
Abstract
Background
While a possible link between assisted reproductive technology (ART) and rare imprinting disorders has been found, it is not clear if this is indicative of subtler disruptions of epigenetic mechanisms. Results from previous studies have been mixed, but some methylation differences have been observed.
Methods
Children conceived through ART and children conceived spontaneously were recruited for this cross-sectional study. Information about reproductive history, demographic factors, birth characteristics, and infertility treatment was obtained from maternal interview and medical records. Peripheral blood lymphocytes and buccal cell samples were collected from participating children. Methylation analysis was performed on five loci using pyrosequencing. Statistical analysis of methylation differences was performed using linear regression with generalized estimating equations. Results are reported as differences with 95% confidence intervals (CI).
Results
A total of 67 ART children and 31 spontaneously conceived (SC) children participated. No significant difference in methylation in lymphocyte samples was observed between groups for any loci. Possible differences were found in buccal cell samples for IGF2 DMR0 (Difference: 2.07; 95% confidence interval (CI): -0.28, 4.42; p = 0.08) and IGF2R (Difference: -2.79; 95% CI: -5.74, 0.16; p = 0.06). Subgroup analysis indicated potential lower methylation in those whose parents used ART for unexplained infertility.
Conclusions
Observed differences in methylation between the ART and SC groups were small for all loci in the two sample types examined and no statistical differences were observed. It is still unclear whether or not small differences observed in several studies represent a real difference between groups and if this difference is biologically meaningful. Larger studies with long term follow-up are needed to fully answer these questions.
Similar content being viewed by others


Background
Use of assisted reproductive technology (ART), including in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI), is rapidly rising in the United States (US) and curxxrently accounts for over one percent of all infants born each year [1]. The potential for epigenetic disruptions in children born after infertility treatment, particularly ART, was first seen in a number of studies showing an increase in imprinting disorders such as Beckwith-Wiedemann syndrome (BWS), Angelman syndrome (AS), and Silver-Russell syndrome (SRS) in these children [2–16]. These studies further indicated that the cause of the imprinting disorders was due to epigenetic disruptions rather than mutations or uniparental disomy [4, 5, 7, 9–11, 13, 17–20].
Imprinting disorders are very rare and, even with a relative increase in incidence of these disorders, most children conceived through ART are healthy. However, a relative increase suggests the possibility for more frequent, but subtler disruptions of epigenetic mechanisms. It has been suggested that such epigenetic disruptions could potentially manifest themselves as an increased propensity for childhood cancer as well as adult onset diseases such as cancer and heart disease that are thought to be epigenetically mediated [21, 22].
Several recent studies have examined differences in methylation in various imprinted gene regions after ART in peripheral blood, placenta tissue, buccal cells, cord blood, chorionic villus samples, and embryos [23–38]. Results have been mixed and difficult to synthesize due to differences in gene regions, tissues examined, and ways of assessing DNA methylation. However, some studies have indicated a difference in DNA methylation or gene expression in the various gene regions [24, 26, 27, 30, 32, 33], although other studies have not observed a difference [25, 28, 29, 31, 34–37].
Given the mixed evidence so far, and since genes in the 11p15 region and the IGF2R gene located at 6q26 have been associated with BWS and SRS and many different types of cancer, we were interested in further exploring these regions for differential methylation. To assess quantitative DNA methylation differences between in children conceived after ART treatment and children conceived spontaneously, we conducted a cross-sectional study and focused on peripheral blood and buccal cell samples. Specifically, we examined quantitative methylation values at the 11p15 region including two CTCF binding sites within H19, one differentially methylated region (DMR) in IGF2, and the imprinting control region KvDMR as well as a DMR in the IGF2R gene located at 6q26. Although some of these sites have been commonly examined (e.g. KvDMR and CTCF binding sites in H19), little information is available about methylation differences in the IGF2 DMR0 and IGF2R regions each having been explored in only one prior study [33, 38].
Methods
Study population
Two groups of children were recruited for this study; one conceived through ART (ART group) and the other born after spontaneous conception (SC group). ART children had to be conceived through IVF or IVF + ICSI with fresh non-donor oocytes. SC children had to have been conceived without the use of any fertility drugs or treatments. In the case of multiple births in either the ART or SC group, only one child was selected for participation in the study. Children diagnosed with BWS, AS, Prader-Willi syndrome, or retinoblastoma were excluded from the study.
