
Abstract
Background
We, and others, have demonstrated previously that there are differences in DNA methylation and transcript levels of a number of genes in cord blood and placenta between children conceived using assisted reproductive technologies (ART) and children conceived in vivo. The source of these differences (the effect of ART versus the underlying infertility) has never been determined in humans. In this study, we have attempted to resolve this issue by comparing placental DNA methylation levels at 37 CpG sites in 16 previously identified candidate genes in independent populations of children conceived in vivo (‘fertile control’ group) with ART children conceived from two groups: either autologous oocytes with infertility in one or both parents (‘infertile ART’ group) or donor oocytes (obtained from young fertile donors) without male infertility (‘donor oocyte ART’ group).
Results
Of the 37 CpG sites analyzed, significant differences between the three groups were found in 11 CpGs (29.73 %), using ANOVA. Tukey’s post hoc test on the significant results indicated that seven (63.63 %) of these differences were significant between the donor oocyte ART and fertile control groups. In addition, 20 of the 37 CpGs analyzed had been identified as differentially methylated between ART and fertile control groups in an independent population in a prior study. Of these 20 CpG sites, 9 also showed significant differences in the present population. An additional 9 CpGs were found to be significantly different between the two groups. Of these 18 candidate CpGs, 12 CpGs (in seven candidate genes) also showed significant differences in placental DNA methylation levels between the donor oocyte ART and fertile control groups.
Conclusions
These data suggest strongly that the DNA methylation differences observed between ART and in vivo conceptions are associated with some aspect of ART protocols, not simply the underlying infertility.
Similar content being viewed by others

Background
Modest but significant differences in DNA methylation have been identified between children conceived in vivo and children conceived by assisted reproductive technology (ART) [1-10]. While there are some inconsistencies between studies [11-20], it seems likely that these differences in methylation between these two groups are genuine. This conclusion is supported by animal studies which find that ART leads to epigenetic changes in offspring [21-29]. At this juncture and given these observations, there are at least two important questions to be answered: 1) Are epigenetic differences in ART pregnancies associated with the observed increase in adverse outcomes in these pregnancies and 2) are the ART-associated differences the result of clinical and/or laboratory interventions used in ART or are they associated with the diagnosis of infertility itself?
In a recent study, we began to address the first question, that is, whether ART-associated epigenetic differences are associated with adverse outcomes by demonstrating that DNA methylation levels at a modest number of loci can explain a large fraction of variance in infant birth weight [30]. It is critical to note that low birth weight (LBW) is the most significant ART-associated adverse perinatal outcome, in terms of number of cases [31-35]. Others have also found that ART is associated with not only adverse neonatal outcomes, such as birth defects, but also long-term outcomes, including growth, gonadal development, physical health, neurological or neurodevelopmental defects and, especially, epigenetic abnormalities [36-52]. Children conceived in vitro are generally at higher risk of small for gestational age (SGA), preterm delivery, perinatal morbidity, and hospital admission than children conceived in vivo [36,37]. In addition, meta-analyses have revealed a 30% to 40% increase in major malformation rates for infants born after ART compared with naturally conceived children [38-40], and children conceived via in vitro fertilization (IVF), who rapidly gained weight during early childhood (1 to 3 years), have been shown to have higher blood pressure levels [41]. Interestingly, even apparently healthy ART-conceived children may have an increased risk of cardiovascular diseases later in life [42-45]. Finally, publications from Europe, the United States, and Australia have all suggested an association between ART and imprinting disorders such as Beckwith-Wiedemann syndrome [37,46-52]. As the number of children born following ART continues to increase, it is critical to understand whether epigenetic changes are responsible for some of the adverse outcomes observed following ART.
