, Diederichsen AC, Munkholm H, Poulsen TS, Tornøe I, et al. Localization of microfibrillar-associated protein 4 (MFAP4) in human tissues: clinical evaluation of serum MFAP4 and its association with various cardiovascular conditions. PLoS One. 2013;8:e82243.
Sun L, Zhang Z, Ma M, Xu G, Liu X. A quantitative assessment of the association between 1425G/A polymorphism in PRKCH and risk of stroke. Neuromolecular Med. 2014;16:814–20.
Chen YC, Huang CJ, Chen P, Wu YR, Shie SS, Chen ST, Lee-Chen GJ, Chen CM. Protein kinase Cη polymorphism and the susceptibility to ischemic stroke in the Taiwan population. Biomed J. 2015;38:433–8.
Ni R, Zheng D, Xiong S, Hill DJ, Sun T, Gardiner RB, Fan GC, Lu Y, Abel ED, Greer PA, Peng T. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes. 2016;65:255–68.
Wheeler E, Huang N, Bochukova E, Keogh JM, Lindsay S, Garg S, Henning E, Blackburn H, Loos RJF, Wareham NJ, et al. Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet. 2013;45:513–7.
Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aïssi D, Wahl S, Meduri E, Morange PE, Gagnon F, Grallert H, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383:1990–8.
Pfeiffer S, Krüger J, Maierhofer A, Böttcher Y, Klöting N, El Hajj N, Schleinitz D, Schön MR, Dietrich A, Fasshauer M, et al. Hypoxia-inducible factor 3A gene expression and methylation in adipose tissue is related to adipose tissue dysfunction. Sci Rep. 2016;6:27969.
Ding GL, Wang FF, Shu J, Tian S, Jiang Y, Zhang D, Wang N, Luo Q, Zhang Y, Jin F, et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes. 2012;61:1133–42.
Ge ZJ, Liang QX, Hou Y, Han ZM, Schatten H, Sun QY, Zhang CL. Maternal obesity and diabetes may cause DNA methylation alteration in the spermatozoa of offspring in mice. Reprod Biol Endocrinol. 2014;12:29.
Hocher B, Schlemm L, Haumann H, Jian L, Rahnenführer J, Guthmann F, Bamberg C, Kalk P, Pfab T, Chen YP. Offspring sex determines the impact of the maternal ACE I/D polymorphism on maternal glycaemic control during the last weeks of pregnancy. J Renin Angiotensin Aldosterone Syst. 2011;12:254–61.
Hejzlarová K, Mráček T, Vrbacký M, Kaplanová V, Karbanová V, Nůsková H, Pecina P, Houštěk J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol Res. 2014;63 Suppl 1:S57–71.
Blindbæk SL, Schlosser A, Green A, Holmskov U, Sorensen GL, Grauslund J. Association between microfibrillar-associated protein 4 (MFAP4) and micro- and macrovascular complications in long-term type 1 diabetes mellitus. Acta Diabetol. 2017;54:367–72.
Zurgil U, Ben-Ari A, Atias K, Isakov N, Apte R, Livneh E. PKCη promotes senescence induced by oxidative stress and chemotherapy. Cell Death Dis. 2014;5:e1531.
Schneider E, Pliushch G, El Hajj N, Galetzka D, Puhl A, Schorsch M, Frauenknecht K, Riepert T, Tresch A, Müller AM, et al. Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic Acids Res. 2010;38:3880–90.
Lee C, Huang CH. LASAGNA-Search: an integrated web tool for transcription factor binding site search and visualization. Biotechniques. 2013;54:141–53.
Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31:2448–60.
Hatanaka M, Shimba S, Sakaue M, Kondo Y, Kagechika H, Kokame K, Miyata T, Hara S. Hypoxia-inducible factor-3alpha functions as an accelerator of 3T3-L1 adipose differentiation. Biol Pharm Bull. 2009;32:1166–72.
Acknowledgements
We thank all pregnant women participating in this study.
Funding
This work was supported by a research grant (HA1374/15-1) from the German Research Foundation.
Availability of data and materials
The methylation array data sets discussed in this article are deposited in NCBI’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE88929). All other data are included within the article and its additional files.
Authors’ contributions
TH designed the study and wrote the manuscript. LH, IN, and NEH performed the experiments. LH, MD, and TM performed the statistical and bioinformatic data analyses. HL collected the fetal cord bloods and clinical information. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable for this study.
Ethics approval and consent to participate
Written informed consent to collect fetal cord blood samples for epigenetic studies was obtained from all participating pregnant women. This study was approved by the Ethics Committee of the Medical Faculty at Würzburg University (no. 100/10).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primers for pyrosequencing. (DOC 52 kb)
Additional file 2: Figure S1.
Estimation of blood cell composition based on 450K methylation array profiles. Blue box plots show the distribution of cell types in GDM cord blood and red box plots in control samples. The median is represented by a horizontal line. The bottom of the box indicates the 25th percentile and the top the 75th percentile. Outliers are shown as circles. (DOC 146 kb)
Additional file 3: Table S2.
Global DNA methylation of different CpG island-related array CpG subsets in control, D-GDM, and I-GDM samples. (DOC 35 kb)
Additional file 4: Table S3.
Multivariate analyses (adjusting for maternal BMI, gestational age, and fetal sex): CpG methylation of candidate genes in GDM versus control FCB samples. (DOC 68 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Haertle, L., El Hajj, N., Dittrich, M. et al. Epigenetic signatures of gestational diabetes mellitus on cord blood methylation. Clin Epigenet 9, 28 (2017). https://doi.org/10.1186/s13148-017-0329-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-017-0329-3