
Abstract
Background
Behçet’s syndrome (BS) is an immune-mediated disease characterized by recurrent oral ulcers, genital ulcers, uveitis, and skin symptoms. HLA-B51, as well as other genetic polymorphisms, has been reported to be associated with BS; however, the pathogenesis of BS and its relationship to genetic risk factors still remain unclear. To address these points, we performed immunophenotyping and transcriptome analysis of immune cells from BS patients and healthy donors.
Methods
ImmuNexUT is a comprehensive database consisting of RNA sequencing data and eQTL database of immune cell subsets from patients with immune-mediated diseases and healthy donors, and flow cytometry data and transcriptome data from 23 BS patients and 28 healthy donors from the ImmuNexUT study were utilized for this study. Differential gene expression analysis and weighted gene co-expression network analysis (WGCNA) were performed to identify genes associated with BS and clinical features of BS. eQTL database was used to assess the relationship between genetic risk factors of BS with those genes.
Results
The frequency of Th17 cells was increased in BS patients, and transcriptome analysis of Th17 cells suggested the activation of the NFκB pathway in Th17 cells of BS patients. Next, WGCNA was used to group genes into modules with similar expression patterns in each subset. Modules of antigen-presenting cells were associated with BS, and pathway analysis suggested the activation of antigen-presenting cells of BS patients. Further examination of genes in BS-associated modules indicated that the expression of YBX3, a member of a plasmacytoid dendritic cell (pDC) gene module associated with BS, is influenced by a BS risk polymorphism, rs2617170, in pDCs, suggesting that YBX3 may be a key molecule connecting genetic risk factors of BS with disease pathogenesis. Furthermore, pathway analysis of modules associated with HLA-B51 indicated that the association of IL-17-associated pathways in memory CD8+ T cells with HLA-B51; therefore, IL-17-producing CD8+ T cells, Tc17 cells, may play a critical role in BS.
Conclusions
Various cells including CD4+ T cells, CD8+ T cells, and antigen-presenting cells are important in the pathogenesis of BS. Tc17 cells and YBX3 may be potential therapeutic targets in BS.
Similar content being viewed by others


Background
Behçet’s syndrome (BS) is an autoimmune disease characterized by recurrent oral aphthous ulcers, genital ulcers, uveitis, and skin symptoms, such as pustular folliculitis and erythema nodosum. Some patients may also develop arthritis, epididymitis, vascular lesions, central nervous system lesions, and gastrointestinal lesions similar to inflammatory bowel disease. The clinical course is characterized by repeated inflammatory attacks with periods of exacerbation and remission. Mucocutaneous manifestations are often self-limiting, but uveitis, intestinal, vascular, or central nervous system diseases are often intractable and severe, impairing the patient’s quality of life. Various immunomodulatory or immunosuppressive drugs are used for the treatment of BS. Mild symptoms may be managed with topical treatment or colchicine, but more severe symptoms may require systemic steroids, immunosuppressants such as cyclosporine and methotrexate, and tumor necrosis factor (TNF)-α inhibitors [1]. However, the current treatment of BS is far from perfect, and many patients have impaired quality of life due to symptoms of BS or due to side effects of treatment [2].
Various immune cells have been implicated in the pathogenesis of BS. BS lesions such as pustular folliculitis are characterized by infiltration of neutrophils [3], suggesting a role for the innate immune system in the pathogenesis of BS. T cells are also seen in BS lesions, and an expansion of Th1 and Th17 cells has been reported in BS patients [4], suggesting a role for the adaptive immune system in the pathogenesis of BS.
BS is considered a multifactorial disease, and both genetic factors and environmental factors are involved in its pathogenesis. Among genetic factors, HLA-B51 has been strongly associated with BS in studies from various ethnic groups. In addition, BS disease-susceptible single nucleotide polymorphisms (SNPs) have been identified in or near IL23R, IL12RB2, IL10, STAT4, IL12A, CCR1, CCR3, KLRC4, ERAP1, IL1B, IRF8, and IFNGR1 [5,6,7,8,9,10]; however, the exact mechanism by which those SNPs contribute to disease pathogenesis and the cell types where they exert their function have not been elucidated.