In the current study, we began to address the question of whether ART-associated differences in DNA methylation are a characteristic of ART-related protocols and/or procedures (clinical or laboratory) or the diagnosis of infertility, per se, by utilizing a group of patients undergoing ART with no infertility diagnosis. In this study, we utilized bisulfite pyrosequencing to analyze placental DNA methylation of genes previously identified by our laboratory as being differentially methylated between in vitro- and in vivo-conceived children using array-based methods. We compared placental DNA methylation in children conceived in vivo (fertile control group) with ART children conceived following fertilization of oocytes from two distinct groups: 1) autologous oocytes with infertility in one or both parents (infertile group); 2) donor oocytes (obtained from young fertile donors) without male factor infertility in the recipient’s partner (donor oocyte group). It is clear that the oocytes, sperm, and the resulting embryos in the donor group, which were obtained from patients without infertility diagnoses, are still subjected to the clinical and laboratory procedures of ART. Therefore, such comparison of the infertile and donor groups should distinguish DNA methylation differences that are the result of ART protocols and/or procedures from effects caused by an infertility diagnosis.
Results
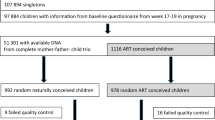
The demographic, cycle, and birth characteristics of the fertile control, infertile ART, and donor oocyte ART groups are shown in Table 1. Mean maternal age, paternal age, gestational age, and birth weight differed significantly between the groups, as expected. Intra-cytoplasmic sperm injection (ICSI) was utilized in 13.6% of the cycles in the infertile ART group for insemination, while ICSI was not used in any of the donor cycles. The placental DNA methylation levels were analyzed by bisulfite conversion and pyrosequencing at 37 candidate CpG sites in 16 genes. These genes were selected based on previous array studies in our laboratory (see ‘Methods’ for details of molecular analysis and CpG site selection). One-way analysis of variance (ANOVA) was used to assess differences between the control, infertile, and donor groups, followed by Tukey’s post hoc honest significant difference (HSD) test. The secondary goal of this study was to validate our previous findings of differences in placental DNA methylation between in vivo- and in vitro-conceived children. We had previously identified differences in 20 of the targeted CpG sites in 15 genes [30]. We therefore compared placental DNA methylation between the fertile control and infertile groups in these 15 genes for independent validation using a one-tailed t-test. Finally, we tested the differences between the donor oocyte ART and fertile control groups at the sites for which differences between the control group and the infertile group existed. The differences between the fertile control group and donor oocyte ART group were tested using a two-tailed t-test.
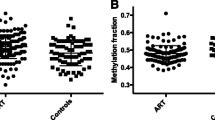
The graphical representation of methylation distribution of the three study groups at specific CpG sites is shown in Figure 1.

Graphs of methylation fractions in different groups- donor oocyte ART, infertile ART, and fertile control of representative CpGs (A) GRIN2C chr17:70368047, (B) CCDC2 chr2:121824853, (C) NDN chr15:21483466, (D) PTPN20B chr10:48448112.
Differences in candidate gene methylation levels between the three study groups (infertile ART, donor oocyte ART, and fertile control)
As we had three study groups (infertile ART, donor oocyte ART, and fertile control, see ‘Methods’), the most stringent approach to determine whether differences exist is ANOVA followed by Tukey’s post hoc HSD test to identify which specific groups are responsible for any significant differences. One-way ANOVA demonstrated that 11 of the 37 CpG sites tested differ between the three groups. When these 11 ANOVA-significant sites were tested for differences between the fertile controls and the two ART groups (infertile and donor oocyte) by Tukey’s post hoc HSD test, 7 of the 11 sites differed between the donor oocyte ART group and the fertile control groups and 2 of the sites differed between the infertile group and the fertile controls. Two of the sites differed between the fertile control group and both ART groups (Table 2). Hence, we identified significant differences in approximately 30% of CpGs using ANOVA, and 64% of these differences were attributed to the methylation differences between the donor oocyte ART and fertile control groups.
Validation of infertile ART vs. fertile control candidate gene methylation differences in an independent population
We performed a one-tailed t-test between the infertile and control groups to validate our previous findings demonstrating differential methylation between ART and control groups at 20 CpG sites in 15 genes [30]. Nine of these twenty CpGs showed differential methylation by bisulfite pyrosequencing between the infertile ART and fertile control groups in this validation cohort (Table 3). This represents a 45% validation rate. In the pyrosequencing assay, additional CpGs not present on the original Illumina Infinium 27 K methylation array (Illumina Inc., Sta. Clara, CA, USA) were also interrogated. Of these new CpGs analyzed, nine additional CpGs were found to differ significantly between the infertile and control groups (Table 3). Hence, a total of 18 CpGs (48.65%) showed methylation differences between the infertile ART and the fertile control groups.