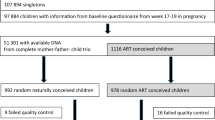
ART children and their mothers were recruited from the University of Minnesota Reproductive Medicine Center. Mothers who conceived through ART and reported a live birth between March 2005 and December 2008 were contacted about participation of their child through a letter and follow-up phone call. Out of 328 women identified as potentially eligible, 99 agreed to participate and 67 children completed the clinic visit.
SC children were recruited through advertisements posted in and around the University of Minnesota. Similar to the ART children, SC children had to have been born between March 2005 and December 2008. A total of 45 women agreed to participate in the study and 31 children completed the clinic visit prior to the close of the study.
This study was approved by the institutional review board at the University of Minnesota. All mothers provided informed consent for themselves and their child prior to participating in the study.
Sample collection
Peripheral blood lymphocytes and buccal cell samples were collected either at a research clinic or at the participant's local clinic. Samples from the research clinic were delivered for processing immediately, while other clinic samples were sent express mail to Dr. Nelson's laboratory and were processed immediately upon receipt (usually within 24 h of sample collection). Up to 6 ml of blood was collected from each child through venipuncture. Buccal cells were collected using Catch-All™ Sample Collection Swabs (Epicentre, Madison, WI, USA). Two swabs were collected from each child.
Data collection
Information about ART and SC mothers and children was collected through a brief questionnaire as well as from delivery records. In addition, for ART mothers, information on specific procedures was obtained from medical records. The questionnaire collected demographic information, as well as maternal reproductive history and the participating child's birth characteristics. All maternal and child characteristics used in the analysis apart from birth weight were based on questionnaire data. Birth weight was taken from the delivery records. If birth weight was not available on the birth record, maternal report from the study questionnaire was used. SC mothers were also asked about time to pregnancy to assess possible infertility that resolved without treatment. Information obtained from infertility clinic records included indication for infertility, number of cycles of treatment, and specific procedure information.
Genetic regions analyzed
Three regions of interest were examined for differential methylation: IGF2/H19, KvDMR, and IGF2R. These regions were selected based on their association with BWS, SRS, and cancer. Specifically, we analyzed IGF DMR0 (3 CpGs), 3 rd (11 CpGs) and 6th (16 CpGs) CTCF-binding site of H19 DMR, IGF2R (15 CpGs), and KvDMR (7 CpGs). The 5th CpG cite in the 6th CTCF-binding site of H19 DMR is known to be polymorphic and so was excluded from the analysis [39].
Methylation analysis
Methylation analysis was performed using pyrosequencing. Genomic DNA was isolated from the lymphocyte and buccal cell samples and treated with sodium bisulfite using the EZ methylation kit (Zymo research). This treatment actively changes unmethylated cytosines to uracils while leaving methylated cytosines unchanged. Primers and procedures for IGF DMR0, 3rd and 6th CTCF-binding site of H19 DMR, IGF2R were the same as in Boissonnas, et al. [39]. The protocol for the KvDMR region was different and based on Bourque, et al. [40]. After PCR amplification, pyrosequencing was performed for all regions using the PyroMark MD system, and analyzed using the accompanying software (Qiagen, Germantown/Gaithersburg, USA).
The percent methylation for each CpG was calculated by taking the peak height of the methylated cytosines divided by the sum of the peak height of the methylated and unmethylated cytosine. Several quality assurance tests were performed to assess the sequence generated by the pyrosequencing reaction against the expected sequence. CpG sites that consistently failed quality assurance tests were excluded from the analysis. We excluded one CpG site from H19 CTCF3, one CpG site from H19 CTCF6 and six CpG sites from IGF2R due to quality and polymorphism issues. Two children were excluded in locus-specific analysis due to unexpected sequences, likely due to polymorphisms, one in the KvDMR locus and the other in the IGF2 DMR0 locus.