In recent years, transcriptome analyses of samples from various connective tissue disease patients have been performed, providing insight into the pathogenesis of those diseases, but reports regarding BS are limited. A microarray analysis comparing the gene expression of the peripheral blood from BS patients and healthy individuals revealed the upregulation of Th17 genes and interferon-related genes in BS [11]. Microarray analysis of CD4+ T cells and CD14+ monocytes from BS patients has also been reported, revealing the activation of the JAK/STAT pathway [12]. An RNA sequencing (RNA-seq) analysis of CD8+ T cells from BS patients has suggested an alteration of cAMP-mediated signaling in CD8+ T cells of BS patients [13]. However, analysis of the whole blood or only certain types of immune cells makes it difficult to identify cell types that are central to disease pathology and to analyze the interaction among the different cell types. To overcome those obstacles and to elucidate the role of each immune cell subset in the pathology of BS, we analyzed the data in ImmuNexUT [14], a comprehensive RNA-seq data of sorted immune cell subsets and eQTL database created by our group, with a focus on BS.
Methods
Patient and control group
Peripheral blood samples were obtained from 23 BS patients and 28 healthy donors. BS patients fulfilled the International Study Group criteria [15]. To detect the differences that are inherent in disease pathology, not the result of excessive inflammation, BS patients in remission or with low disease activity (the Behçet’s Disease Current Activity Form scores less than 3) were recruited [16]. As transcriptomic studies of the peripheral blood from patients with other autoimmune diseases have indicated that biologics induce profound changes in the transcriptome [17, 18], patients receiving biologics were excluded. The control group consisted of 28 healthy people, age- and gender-matched to the patient group. Written consent was obtained from the participants according to the Declaration of Helsinki principle, and the study was approved by the ethics committee of the University of Tokyo (G10095 [4]).
Isolation and flow cytometric analysis (FCM) of the immune cells
Samples were collected as described previously [14]. Briefly, PBMCs were isolated from the peripheral blood by density gradient centrifugation with Ficoll-Paque Plus (GE Healthcare), and erythrocytes were lysed using ammonium chloride potassium buffer, as ammonium chloride potassium buffer has been reported to lyse erythrocytes efficiently without affecting the proportion of immune cells [19]. After blocking the non-specific binding of antibodies with Fc receptor binding inhibitor (eBioscience), the cells were stained with antibodies shown in Additional file 1The cells were analyzed and sorted using 8-color MoFlo XDP (Beckman Coulter) following the cell surface antigen-based definitions shown in Additional file 2. The results were analyzed using FlowJo version 10.5.0 (TreeStar Software). Neutrophils were isolated from EDTA anti-coagulated blood using MACSxpress Neutrophil Isolation Kit human and MACSxpress Erythrocyte Depletion Kit (Miltenyi Biotec).
RNA-seq
PBMC subsets were collected into RLT (QIAGEN), and neutrophils were collected into TRIzol LS Reagent (Invitrogen). Total RNA was extracted using the RNeasy micro kit (QIAGEN). The SMART-seq v4 Ultra Low Input RNA Kit for Sequencing (Clontech) was used for cDNA library preparation, and sequencing was performed with HiSeq 2500 (Illumina) with a pair-end read of 100 base pairs. Sequencing data in BCL format was converted to FASTQ format using bcl2fastq2 v2.17.
Quality control of RNA-seq data, mapping, and counting of reads
Adapter sequences were first removed from FASTQ format data using Cutadapt version 1.14. Next, using FASTX-Toolkit version 0.0.14, reads with Phred quality score below 20 were excluded. Mapping was performed using STAR version 2.5.3a with UCSC human genome 38 as the reference sequence [20]. Reads were counted using HTSeq version 0.9.1 [21]. Samples with uniquely mapped rates less than 80% or with uniquely mapped counts less than 5.00 × 106 reads were removed from the analysis. The correlation coefficient between every pair of two samples from the same cell subset was calculated, and if the average of those correlation coefficients (Di) was less than 0.9, the sample was also removed from the analysis.
Identification of differentially expressed genes (DEG)
Genes with a read count of less than 10 in 90% or more of the samples of the same subset were excluded, and normalization was performed using R package TCC version 1.22.0 [22]. After removing the batch effect using R package RUVseq version 3.24.0 [23], comparisons between the two groups were performed, using quasi-likelihood F-test in edge R version 1.16.0 [24]. Genes with a false discovery rate (FDR) less than 0.05 were considered significant.