Gene methylation levels in fertile compared to donor group
As we had no prior knowledge about the direction of results for donor oocyte ART versus fertile control groups, we used a two-tailed t-test to study these methylation differences. Among the CpGs differing between infertile ART and fertile control groups in the validation (Table 3), 12 CpGs also differed significantly between fertile control and donor oocyte ART groups. These 12 CpGs are shown in Table 4, along with the direction of difference in the Illumina Infinium 27 K array [30] and the pyrosequencing results in the present study for infertile ART and fertile control groups. All significant differences were in the same direction in both the original (array-based profiling) [30] and the validation (bisulfite pyrosequencing) populations. In other words, 67% of the total methylation differences observed between infertile ART and fertile control groups also existed between the donor oocyte ART and fertile control groups.
Discussion
Our analysis of placental DNA methylation levels at 37 CpG sites in 16 genes demonstrates that there are methylation differences in children conceived in vivo compared to children conceived using ART, both in the presence and absence of parental infertility. We observed significant differences in 30% of CpGs between the control, infertile, and donor groups. Furthermore, the majority of these differences (63.63%) could be attributed to the difference between donor oocyte ART and fertile control groups. Additionally, nearly 67% of the differences between ART and control placentas were present in both the infertile and donor groups. Therefore, these results suggest that many of the site-specific DNA methylation differences observed by us and others [1-10] are associated with some aspects of ART clinical and/or laboratory practice(s) rather than some aspect of infertility itself. We also observed that in a subset of genes in Table 2, placental DNA methylation appears to differ between the donor and infertile groups and we are currently validating these findings and investigating the potential role of these changes in the infertility diagnosis. However, it should be noted that in all of these cases, the methylation of the donor oocyte ART group is even more significantly different than the control group when compared to the methylation of the infertile group. This observation strengthens our interpretation of the data that the donor oocyte ART group methylation effects are not due to the infertility, per se, but to some aspect of the ART procedures.
Our findings are strengthened by the fact that our donor oocyte ART population had no additional male factor infertility, eliminating the possibility of infertility in either parent as the cause of the observed methylation differences between the donor oocyte ART group and fertile controls. These data are consistent with animal studies that have demonstrated epigenetic changes in domestic cattle (reviewed by [53]), sheep [23], and mice (reviewed by [54]), where no infertility is present. Therefore, the observed changes in animals are attributed to some aspect(s) of the ART procedure(s).
Additionally, we successfully validated placental DNA methylation differences in an independent population for 45% of the CpGs in which we had found differences previously [30]. Given the relatively small differences in mean methylation levels, small sample size (Table 1), and the relatively large variance observed within groups for methylation levels at these specific CpG sites [1,4], we regard the validation of 45% of differences in completely independent populations of ART- and in vivo-conceived children as a substantial success rate.
It should be noted that the absolute magnitude of the methylation differences observed between groups is small (Tables 3 and 4) and one might question its clinical significance. However, although the mechanism by which such differences might act is unclear, methylation differences of this magnitude are associated with significant differences in transcript level at multiple genes (Table four and Figures two and three in [1]; Tables one and five in [4]). In the case of the genes interrogated in the present study, several are associated with clinical phenotypes. Necdin (NDN), an imprinted gene, is associated with Prader-Willi syndrome [55] and prostate cancer [56]; similarly, CCDC62 acts as coactivator of the estrogen receptor in prostate cancer cell lines [57]; GRIN2C, a member of the NMDA glutamate receptor family, is associated with a number of neurological disorders [58,59]; Laeverin, the gene product of FLJ90650 gene, has been found to have differential expression in the placentas from patients with pre-eclampsia [60], and methylation levels of GRB10 are correlated with birth weight [30]. Therefore, we suggest that the small methylation differences observed may prove to be related to a pathophysiologic phenotype in the offspring at a later time and therefore should be considered as important rather than being dismissed as too small to have a clinically relevant significance.