To assess the reliability of the methylation assays, we obtained lymphocyte and buccal cell samples on nine healthy adults. Samples were split and processed on different days for three loci (KvDMR, H19 CTCF3, and IGF2R). Reliability was assessed using an intraclass correlation coefficient (ICC). While there were a few low ICC values for individual CpG sites, most of the sites had high ICC values (Additional file 2). We found that there was poor reliability for methylation analysis over amplifications for the KvDMR region in lymphocyte samples (ICC median (Interquartile range (IQR)): 0.54 (0.44-0.56)). However, this lower reliability was due to an overall lower methylation level for one of the three amplifications. Since a linear transformation of one of the three amplifications greatly improved reliability (ICC median (IQR): 0.78 (0.62-0.81)) accounting for amplification in our analysis will adjust our data for possible similar discrepancies. Reliability was goo d for the KvDMR region in buccal samples with all individual CpG sites found to have an ICC greater than 0.7 (ICC median (IQR): 0.87 (0.84-0.87)). The H19 CTCF3 region had good reliability for both buccal cells (ICC median (IQR): 0.73 (0.66-0.77)) and lymphocytes (ICC median (IQR): 0.76 (0.73-0.81)). The IGF2R region also had good reliability for both buccal cells (ICC median (IQR): 0.78 (0.59-0.81)) and lymphocytes (ICC median (IQR): 0.88 (0.70-0.89)).
Data analysis
Descriptive data were compared between the two groups using Fisher's exact tests for categorical variables, and t-tests for continuous variables. Differences in methylation between groups were analyzed using linear models with generalized estimating equations (GEE). The GEE model accounts for the correlation between CpG cites within an individual. Each locus was considered separately. Adjusted models were constructed using variables related to use of ART: maternal age (continuous), maternal education (some college or less, college graduate, advanced degree), household income (< $40,000, $40,000-$79,999, ≥ $80,000) and child's birth weight (continuous); and variables possibly associated with methylation: child's age (continuous) and sex (male, female). All models also included a variable indicating day of the pyrosequencing run to control for any amplification effects. Subgroup analysis was performed within the ART group to examine differences in methylation by type of infertility (female only, male only, both male and female, unexplained). Results are reported as group means and 95% confidence intervals (CI) for the difference between groups. Sensitivity analysis was performed excluding the samples that were rerun after assay failure. All analysis was performed using SAS 9.2 (SAS Institute, Inc, Cary, NC).
Results
A total of 67 children were included in the ART group and 31 in the SC group. Of these, 53 ART children and 27 SC children provided a blood sample while 67 ART children and 30 SC children provided a buccal cell sample. Demographic factors of the two groups are presented in Table 1. Many factors were different between the two groups, with the ART group tending to have higher household income, increased maternal age, and greater frequency of multiple births. Children in the ART group tended to be younger and have lower birth weights.
Medical records for infertility treatment could not be obtained for six women, leaving 61 subjects for analysis of specific infertility diagnoses. Infertility diagnoses were fairly equally divided between female factor only (n = 21, 34%), male factor only (n = 17, 28%), and both male and female factors (n = 16, 26%). No medical explanation for infertility was found in 10% (n = 6) of couples and one couple was seeking treatment for reasons other than fertility. Most couples used ICSI for at least some of the embryos (84%) and over 70% had two embryos transferred.
No large differences in methylation were found between the ART and SC groups in either lymphocyte or buccal cell samples. Table 2 displays the average differences and 95% CI for the adjusted models. A possible, but statistically non-significant difference was seen in buccal cells only at IGF2 DMR0, with the ART group having higher methylation levels compared to the SC group (Difference: 2.07; 95% CI: -0.28, 4.42; p = 0.08). Larger, but non-significant, differences were also observed for the IGF2R region; for both buccal cells (Difference: -2.79; 95% CI: -5.74, 0.16; p = 0.06) and lymphocytes (Difference: -4.41; 95% CI: -9.49, 0.66; p = 0.09) there was an indication of lower levels of methylation in the ART group. Estimates were similar when limited to those samples that were not rerun due to assay failure (data not shown).
In the subgroup analysis, couples with unexplained infertility had children that tended to have lower methylation levels compared with couples in which both partners had an identified cause of infertility (Table 3). Sample size was small, however, and no overall group comparison was statistically significant.