Weighted gene co-expression network analysis (WGCNA)
For weighted gene co-expression network analysis (WGCNA), genes with read counts less than 10 in 90% or more of the samples of the same subset were excluded. Count data was normalized with iterative DEGES/edgeR and converted to log 2 (CPM + 1) using R package TCC version 1.22.0. WGCNA was performed using WGCNA package version 1.64.1 [25] and iterative WGCNA version 1.1.6. VisNetwork was used to visualize the results (https://CRAN.R-project.org/package=visNetwork).
eQTL analysis
The eQTL analysis was performed as described elsewhere [14]. After filtering out genes expressed at low levels in each cell subset (< 5 count in more than 80% samples or < 0.5 CPM in more than 80% samples), the expression data were normalized between samples with TMM, converted to CPM, and then normalized across samples using an inverse normal transform. A probabilistic estimation of expression residuals (PEER) method [26] was used to find hidden covariates. For each cell subset, a QTLtools permutation pass with 10,000 permutations was used to obtain gene-level nominal p-value thresholds corresponding to FDR < 0.05. A forward-backward stepwise regression eQTL analysis was subsequently preformed with a QTLtools conditional pass.
Pathway analysis
ClusterProfiler was used to assess the enrichment of genes in modules with correlation with the diagnosis of BS to the KEGG pathways [27]. Pathways related to biological processes are depicted in the dotplot, and those related to diseases were omitted. Pathway analysis of modules with correlation to HLA-B51 was performed using Ingenuity Pathway Analysis 2.3 (QIAGEN Inc.).
Tc17 score
Tc17 score was calculated using GSVA [28]. Genes included in the gene set are the following genes reported to be upregulated in Tc17 cells [29], especially pathogenic Tc17 cells [30]: RORC, CCR6, IL23R, CD58, MAF, BLK, IL1R1, SOX13, SOCS3, CCL20, TCF7, RORA, NFKB1, and GZMB.
Statistical analysis
Categorical data were analyzed using Fisher’s exact test. Quantitative variables were compared using the Mann-Whitney U test. Correlation between two variables was evaluated by Pearson’s correlation coefficient or Spearman’s correlation coefficient. In multiple tests, Bonferroni’s method was used to correct p-values, and p-value < 0.05 was judged to be statistically significant. Statistical significance was separately described if other criteria were adopted. Graphs were generated using R version 3.5.1, R version 4.0.3, or GraphPad Prism version 9.0.0.
Results
Clinical characteristics of the participants
The clinical characteristics of participants are shown in Table 1. About half of the BS patients (47%) were HLA-B51-positive. At the time of participation in the study, 69% of the patients were taking colchicine, and 39% were taking steroids but the dosage was generally low. About half of the patients (56%) had at least one active symptom, most commonly skin lesions, arthralgia, or oral aphthous ulcers (Table 1). During the entire disease course, more than half of the patients had oral aphthous ulcerations, genital ulcers, uveitis, cutaneous lesions, and arthritis/arthralgia, but patients with intestinal lesions, vascular lesions, and neurological involvement were limited (Additional file 3).
Th17 cells are increased in the peripheral blood of BS patients
First, we compared the frequency of peripheral blood immune cells between HC and BS. The frequency of Th17 cells, defined as CD3+CD4+CD25−CD45RA−CXCR5−CCR6+CXCR3− cells, was significantly increased in BS compared to HC, while no significant differences were found in CD8+ T cells, B cells, monocytes, or dendritic cells (Table 2, Additional file 4). We then compared the gene expression profile of immune cells between BS and HC (Additional files 5 and 6). DEG in many subsets included many immunologically important genes, including CXCL8, RGS1, STAT6, and cytokine receptors (Additional file 7). For Th17 cells, we identified 58 DEG, and those genes made a clear distinction between BS and HC (Additional file 8). Pathway analysis of the DEG suggested activation of the NFκB pathway in Th17 cells in BS (Additional file 9).
Gene expression profiles of various immune cells show correlation with clinical features of BS
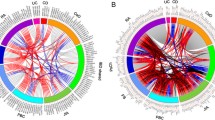
Next, we sought to investigate the association of gene expression profiles in various immune cell subsets with clinical parameters. We first performed WGCNA to identify gene modules consisting of genes with similar co-expression patterns. We then assessed the correlation of those modules with clinical parameters. A total of 247 modules were identified in 20 subsets (Additional files 10 and 11). Modules from various immune cells, including CD4+ T cells, CD8+ T cells, B cells, NK cells, monocytes, dendritic cells (DCs), and neutrophils, showed significant correlation with either the diagnosis of BS or clinical features of BS patients, suggesting the contribution of various immune cells to the pathogenesis of BS (Fig. 1), and some of those modules were further examined.