Our study has several limitations. An effect of IVF cycle type (fresh or frozen) on the observed differences cannot be ruled out. Our study group has less than 20% (Table 1) frozen/thawed embryo transfer cycles. Following exclusion of these samples, the analysis of the results of our study remains unchanged (data not shown). An additional limitation is that methylation levels were measured at only a single placental site (directly behind the umbilical cord) and there is some evidence for intra-placental epigenetic variability in humans [4,61]. However, we have measured (see Figures one and three in [4]) DNA methylation levels in five sections of placenta (one tissue sample from each quadrant plus one central sample from directly behind the umbilical cord) from 54 individuals and shown that there is a strong correlation in IGF2/H19 and IGF2R methylation between biopsy locations within the same placenta, even though there can be substantial differences between individuals. An additional limitation is that we did not stratify our results by sex because of sample size constraints. It would be interesting to evaluate such methylation differences in male and female placentas, separately; however, the small numbers in the present cohorts, and especially in the donor oocyte ART group, might result in misleading conclusions. Finally, bisulfite conversion and pyrosequencing as a method of determining DNA methylation has limitations, including the possibility of incomplete bisulfite conversion and biased amplification. However, it should be noted that the genes chosen had previously been analyzed by a methylation array, and our study validated many of these previous findings.
Conclusions
Overall, our data indicate strongly that the majority of the placental DNA methylation differences we observed are associated with ART procedures rather than the diagnosis of infertility per se. This is the first study, to our knowledge, that has examined placental DNA methylation following ART in the absence of any known diagnosis of parental infertility in humans. Further studies on larger numbers of patients are needed to evaluate expression level differences, given the much greater inter-individual variability in transcript than in methylation levels (1, 4). Our findings should also direct future study of the underlying mechanism(s) behind the observed differences and focus on the question of which factors might be associated with the observed methylation differences. Elucidating such potentially modifiable factors related to ART-associated methylation differences should guide alterations to clinical and/or laboratory practice protocols, ultimately leading to an improvement in neonatal and long-term outcomes following ART.
Methods
Ethics statement
University of Pennsylvania’s Institutional Review Board approved the study (IRB approved protocol no. 804530).
Samples and sample preparation
Placenta biopsies were collected from in vivo-conceived controls (fertile control group) and ART children conceived following fertilization of oocytes from two groups: 1) autologous oocytes with infertility in one or both parents (infertile ART group) and 2) donor oocytes (obtained from young fertile donors) without male factor infertility (donor oocyte ART group). Genomic DNA was prepared as described [1,4]. All IVF cycles were performed at a single center and the clinical and laboratory procedures are uniform for all the samples. The fertile control group was conceived without medical assistance (infertility medications or treatments) and the parents had no prior history of infertility. All of the biopsies used in the present study were taken from the same location in each placenta, directly behind the cord on the maternal side. Eighty-eight ART placental samples (22 in the donor oocyte ART group and 66 in the female infertile ART group) and 49 controls were analyzed for all CpGs. Mean maternal age (the maternal age of recipient was included in case of the donor group), mean gestational age, and mean birth weight, as well as sex of controls and ART children, are also described in Table 1. All donor oocytes were obtained from young healthy women between the ages 21 and 33.
Selection of candidate genes
We selected candidate genes for validation of DNA methylation differences between ART and controls from cord blood and placenta samples assayed on an Illumina Human Methylation27 Bead-Chip array that was analyzed in a previous study [30]. The candidate genes whose methylation differed significantly between ART (n = 24) and control (n = 24) individuals were selected based on the following: 1) differential methylation of two or more CpG sites within the same gene (P < 0.05 at each, two-tailed t-test); 2) absolute magnitude of mean methylation differences, those with differences >0.07 were retained for further consideration; 3) ability to design high-quality pyrosequencing assays. Using these criteria, seven genes (GRIN2C, PCDHGB7, PTPN20B, MEST, TCF2, FLJ10260, NDN) were selected (additional genes fulfilling these criteria were available; however, high-quality pyrosequencing assays could not be designed or the assays failed). We considered further those candidate genes represented by only a single CpG on the array or for which only a single CpG was significant but that the mean absolute magnitude of difference was >0.07. Under these criteria, six additional genes (CRTAM, TTR, SNRPN, IL5, LYST, FLJ90650) were selected. The imprinted genes H19 and GRB10 were also selected because we had observed differences for multiple CpGs at both loci in previous studies [1,30], and 7 of the 16 H19 CpGs and 8 of the 12 GRB10 CpGs differed significantly on the array. Lastly, CCDC62 was selected because of the large mean difference between ART and control groups for both CpGs on the array (−0.079 and −0.062). Even though these differences did not reach statistical significance, the very large variance and absolute magnitude of difference in maximum/minimum beta values (>0.4) suggested that the significance might be reached on examining a larger number of individuals.