Discussion
Overall, there was little difference in methylation between children conceived through ART and children conceived spontaneously. In subgroup analysis, couples with unexplained infertility tended to have lower methylation levels compared to couples in which both partners had an identified cause of infertility.
The observed differences in methylation between the ART and SC group were very small, indicating that our failure to detect moderate differences was not due to a lack of statistical power even though variability was increased due to the small sample size and imperfect assay reliability [41]. Larger samples or samples with increased reliability could be used to detect smaller differences; however, we were able to rule out differences in methylation greater than about 7% in all analysis. For lymphocyte samples, average differences were around 1% or less between groups. Based on the variability of the samples, our analysis suggested that differences greater than 2-4 percentage points for all loci except IGF2R are unlikely. Methylation in buccal cell samples was more variable, with an average difference of around 2%. For these samples, differences greater than 5-7 percentage points appeared unlikely. It should be noted that it is unknown if very small differences in methylation can lead to difference in gene expression levels; so, although it seems unlikely that there is an effect for a few percentage points difference in methylation, it is possible. One study did find that small differences in DNA methylation resulted in differences in transcript levels, suggesting that these small differences could be biologically relevant [26].
Other studies have examined differential methylation in various tissues of children conceived through ART with normal phenotypes (Table 4). Small, non-significant differences between groups, such as those seen in our study, have been observed in many studies (Additional file 1). The most common region that has been examined is the IGF2/H19 imprinting region. In placenta tissue, two studies have indicated a potential difference in methylation and expression in H19 and IGF2 [33] and expression only in H19 but not IGF2 [30] while another study found no difference in methylation [34]. Two studies examined possible difference in this region in miscarriages, abortions, and stillbirths with one finding a possible difference but more extreme values in the control group [35] and the other finding six cases with hypomethylation in the ART group and none in the control group [27]. In addition, a study on embryos from ART patients found that close to 19% of these embryos had hypomethylation or demethylation in the H19 DMR [23]. Overall, there is some evidence for possible hypomethylation or reduced methylation in children conceived through ART in several studies [23, 27, 31, 35]; however, the majority of studies have not found a statistically significant difference between groups [25, 26, 28, 31, 32, 34, 35, 38].
For the KvDMR region, some expression differences have been seen in placenta for CDKN1C, but not KCNQ1OT1 [30]. Other studies have found no differences within this region [27, 32, 35, 36], although one did note hypomethylation in three children conceived through ART [24] and another found hypermethylation of KCNQ1OT1 in a child conceived using ART [25]. As with the IGF2/H19 region while there is some evidence of possible hypomethylation [24, 27, 28, 35], most studies have not found a statistically significant difference [24, 26, 28, 32, 36, 38].
Two regions examined in this study are relatively novel, having only been examined in one prior study. The DMR in the IGF2R gene is of particular interest since it has not be well studied, and a recent study associated gain of methylation at this locus with growth restriction [42]. We found a non-significant decrease in methylation within the IGF2R region for both lymphocyte and buccal cell samples, which would be in contrast to the growth restriction study, since children conceived through ART tend to be smaller at birth [43]. The only other study examining this locus in children conceived through ART also did not find a difference in DNA methylation in cord blood, cord samples, or placenta, but quantitative results are difficult to compare since that study used the ratio of maternal to paternal allele methylation as their primary outcome [33]. The IGF2 DMR0 was only recently examined in a study by Oliver, et al., who found no difference in DNA methylation at this region in peripheral blood samples [38]. While our study concurs with these finding for peripheral blood, we did find non-significant increase in DNA methylation at this region in buccal cells of children conceived through ART, so small differences in DNA methylation cannot be ruled out for this locus.
Another study used a methylation bead-array platform and examined 1536 CpG sites in over 700 genes using placenta and cord blood samples and found an overall lower level of methylation at CpG sites in the placenta samples of in vitro conceived children and a higher level of methylation in cord blood of in vitro conceived children [26]. This study also found many individual genes that had differential methylation between the two groups. However, none of the genes analyzed in our study were specifically identified.