Gene expression profiles of various immune cells show a correlation with clinical features of BS. Gene modules consisting of genes with similar co-expression patterns in each cell subset were identified using WGCNA. Each circle represents a module with the colors indicating the cell subset of the module and the numbers indicating the module number. Modules with significant correlation with clinical parameters are visualized. Blue lines represent the negative correlation, and red lines represent the positive correlation with line widths indicating the absolute value of correlation coefficients. The squares indicated clinical parameters. Int/Vasc/Neuro, BS patients with intestinal, vascular, and neurological involvement compared to BS patients without those organs involved. Duration: disease duration
Pathways of inflammatory chemokines and cytokines are activated in antigen-presenting cells of BS patients
To further examine the relationship between gene modules and clinical parameters, we first focused on modules that were upregulated in BS patients compared to HC. Modules in antigen-presenting cells such as myeloid DC, plasmacytoid DC (pDC), CD16+ monocyte, and B cells (unswitched memory B cells, double-negative B cells, naïve B cells, switched memory B cells) showed a significant correlation with the diagnosis of BS, and eigengenes of these modules were higher in BS patients compared to healthy controls (Fig. 2A). Pathway enrichment analysis of those modules suggested that inflammatory cytokines, MAPK pathways, and NFkB pathways were upregulated in these subsets (Fig. 2B). These results suggest the activation of pathways related to chemokines and inflammatory cytokines and pathways mediated by MAPK and NFκB in antigen presenting cells of BS patients.

Pathways of inflammatory chemokines and cytokines are activated in antigen-presenting cells of BS patients. A Eigengenes of modules with a positive correlation of BS in healthy controls and BS patients. B Pathway analysis of modules with positive correlation with the diagnosis of BS
Genetic risk factors may account for the differences in the expression of BS-associated modules
To elucidate the relationship between genetic risk factors and the changes in the transcriptome that we observed in BS patients, we examined the eQTL catalog in ImmuNexUT [14]. The IL10 locus has been associated with BS, and it has been reported that the BS risk allele at rs1518111 decreases the production of IL10 in monocytes and macrophages [5, 31]. Our eQTL analysis was consistent with these results (Additional file 12). Furthermore, rs4683184, a BS risk SNP located in the CCR1-CCR3 locus, had an eQTL effect on multiple chemokine receptors (Additional file 13).
In addition, rs2617170, which has been reported to be associated with BS [6, 10], is a missense SNP in KLRC4, and KLRC4 has been reported as the responsible gene at this locus (Fig. 3A). However, this SNP has pleotropic effects and influences the expression of various genes, including KLRC4, KLRC1, KLRK1, and YBX3 (Fig. 3B, Additional file 14). YBX3, whose expression was increased in individuals with the risk allele, is a member of the pDC_15 module (Additional file 15) associated with the diagnosis of BS. Thus, the genetic risk for BS increases the expression of YBX3; therefore, in addition to the changes in the KLRC4 protein induced by rs2617170, rs2617160 may contribute to disease pathogenesis by altering the expression pattern of YBX3.

BS risk SNP rs2617170 modulates the expression of YBX3. A Relationship of rs2617170 with nearby genes. B eQTL effect of rs2617170 on YBX3. Residuals after normalization are plotted by genotype
A Memory CD8+ T cell module with IL-17-associated genes shows correlation with HLA-B51 positivity
HLA-B51 positivity is a strong risk factor for BS, but the role of HLA-B51 in the pathogenesis of BS has not been elucidated. To investigate this point, we focused on modules that showed a correlation with HLA-B51 positivity in BS. A module from memory CD8+ T cells “MCD8_08” showed the greatest correlation with HLA-B51 positivity in BS patients (Fig. 4A). This module included genes associated with IL-17 producing CD8+ T cells, Tc17 cells, for example, RORC, IL23R, and CCR6 [32] (Additional file 14), and Tc17-associated gene expression score (Tc17 score) was higher in HLA-B51-positive patients compared to HLA-B51-negative patients (Fig. 4B). Pathway analysis for the module “MCD8_08” suggested activation of NF-κB signaling, Th17 activation pathway, Th1 pathway, and STAT3 pathway (Fig. 4C).