It is a common observation (for example, Figure two in [1]; Figure two in [4]; Figures one and two in [62]) that within a CpG island, methylation at one site is highly correlated with methylation at other sites within an individual. Hence, we selected one CpG site per CpG island for methylation analysis, except in the cases where multiple CpG sites could be analyzed in a single pyrosequencing assay. Hence, a total of 22 CpGs in 16 genes were selected for designing of pyrosequencing assays.
Design and performance of pyrosequencing assays
The designing of pyrosequencing assays for the 22 CpGs thus selected resulted in inclusion of 15 additional CpGs that were in close proximity to the selected CpGs. The genomic location of the 37 CpGs interrogated in the 16 genes by the bisulfite pyrosequencing assays and the primer sequences used are in Additional file 1.
Statistical analysis
One-way ANOVA was used to estimate differences between the three groups (infertile ART, donor oocyte ART, and fertile control). The significant results of ANOVA were analyzed using Tukey’s HSD test. One-tailed t-tests were used in comparing mean methylation levels between infertile ART and fertile control groups for the validation of our previous array findings [1,30] as we had a prior expectation for the direction of change and the use of one-sided tests is an accepted statistical procedure in validation studies [63]. Two-tailed t-tests were used for comparing fertile control and donor oocyte ART groups. A P-value of <0.05 was considered significant, without correction for multiple testing, because we were validating results for candidate genes shown previously to differ between ART- and in vivo-conceived children.
References
Katari S, Turan N, Bibikova M, Erinle O, Chalian R, Foster M, et al. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet. 2009;18:3769–78.
Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, et al. DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet. 2009;17(12):1582–91.
Gomes MV, Huber J, Ferriani RA, Amaral Neto AM, Ramos ES. Abnormal methylation at the KvDMR1 imprinting control region in clinically normal children conceived by assisted reproductive technologies. Mol Hum Reprod. 2009;15:471–7.
Turan N, Katari S, Gerson LF, Chalian R, Foster MW, Gaughan JP, et al. Inter- and intraindividual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS Genet. 2010;6:e1001033.
Katagiri Y, Aoki C, Tamaki-Ishihara Y, Fukuda Y, Kitamura M, Matsue Y, et al. Effects of assisted reproduction technology on placental imprinted gene expression. Obstet Gynecol Int. 2010;2010:pii: 437528.
Santos F, Hyslop L, Stojkovic P, Leary C, Murdoch A, Reik W, et al. Evaluation of epigenetic marks in human embryos derived from IVF and ICSI. Hum Reprod. 2010;25(9):2387–95.
Shi X, Ni Y, Zheng H, Chen S, Zhong M, Wu F, et al. Abnormal methylation patterns at the IGF2/H19 imprinting control region in phenotypically normal babies conceived by assisted reproductive technologies. Eur J Obstet Gynecol Reprod Biol. 2011;158(1):52–5.
Nelissen EC, Dumoulin JC, Daunay A, Evers JL, Tost J, Van Montfoort AP. Placentas from pregnancies conceived by IVF/ICSI have a reduced DNA methylation level at the H19 and MEST differentially methylated regions. Hum Reprod. 2013;28(4):1117–26.
Zheng HY, Tang Y, Niu J, Li P, Ye DS, Chen X, et al. Aberrant DNA methylation of imprinted loci in human spontaneous abortions after assisted reproduction techniques and natural conception. Hum Reprod. 2013;28(1):265–73.
Lou H, Le F, Zheng Y, Li L, Wang L, Wang N, et al. Assisted reproductive technologies impair the expression and methylation of insulin-induced gene 1 and sterol regulatory element-binding factor 1 in the fetus and placenta. Fertil Steril. 2014;101(4):974–80.