Other regions that have been explored in multiple studies include SNRPN, MEG1/GTL2, PEG1/MEST, and PEG3. No studies have found evidence of a difference in DNA methylation in the SNRPN region in children conceived through ART compared with an SC group [26, 27, 29, 30, 32, 35, 36, 38]. Differences have also not been observed for the MEG1/GTL2 region in multiple studies [25, 26, 32, 35, 36]. Methylation differences were found between the ART and SC group in CpG sites associated with the PEG1/MEST region in both cord blood and placenta [26]. A second study support this finding, but also found differences in methylation in maternal blood, suggesting that the pattern seen in offspring was not due to the infertility treatment [32]. Other studies have not found a quantitative difference [28, 35, 37]; however one study found a case of hypermethylation in buccal cells [25] and one found a case of hypomethylation in extra-embryonic tissue [27]. PEG3 was also identified as have different methylation patterns between the ART and SC group from the genome-wide study both in cord blood and placenta [26], but other studies have not confirmed this finding [25, 35]. One study found evidence of hypomethylation in one ART case in this region [27]. Other regions have been identified in single studies, but have not been confirmed.
The current study adds to the growing body of literature examining methylation differences in non-syndromic children conceived through ART and children conceived spontaneously. This study explored several specific loci associated with growth, cancer, and BWS some of which have been rarely studied in this population. Using a quantitative measure of DNA methylation is sensitive to detect subtle differences between groups which may influence transcription and gene expression [26]. It also included two sample types easily available after birth to permit broad follow-up studies. Finally, even though our sample size was not large, it is unlikely that we missed substantial differences in methylation based on our confidence intervals.
There are several caveats that should be addressed in this study. First, methylation abnormalities such as the type we are looking for here could be tissue specific rather than a global phenomenon. While it would be interesting to examine other tissues, it would not be ethical to obtain other tissue/sample types in otherwise healthy children since most collection procedures would be invasive. Unexposed individuals were a convenience sample rather than a random sample from a particular population. Women who use ART are more likely to be white, have higher incomes, and be better educated compared to infertile women who choose other or no treatment [44]. Although we observed some demographic differences between the ART and SC groups, mothers in the SC group were more likely to be white and have higher income and education levels compared to the US population and thus may represent a good comparison group. Finally, a limited number of genetic loci were evaluated in this study. Only those with the most a priori likelihood of an association were examined to maximize the potential for finding important associations.
Conclusions
Overall, very small differences were observed in methylation level at all loci between groups. Some possible but non-significant differences in buccal cells and possible differences by infertility diagnosis may warrant additional follow up in other studies. The bulk of studies performed so far indicate that large differences in methylation are unlikely. However, it is not known the potential biological effect that small differences in methylation could have and there is some evidence that these might be relevant. Although results of this study are reassuring, additional large studies that include a broad range of genes as well as long term follow-up of children conceived through ART are needed to fully assess possible epigenetic differences and the potential impact of small differences on future disease in this population.
Abbreviations
- AS:
-
Angelman syndrome
- ART:
-
Assisted reproductive technology
- BWS:
-
Beckwith-Wiedemann syndrome
- CI:
-
Confidence interval
- GEE:
-
Generalized estimating equations
- ICC:
-
Intraclass correlation coefficient
- ICSI:
-
Intracytoplasmic sperm injection
- IQR:
-
Interquartile range
- IVF:
-
In vitro fertilization
- SRS:
-
Silver-Russell syndrome
- SC:
-
Spontaneously conceived
- US:
-
United States.
References
Centers for Disease Control and Prevention: American Society for Reproductive Medicine, Society for Assisted Reproductive Technology: 2007 Assisted Reproductive Technology Success Rates: National Summary and Fertility Clinic Reports. 2009, Atlanta, GA: U.S. Department of Health and Human Services
Bowdin S, Allen C, Kirby G, Brueton L, Afnan M, Barratt C, Kirkman-Brown J, Harrison R, Maher ER, Reardon W: A survey of assisted reproductive technology births and imprinting disorders. Hum Reprod. 2007, 22: 3237-3240. 10.1093/humrep/dem268.
Chang AS, Moley KH, Wangler M, Feinberg AP, Debaun MR: Association between Beckwith-Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients. Fertil Steril. 2005, 83: 349-354. 10.1016/j.fertnstert.2004.07.964.
