
Abstract
Background
Integration of RNA-seq expression data with knowledge on chromatin accessibility, histone modifications, DNA methylation, and transcription factor binding has been instrumental for the unveiling of cell-specific local and long-range regulatory patterns, facilitating further investigation on the underlying rules of transcription regulation at an individual and allele-specific level. However, full genome transcriptome characterization has been partially limited by the complexity and increased time-requirements of available RNA-seq library construction protocols.
Findings
Use of the SX-8G IP-Star® Compact System significantly reduces the hands-on time for RNA-seq library synthesis, adenylation, and adaptor ligation providing with high quality RNA-seq libraries tailored for Illumina high-throughput next-generation sequencing. Generated data exhibits high technical reproducibility compared to data from RNA-seq libraries synthesized manually for the same samples. Obtained results are consistent regardless the researcher, day of the experiment, and experimental run.
Conclusions
Overall, the SX-8G IP-Star® Compact System proves an efficient, fast and reliable tool for the construction of next-generation RNA-seq libraries especially for trancriptome-based annotation of larger genomes.
Similar content being viewed by others


Findings
Background
Deciphering the underlying determinants of transcriptional regulation in relation to cell differentiation, functional diversification, environmental signaling, and disease development remains a central question in biology today. Integration of expression data with knowledge on chromatin accessibility, histone modifications, DNA methylation, and transcription factor binding, has been instrumental for the unveiling of cell-specific local and long-range regulatory patterns, facilitating further investigation on the underlying rules of transcription regulation at an individual and allele-specific level. Current interest by large collaborative projects, such as the ENCODE [1], the NIH Roadmap Epigenomics Mapping Consortium [2,3], and the C. elegans and D. melanogaster modENCODE [4], has been placed on generating genome-wide gene expression maps to locate gene expression changes that accompany important developmental and disease development processes. The pairing of traditional expression assays with high-throughput sequencing (RNA-seq) has allowed the generation of genome-wide gene expression data with unparalleled specificity, throughput, and sensitivity delivering a detailed representation of the transcriptome.
However, full genome transcriptional gene characterization has been partially limited by the complexity and increased time-requirements of available RNA-seq library construction protocols. Here we report the successful application of the SX-8G IP-Star® Compact System (Diagenode) for the easy, rapid, and reproducible RNA-seq library construction of five Mus musculus (mouse) samples. Use of the SX-8G IP-Star® Compact System significantly reduced the hands-on time for RNA-seq library synthesis, adenylation, and adaptor ligation providing with high quality RNA-seq libraries tailored for Illumina high-throughput next-generation sequencing. Generated data exhibited high technical reproducibility compared to data from RNA-seq libraries synthesized manually for the same samples. Obtained results are consistent regardless the researcher, day of the experiment, and experimental run. Overall, the SX-8G IP-Star® Compact System proves an efficient and reliable tool for the construction of next-generation RNA-seq libraries especially for trancriptome-based annotation of larger genomes.
Methods
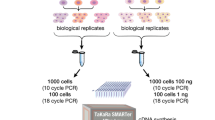
A schematic step-wise representation of the two tested protocols is presented in Figure 1. Specifically, we tested application of the SX-8G IP-Star® Compact System for the construction of RNA-seq libraries of five mouse (Mm_1-5_Auto) samples in comparison to a manual protocol routinely used in our laboratory. The two protocols were compared using the same thermocycling machines and reagents. Total RNA integrity value following isolation was measured using the Agilent Technologies 2100 Bioanalyzer and was equal to eight for all tested samples. For the manual protocol mRNA preparation, library construction, and purification were done according to the TruSeq™ RNA Sample Preparation v2 low sample (LS) protocol (Illumina). Briefly, mRNA was extracted from 0.2 μg of total RNA for each sample using 5 min incubation with 50 μl of RNA Purification Beads (TruSeq™ RNA Sample Preparation Kit v2; Illumina) at 65°C, followed by 5 min incubation at room temperature. Following washing and elution of the mRNA denaturation reaction, mRNA was fragmented using 8 min incubation with 19.5 μl of the Elute, Prime, Fragment Mix (TruSeq™ RNA Sample Preparation Kit v2) at 94°C. First Strand Synthesis was performed using thermocycling with 8 μl of First Strand Master Mix (TruSeq™ RNA Sample Preparation Kit v2) and SuperScript II Reverse Transcriptase (Invitrogen) at 25°C for 10 min, 42°C for 50 min and 70°C for 15 min. For second strand synthesis samples were incubated with 25 μl of Second Strand Master Mix (TruSeq™ RNA Sample Preparation Kit v2) at 16°C for 1 hour. Reactions were cleaned up with Agencourt AMPure XP beads (Beckman Coulter Genomics). Libraries were end-repaired, adenylated at the 3’ end, ligated with adapters and amplified according to the TruSeq™ RNA Sample Preparation v2 LS protocol. Constructed RNA-seq libraries were purified with Agencourt AMPure XP beads and quantified using the Quant-iT™ PicoGreen® ds DNA Assay Kit (Invitrogen) and the KAPA Library Quantification Kit (KAPABIOSYSTEMS) using qPCR. Library quality control was performed with the Agilent Technologies 2100 Bioanalyzer. Libraries were normalized and pooled using the TruSeq™ Cluster Kit v3 (Illumina) based on the qPCR values. Pooled samples were sequenced using the HiSeq 2500 v3 sequencer (Illumina). For the automated protocol the assay was performed as above except that the most time-consuming stage of library preparation, synthesis, and adaptor ligation was performed using the SX-8G IP-Star® Compact System. The only required actions for this purpose were to select the appropriate Diagenode Library Preparation protocol (Illumina_TruSeq_DNA_SamplePrep_v2) for the corresponding sample number and to set up the necessary reagents and consumables following the robot’s user-friendly and simple interface.