Tierling S, Souren NY, Gries J, Loporto C, Groth M, Lutsik P, et al. Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human. J Med Genet. 2010;47:371–6.
Zechner U, Pliushch G, Schneider E, El Hajj N, Tresch A, Shufaro Y, et al. Quantitative methylation analysis of developmentally important genes in human pregnancy losses after ART and spontaneous conception. Mol Hum Reprod. 2010;16(9):704–13.
Li L, Wang L, Le F, Liu X, Yu P, Sheng J, et al. Evaluation of DNA methylation status ay differentially methylated regions in IVF-conceived newborn twins. Fertil Steril. 2011;95(6):1975–9.
Wong EC, Hatakeyama C, Robinson WP, Ma S. DNA methylation at H19/IGF2 ICR1 in the placenta of pregnancies conceived by in vitro fertilization and intracytoplasmic sperm injection. Fertil Steril. 2011;95:2524–6.
Feng C, Tian S, Zhang Y, He J, Zhu XM, Zhang D, et al. General imprinting status is stable in assisted reproduction conceived offspring. Fertil Steril. 2011;96:1417–23.
Zheng HY, Shi XY, Wang LL, Wu YQ, Chen SL, Zhang L. Study of DNA methylation patterns of imprinted genes in children born after assisted reproductive technologies reveals no imprinting errors: a pilot study. Exp Ther Med. 2011;2(4):751–5.
Oliver VF, Miles HL, Cutfield WS, Hofman PL, Ludgate JL, Morison IM. Defects in imprinting and genome-wide DNA methylation are not common in the in the in vitro fertilization population. Fertil Steril. 2012;97(1):147–53.
Puumala SE, Nelson HH, Ross JA, Nguyen RH, Damario MA, Spector LG. Similar DNA methylation levels in specific imprinting control regions in children conceived with and without assisted reproductive technology: a cross-sectional study. BMC Pediatr. 2012;12:33.
Rancourt RC, Harris HR, Michels KB. Methylation levels at imprinting control regions are not altered with ovulation induction or in vitro fertilization in a birth cohort. Hum Reprod. 2012;27:2208–16.
Camprubí C, Iglesias-Platas I, Martin T, Salvador-Alarcon C, Rodriguez MA, Barredo DR, et al. Stability of genomic imprinting and gestational-age dynamic methylation in complicated pregnancies conceived following assisted reproductive technologies. Biol Reprod. 2013;89(3):50.
Bloise E, Lin W, Liu X, Simbulan R, Kolahi KS, Petraglia F, et al. Impaired placental nutrient transport in mice generated by in vitro fertilization. Endocrinology. 2012;153(7):3457–67.
Doherty AS, Mann MR, Tremblay KD, Bartolomei MS, Schultz RM. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biol Reprod. 2000;62:1526–35.
Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, et al. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet. 2001;27:153–4.
Mann MR, Lee SS, Doherty AS, Verona RL, Nolen LD, Schultz RM, et al. Selective loss of imprinting in the placenta following preimplantation development in culture. Development. 2004;131:3727–35.
Farin PW, Piedrahita JA, Farin CE. Errors in development of fetuses and placentas from in vitro-produced bovine embryos. Theriogenology. 2006;65(1):178–91.
Rivera RM, Stein P, Weaver JR, Mager J, Schultz RM, Bartolomei MS. Manipulations of mouse embryos prior to implantation result in aberrant expression of imprinted genes on day 95 of development. Hum Mol Genet. 2008;17:1–14.
Fortier AL, Lopes FL, Darricarrère N, Martel J, Trasler JM. Superovulation alters the expression of imprinted genes in the midgestation mouse placenta. Hum Mol Genet. 2008;17(11):1653–65.
Li L, Wang L, Xu X, Lou H, Le F, Li L, et al. Genome-wide DNA methylation patterns in IVF-conceived mice and their progeny: a putative model for ART-conceived humans. Reprod Toxicol. 2011;32:98–105.
Wright K, Brown L, Brown G, Casson P, Brown S. Microarray assessment of methylation in individual mouse blastocyst stage embryos shows that in vitro culture may have widespread genomic effects. Hum Reprod. 2011;26:2576–85.