Cox GF, Burger J, Lip V, Mau UA, Sperling K, Wu BL, Horsthemke B: Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am J Hum Genet. 2002, 71: 162-164. 10.1086/341096.
DeBaun MR, Niemitz EL, Feinberg AP: Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003, 72: 156-160. 10.1086/346031.
Doornbos ME, Maas SM, McDonnell J, Vermeiden JP, Hennekam RC: Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum Reprod. 2007, 22: 2476-2480. 10.1093/humrep/dem172.
Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le Bouc Y: In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003, 72: 1338-1341. 10.1086/374824.
Olivennes F, Mannaerts B, Struijs M, Bonduelle M, Devroey P: Perinatal outcome of pregnancy after GnRH antagonist (ganirelix) treatment during ovarian stimulation for conventional IVF or ICSI: a preliminary report. Hum Reprod. 2001, 16: 1588-1591. 10.1093/humrep/16.8.1588.
Sutcliffe AG, Peters CJ, Bowdin S, Temple K, Reardon W, Wilson L, Clayton-Smith J, Brueton LA, Bannister W, Maher ER: Assisted reproductive therapies and imprinting disorders-a preliminary British survey. Hum Reprod. 2006, 21: 1009-1011.
Halliday J, Oke K, Breheny S, Algar E: D JA: Beckwith-Wiedemann syndrome and IVF: a case-control study. Am J Hum Genet. 2004, 75: 526-528. 10.1086/423902.
Maher ER, Brueton LA, Bowdin SC, Luharia A, Cooper W, Cole TR, Macdonald F, Sampson JR, Barratt CL, Reik W, et al: Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J Med Genet. 2003, 40: 62-64. 10.1136/jmg.40.1.62.
Galli-Tsinopoulou A, Emmanouilidou E, Karagianni P, Grigoriadou M, Kirkos J, Varlamis GS: A female infant with Silver Russell Syndrome, mesocardia and enlargement of the clitoris. Hormones (Athens, Greece). 2008, 7: 77-81.
Kagami M, Nagai T, Fukami M, Yamazawa K, Ogata T: Silver-Russell syndrome in a girl born after in vitro fertilization: partial hypermethylation at the differentially methylated region of PEG1/MEST. Journal of assisted reproduction and genetics. 2007, 24: 131-136. 10.1007/s10815-006-9096-3.
Kallen B, Finnstrom O, Nygren KG, Olausson PO: In vitro fertilization (IVF) in Sweden: risk for congenital malformations after different IVF methods. Birth defects research. 2005, 73: 162-169. 10.1002/bdra.20107.
Svensson J, Bjornstahl A, Ivarsson SA: Increased risk of Silver-Russell syndrome after in vitro fertilization?. Acta Paediatr. 2005, 94: 1163-1165. 10.1080/08035250510030125.
Ludwig M, Katalinic A, Gross S, Sutcliffe A, Varon R, Horsthemke B: Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J Med Genet. 2005, 42: 289-291. 10.1136/jmg.2004.026930.
Gomes MV, Gomes CC, Pinto W, Ramos ES: Methylation pattern at the KvDMR in a child with Beckwith-Wiedemann syndrome conceived by ICSI. American journal of medical genetics. 2007, 143: 625-629.
Rossignol S, Steunou V, Chalas C, Kerjean A, Rigolet M, Viegas-Pequignot E, Jouannet P, Le Bouc Y, Gicquel C: The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J Med Genet. 2006, 43: 902-907. 10.1136/jmg.2006.042135.
Lim D, Bowdin SC, Tee L, Kirby GA, Blair E, Fryer A, Lam W, Oley C, Cole T, Brueton LA, et al: Clinical and molecular genetic features of Beckwith-Wiedemann syndrome associated with assisted reproductive technologies. Hum Reprod. 2009, 24: 741-747.
Bliek J, Terhal P, van den Bogaard MJ, Maas S, Hamel B, Salieb-Beugelaar G, Simon M, Letteboer T, van der Smagt J, Kroes H, et al: Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet. 2006, 78: 604-614. 10.1086/502981.