A schematic representation of the sample preparation workflow. The processes of the TruSeq™ RNA Sample Preparation v2 low sample (LS) protocol (Illumina) performed manually and adopted for automated use with the SX-8G IP-Star® Compact System are illustrated. The automated protocol minimizes the hands-on time required for the error-prone manual steps of RNA-seq library synthesis, adenylation, and adaptor ligation including all related clean up steps and allows experimental multitasking for the researcher in task.
RNA-seq data generated using the manual and automated protocols were aligned against the Mus musculus GRCm38/mm10 genome using TopHat 2.0.7 [5]. Following extraction of known transcripts, based on the most parsimonious trancriptome assembly, Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values for each sample processed with the automated (Mm_1-5_Auto) and manual protocol (Mm_1-5_Man) were generated using the open-source software package Cufflinks 2.1.1 [6,7] to estimate relative transcript abundance. Transcripts from unexpressed genes with FPKM values equal to or less than 0.01 were excluded from subsequent analysis. Heat map plots and correlation coefficient values (r2, linear regression model) based on FPKM values of each sample and corresponding technical replicate were generated using the statistical language R. Data visualization, density distribution of FPKM values and cluster analysis were performed using the CummeRbund 2.7.1 R package (http://compbio.mit.edu/cummeRbund/).
Results
Application of the SX-8G IP-Star® Compact System for the RNA-seq library construction of five mouse samples, significantly reduced the amount of hands-on time required for the most time-demanding stages of library synthesis, adenylation, and adaptor ligation including all related clean up steps. Specifically, manual library construction with the protocol routinely used in our laboratory typically takes an average of four hours of hands-on time whereas Diagenode automated library construction with the same reagents and samples required only 30 minutes. This corresponds to a 8-fold decrease in the amount of time the researcher has to be directly involved with the procedure, offering substantial flexibility for experimental multitasking.
Notably, generated data with the automated protocol exhibited high technical reproducibility compared to data from RNA-seq libraries synthesized manually for the same samples regardless operator and experimental run. Specifically, density distributions of FPKM values demonstrated high data concordance among samples and technical replicates (Figure 2). Correlation coefficient values r2 obtained using the linear regression model in R for the five mouse samples and corresponding technical replicates ranged from 0.97-0.98, confirming that the SX-8G IP-Star® Compact System can be reliably used for the efficient and accurate construction of RNA-seq libraries (Figure 3). Cluster analysis illustrated tight clustering between samples and technical replicates, further supporting high technical reproducibility between the two tested protocols (Figure 4).

Comparison of distributions of FPKM values. Density distributions of FPKM values created using the CummeRbund 2.7.1 R package, support high data concordance among samples and corresponding technical replicates. Mm_1-5_Auto and Mm_1-5_Man correspond to mouse samples processed with the automated and manual protocols respectively.

Correlation analysis of FPKM values. Heat map plots generated based on FPKM data from samples processed with the SX-8G IP-Star® Compact System (Mm_1-5_Auto) and their corresponding technical replicates (Mm_1-5_Man). Transcripts from unexpressed genes were excluded using a cut-off FPKM value equal to or less than 0.01. Correlation coefficient r2 between each sample and technical replicate was estimated using the linear regression model in R and ranged from 0.97-0.98, confirming the high technical reproducibility between the two tested protocols.

Cluster analysis of FPKM values. Analysis exhibits tight clustering of the tested samples (Mm_1-5_Auto) with the corresponding technical replicates (Mm_1-5_Man) confirming high technical reproducibility between the two protocols under study.
Conclusions
Overall, the SX-8G IP-Star® Compact System proves an efficient, reliable and accurate tool for the construction of next-generation RNA-seq libraries, especially for trancriptome-based annotation of larger genomes. We foresee that incorporation of this technology in Next-Generation Sequencing Cores or Genomics Laboratories will prove an indispensable tool for high-throughput RNA-seq library construction, significantly saving on-hands experimentation time, related costs and error-prone manual steps. Added benefits of the automated protocol include ease of operation and generation of consistent data regardless of human variability and experimental run. Adaptation of this technology should support the unveiling of the mechanisms governing differential gene expression and transcription processing genome-wide, leading to a better understanding of genetic and epigenetic regulation and inheritance in a time-efficient manner.
Abbreviations
- RNA-Seq:
-
Ribonucleic acid next-generation sequencing
- mRNA:
-
Messenger ribonucleic acid
- qPCR:
-
Quantitative polymerase chain reaction
- FPKM:
-
Fragments per kilobase of transcript per million mapped reads
References
Consortium EP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–40.
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045–8.
Chadwick LRNA-SEQ. The NIH roadmap epigenomics program data resource. Epigenomics. 2012;4(3):317–24.
Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, et al. Unlocking the secrets of the genome. Nature. 2009;459(7249):927–30.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78.
Acknowledgements
This research was supported by NY State Department of Health (C026714 to MJB). Sequencing and bioinformatics was performed at the UB Genomics & Bioinformatics Core.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MT participated in the conception of the study, its design and coordination, and wrote the manuscript. SV participated in the conception and design of the study and the execution of the RNA-seq experiments. JB and BM performed the data analysis. NN and MJB supervised all aspects of the project. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Tsompana, M., Valiyaparambil, S., Bard, J. et al. An automated method for efficient, accurate and reproducible construction of RNA-seq libraries. BMC Res Notes 8, 124 (2015). https://doi.org/10.1186/s13104-015-1089-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-015-1089-9