Turan N, Ghalwash MF, Katari S, Coutifaris C, Obradovic Z, Sapienza C. DNA methylation differences at growth related genes correlate with birth weight: a molecular signature linked to developmental origins of adult disease? BMC Med Genomics. 2012;5:10.
Schieve LA, Rasmussen SA, Buck GM, Schendel DE, Reynolds MA, Wright VC. Are children born after assisted reproductive technology at increased risk for adverse health outcomes? Obstet Gynecol. 2004;103(6):1154–63.
Helmerhorst FM, Perquin DA, Donker D, Keirse MJ. Perinatal outcome of singletons and twins after assisted conception: a systematic review of controlled studies. Br Med J. 2004;328:261.
Jackson RA, Gibson KA, Wu YW, Croughan MS. Perinatal outcomes in singletons following in vitro fertilization: a meta-analysis. Obstet Gynecol. 2004;103:551–63.
Sunderam S, Chang J, Flowers L, Kulkarni A, Sentelle G, Jeng G, et al. Assisted reproductive technology surveillance - United States, 2006. Morb Mortal Wkly Rep. 2009;58:1–25.
McDonald SD, Han Z, Mulla S, Murphy KE, Beyene J, Ohlsson A. Preterm birth and low birth weight among in vitro fertilization singletons: a systematic review and meta-analyses. Eur J Obstet Gynecol Reprod Biol. 2009;146:138–48.
Schieve LA, Meikle SF, Ferre C, Peterson HB, Jeng G, Wilcox LS. Low and very low birth weight in infants conceived with use of assisted reproductive technology. N Engl J Med. 2002;346(10):731–7.
Kallen B, Finnstrom O, Nygren KG, Olausson PO. In vitro fertilization (IVF) in Sweden: risk for congenital malformations after different IVF methods. Birth Defects Res A Clin Mol Teratol. 2005;73:162–9.
Rimm AA, Katayama AC, Diaz M, Katayama KP. A meta-analysis of controlled studies comparing major malformation rates in IVF and ICSI infants with naturally conceived children. J Assist Reprod Genet. 2004;21(12):437–43.
Hansen M, Bower C, Milne E, de Klerk N, Kurinczuk JJ. Assisted reproductive technologies and the risk of birth defects - a systematic review. Hum Reprod. 2005;20(2):328–38.
Wen J, Jiang J, Ding C, Dai J, Liu Y, Xia Y, et al. Birth defects in children conceived by in vitro fertilization and intracytoplasmic sperm injection: a meta-analysis. Fertil Steril. 2012;97(6):1331–7.
Ceelen M, van Weissenbruch MM, Prein J, Smit JJ, Vermeiden JP, Spreeuwenberg M, et al. Delemarre-van de Waal HA. Growth during infancy and early childhood in relation to blood pressure and body fat measures at age 8–18 years of IVF children and spontaneously conceived controls born to subfertile parents. Hum Reprod. 2009;24(11):2788–95.
Ceelen M, van Weissenbruch MM, Vermeiden JP, van Leeuwen FE, de Waal HA D-v. Cardiometabolic differences in children born after in vitro fertilization: follow-up study. J Clin Endocrinol Metab. 2008;93(5):1682–8.
Wikstrand MH, Niklasson A, Strömland K, Hellström A. Abnormal vessel morphology in boys born after intracytoplasmic sperm injection. Acta Paediatr. 2008;97(11):1512–7.
Scott KA, Yamazaki Y, Yamamoto M, Lin Y, Melhorn SJ, Krause EG, et al. Glucose parameters are altered in mouse offspring produced by assisted reproductive technologies and somatic cell nuclear transfer. Biol Reprod. 2010;83(2):220–7.
Scherrer U, Rimoldi SF, Rexhaj E, Stuber T, Duplain H, Garcin S, et al. Systemic and pulmonary vascular dysfunction in children conceived by assisted reproductive technologies. Circulation. 2012;125(15):1890–6.
DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alteration of LIT1 and H19. Am J Hum Genet. 2003;72:156–60.
Maher ER, Brueton LA, Bowdin SC, Luharia A, Cooper W, Cole TR, et al. Beckwith-Wiedemann syndrome and assisted reproductive technology (ART). J Med Genet. 2003;40:62–4.
Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le Bouc Y. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCNQ1OT gene. Am J Hum Genet. 2003;72:1338–41.
Halliday J, Oke K, Breheny S, Algar E, Amor DJ. Beckwith-Wiedemann syndrome and IVF: a case-control study. Am J Hum Genet. 2004;75:526–8.
Weksberg R, Shuman C, Smith AC. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2005;137:12–23.
Sutcliffe AG, Peters CJ, Bowdin S, Temple K, Reardon W, Wilson L, et al. Assisted reproductive therapies and imprinting disorders - a preliminary British survey. Hum Reprod. 2006;21:1009–11.
Doornbos ME, Maas SM, McDonnell J, Vermeiden JPW, Hennekam RCM. Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum Reprod. 2007;22:2476–80.
Urrego R, Rodriguez-Osorio N, Niemann H. Epigenetic disorders and altered gene expression after use of assisted reproductive technologies in domestic cattle. Epigenetics. 2014;9(6):803–15.
van Montfoort APA, Hanssen LLP, de Sutter P, Viville S, Geraedts JPM, de Boer P. Assisted reproduction treatment and epigenetic inheritance. Hum Reprod. 2012;18(2):171–97.
Rieusset A, Schaller F, Unmehopa U, Matarazzo V, Watrin F, Linke M, et al. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse and humans with Prader-Willi syndrome, has functional consequences. PLoS Genet. 2013;9(9):e1003752.
Ribarska T, Goering W, Droop J, Bastian KM, Ingenwerth M, Schulz WA. Deregulation of an imprinted gene network in prostate cancer. Epigenetics. 2014;9(5):704–17.
Chen M, Ni J, Chang HC, Lin CY, Muyan M, Yeh S. CCDC62/ERAP75 functions as a coactivator to enhance estrogen receptor beta-mediated transactivation and target gene expression in prostate cancer cells. Carcinogenesis. 2009;30(5):841–50.
Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1:e55.
Chandley MJ, Szebeni A, Szebeni K, Crawford JD, Stockmeier CA, Turecki G, et al. Elevated gene expression of glutamate receptors in noradrenergic neurons from the locus coeruleus in major depression. Int J Neuropsychopharmacol. 2014;17(10):1569–78.
Nystad M, Sitras V, Larsen M, Acharya G. Placental expression of aminopeptidase-Q (laeverin) and its role in pathophysiology of pre-eclampsia. Am J Obstet Gynecol. 2014;211(6):686.
Looijenga LH, Gillis AJ, Verkerk AJ, Van Putten WL, Oosterhuis JW. Heterogeneous X inactivation in trophoblastic cells of human full-term female placentas. Am J Hum Genet. 1999;64:1445–52.
Silviera ML, Smith BP, Powell J, Sapienza C. Epigenetic differences in normal colon mucosa of cancer patients suggests altered dietary metabolic pathways. Cancer Prev Res. 2012;5(3):374–84.
Cesaroni M, Powell J, Sapienza C. Validation of methylation biomarkers that distinguish normal colon mucosa of cancer patients from normal colon mucosa of patients without cancer. Cancer Prev Res (Phila). 2014;7(7):717–26.
Acknowledgements
This work was supported by National Institute of Health (grant numbers P50-HD-068157 to CC and CS, T32-HD-040135 to RW).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS, JG, NT, and MT participated in the sample preparation, selection of candidate genes, bisulfite pyrosequencing, and methylation data analysis. SS, JG, CC, and CS participated in data interpretation and manuscript preparation. MM, RW, and CC participated in patient selection, collection and evaluation of clinical data for subgroup analysis. CC and CS designed the study, supervised all aspects of the work, and participated in the manuscript preparation. All authors read and approved the final manuscript.
Authors’ information
SS and JG are joint first authors. CC and CS are co-senior authors and contributed equally in formulating and overseeing the research described in this manuscript.
Additional file
Additional file 1:
Primer sequences for the pyrosequencing assays.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Song, S., Ghosh, J., Mainigi, M. et al. DNA methylation differences between in vitro- and in vivo-conceived children are associated with ART procedures rather than infertility. Clin Epigenet 7, 41 (2015). https://doi.org/10.1186/s13148-015-0071-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-015-0071-7