Sharma P, Kumar J, Garg G, Kumar A, Patowary A, Karthikeyan G, Ramakrishnan L, Brahmachari V, Sengupta S: Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol. 2008, 27: 357-365. 10.1089/dna.2007.0694.
Feinberg AP, Tycko B: The history of cancer epigenetics. Nat Rev Cancer. 2004, 4: 143-153. 10.1038/nrc1279.
Chen SL, Shi XY, Zheng HY, Wu FR, Luo C: Aberrant DNA methylation of imprinted H19 gene in human preimplantation embryos. Fertil Steril. 2010, 94: 2356-2358. 10.1016/j.fertnstert.2010.01.120. 2358 e2351,
Gomes MV, Huber J, Ferriani RA: Amaral Neto AM, Ramos ES: Abnormal methylation at the KvDMR1 imprinting control region in clinically normal children conceived by assisted reproductive technologies. Mol Hum Reprod. 2009, 15: 471-477. 10.1093/molehr/gap038.
Kanber D, Buiting K, Zeschnigk M, Ludwig M, Horsthemke B: Low frequency of imprinting defects in ICSI children born small for gestational age. Eur J Hum Genet. 2009, 17: 22-29. 10.1038/ejhg.2008.177.
Katari S, Turan N, Bibikova M, Erinle O, Chalian R, Foster M, Gaughan JP, Coutifaris C, Sapienza C: DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet. 2009, 18: 3769-3778. 10.1093/hmg/ddp319.
Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, Suzuki R, Suzuki F, Hayashi C, Utsunomiya T, et al: DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet. 2009, 17: 1582-1591. 10.1038/ejhg.2009.68.
Li L, Wang L, Le F, Liu X, Yu P, Sheng J, Huang H, Jin F: Evaluation of DNA methylation status at differentially methylated regions in IVF-conceived newborn twins. Fertil Steril. 2011, 95: 1975-1979. 10.1016/j.fertnstert.2011.01.173.
Manning M, Lissens W, Bonduelle M, Camus M, De Rijcke M, Liebaers I, Van Steirteghem A: Study of DNA-methylation patterns at chromosome 15q11-q13 in children born after ICSI reveals no imprinting defects. Mol Hum Reprod. 2000, 6: 1049-1053. 10.1093/molehr/6.11.1049.
Palermo GD, Neri QV, Takeuchi T, Squires J, Moy F, Rosenwaks Z: Genetic and epigenetic characteristics of ICSI children. Reproductive biomedicine online. 2008, 17: 820-833. 10.1016/S1472-6483(10)60411-7.
Shi X, Ni Y, Zheng H, Chen S, Zhong M, Wu F, Xia R, Luo Y: Abnormal methylation patterns at the IGF2/H19 imprinting control region in phenotypically normal babies conceived by assisted reproductive technologies. Eur J Obstet Gynecol Reprod Biol. 2011, 158: 52-55. 10.1016/j.ejogrb.2011.04.001.
Tierling S, Souren NY, Gries J, Loporto C, Groth M, Lutsik P, Neitzel H, Utz-Billing I, Gillessen-Kaesbach G, Kentenich H, et al: Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human. J Med Genet. 2010, 47: 371-376. 10.1136/jmg.2009.073189.
Turan N, Katari S, Gerson LF, Chalian R, Foster MW, Gaughan JP, Coutifaris C, Sapienza C: Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS genetics. 2010, 6: e1001033-10.1371/journal.pgen.1001033.
Wong EC, Hatakeyama C, Robinson WP, Ma S: DNA methylation at H19/IGF2 ICR1 in the placenta of pregnancies conceived by in vitro fertilization and intracytoplasmic sperm injection. Fertil Steril. 2011, 95: 2524-2526. 10.1016/j.fertnstert.2011.05.047. e2521-2523,
Zechner U, Pliushch G, Schneider E: El Hajj N, Tresch A, Shufaro Y, Seidmann L, Coerdt W, Muller AM, Haaf T: Quantitative methylation analysis of developmentally important genes in human pregnancy losses after ART and spontaneous conception. Mol Hum Reprod. 2010, 16: 704-713. 10.1093/molehr/gap107.
Zheng H, Shi X, Wang L, Wu Y, Chen S, Zhang L: Study of DNA methylation patterns of imprinted genes in children born after assisted reproductive technologies reveals no imprinting errors: A pilot study. Exp Ther Med. 2011, 2: 751-755.
Zheng HY, Shi XY, Wu FR, Wu YQ, Wang LL, Chen SL: Assisted reproductive technologies do not increase risk of abnormal methylation of PEG1/MEST in human early pregnancy loss. Fertil Steril. 2011, 96: 84-89. 10.1016/j.fertnstert.2011.04.021. e82
Oliver VF, Miles HL, Cutfield WS, Hofman PL, Ludgate JL, Morison IM: Defects in imprinting and genome-wide DNA methylation are not common in the in vitro fertilization population. Fertil Steril. 2012, 97: 147-153. 10.1016/j.fertnstert.2011.10.027. e147,
Boissonnas CC, Abdalaoui HE, Haelewyn V, Fauque P, Dupont JM, Gut I, Vaiman D, Jouannet P, Tost J, Jammes H: Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur J Hum Genet. 2010, 18: 73-80. 10.1038/ejhg.2009.117.
Bourque DK, Avila L, Penaherrera M, von Dadelszen P, Robinson WP: Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta. 2010, 31: 197-202. 10.1016/j.placenta.2009.12.003.
Lachin JM: The role of measurement reliability in clinical trials. Clin Trials. 2004, 1: 553-566. 10.1191/1740774504cn057oa.
Turner CL, Mackay DM, Callaway JL, Docherty LE, Poole RL, Bullman H, Lever M, Castle BM, Kivuva EC, Turnpenny PD, et al: Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur J Hum Genet. 2010, 18: 648-655. 10.1038/ejhg.2009.246.
Schieve LA, Ferre C, Peterson HB, Macaluso M, Reynolds MA, Wright VC: Perinatal outcome among singleton infants conceived through assisted reproductive technology in the United States. Obstet Gynecol. 2004, 103: 1144-1153. 10.1097/01.AOG.0000127037.12652.76.
Farley Ordovensky Staniec J, Webb NJ: Utilization of infertility services: how much does money matter?. Health Serv Res. 2007, 42: 971-989. 10.1111/j.1475-6773.2006.00640.x.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2431/12/33/prepub
Acknowledgements
The authors thank Trina Kuriger, Crystal Blommer, and AJ Hooten for their outstanding work processing and analyzing study samples and their assistance in specifying and clarifying technical details; and Rachel Radman for all of her efforts in study recruitment. This work was supported by the National Institutes of Health [K05 CA157439 to Dr. Ross]; and the Viking Children's Fund (Minneapolis, Minnesota).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SEP designed the study, directed its implementation, performed the study analysis, and prepared and revised the text. HHN participated in the study design, supervised the DNA methylation analysis, and critically reviewed and revised the text. JAR participated in the study design, supervised sample preparation and DNA extraction, and critically reviewed and revised the text. RHNN assisted in the study design and critically reviewed and revised the text. MAD assisted in case enrollment and critically reviewed and revised the text. LGS assisted in the study design and implementation and helped prepare and revise all sections of the text. All authors read and approved the final manuscript.
Electronic supplementary material
12887_2011_556_MOESM1_ESM.XLS
Additional file 1: Detailed summary of the literature on DNA methylation in children after ART. Two tables are included providing qualitative (hypomethylation or hypermethyation) and quantitative methylation differences between children conceived through ART and those conceived spontaneously. One table presents the data for studies examining blood or buccal cells and the other presents the same data for extra-embryonic tissues. (XLS 74 KB)
12887_2011_556_MOESM2_ESM.XLS
Additional file 2: Intraclass correlation coefficients for individual CpG sites. Description: Table of the intraclass correlation coefficients for the reliability analysis for the individual CpG sites. (XLS 28 KB)
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Puumala, S.E., Nelson, H.H., Ross, J.A. et al. Similar DNA methylation levels in specific imprinting control regions in children conceived with and without assisted reproductive technology: a cross-sectional study. BMC Pediatr 12, 33 (2012). https://doi.org/10.1186/1471-2431-12-33
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2431-12-33