A memory CD8+ T cell module with IL-17-associated genes shows a correlation with HLA-B51 positivity. A An enlarged view of modules showing correlation with HLA-B51 positivity in BS patients. B Tc17 score in HLA-B51-positive patients (n = 7) compared to HLA-B51-negative patients (n = 17). C Pathway analysis of “MCD8_08,” the module with the strongest positive correlation with HLA-B51 positivity. Pathways with absolute z scores ≧ 2 are shown. D Pathway analysis of “NCD8_06,” the module with the strongest negative correlation with HLA-B51 positivity. Pathways with absolute z scores ≧ 5 are shown
In contrast to modules from memory CD8+ T cells showing a positive correlation with HLA-B51 positivity in BS patients, modules of naïve CD8+ T cells showed a negative correlation with HLA-B51 positivity in BS. Pathway analysis of the module “NCD8_06” showed possible activation of pathways of integrins and chemokines in naïve CD8+ T cells in HLA-B51-negative BS patients (Fig. 4D). These results suggest that CD8+ T cells may play a different role in HLA-B51-positive BS and HLA-B51-negative BS.
Discussion
We herein reported our FCM and transcriptome analysis of immune cells of BS patients. FCM analysis revealed an increase in Th17 cells in the peripheral blood of BS patients. Various reports have implicated a role for IL-17 and Th17 cells in BS [33,34,35], and our findings are consistent with those reports. Our analysis of the transcriptome of Th17 cells from BS patients suggested the activation of the NFκB pathway in Th17 cells of BS patients. NFκB transcription factor c-Rel has been reported to be essential for the differentiation of Th17 cells [36], and the activation of the NFκB pathway seen in Th17 cells of BS patients may contribute to the increased number of Th17 cells in BS patients. An increase in Th17 cells has also been reported in other autoimmune diseases, including early systemic sclerosis [37], suggesting that the role of Th17 cells in autoimmunity may be a common theme in many autoimmune diseases, not just BS.
In addition, transcriptome analysis of peripheral blood immune cell subsets using WGCNA indicated that an increase in the expression of genes related to IL-17 production in CD8+ memory cells of HLA-B51-positive BS patients, and this may reflect an increase in the number or function of Tc17 cells, IL-17-producing CD8+ cells. Previous reports [33,34,35] regarding the role of IL-17 in BS have mainly focused on CD4+ IL-17-producing cells, Th17 cells; however, it has been reported that a significant proportion of IL-17-producing cells in BS patients are CD4-negative [33], and our results suggest that these cells may be Tc17 cells. Despite being well known as a strong genetic risk factor for BS, the role of HLA-B51 in the pathogenesis of BS has remained elusive. Some have suggested that aberrant activation of CD8+ T cells due to the HLA-B51 contributes to the pathogenesis of BS [38], while others emphasize the role of interactions with other immune cells, such as NK cells, and HLA-B51 [39]. Our data suggests the importance of CD8+ T cells in the disease pathology of HLA-B51 patients. That is, in HLA-B51-positive patients, genetic factors may promote the differentiation of Tc17 cells, and these cells may serve as a potential therapeutic target in BS.
Another important finding from the transcriptomic analysis in this study is the association of gene expression profiles in antigen-presenting cells, such as myeloid DC, pDC, CD16-positive monocyte, and B cells, with BS. Pathway analysis of those modules indicated that pathways related to inflammatory cytokines, as well as the MAPK pathway and NFκB pathway, are upregulated in those cell subsets in BS. It has been reported that activated macrophages play an important role in the differentiation of human Th17 cells [40], and the activation of DCs may promote the differentiation of Th17 cells and contribute to the increase in Th17 cells in BS patients.
Analysis of transcriptomic data together with an eQTL database, ImmuNexUT, allowed us to identify possible links between the gene expression and genetic risk factors for BS. For example, rs2617170 is a missense SNP in KLRC4, and in addition to altering the amino acid sequence, the eQTL catalog in ImmuNexUT indicated that it also acts as an eQTL for KLRC4, consistent with a previous report by Yang et al. [10]. Furthermore, we observed eQTL effects on multiple genes in multiple cell types, including the expression of YBX3 in pDCs. The pleotropic effect of rs2617170 may be due to it acting as an enhancer for these genes, in addition to being a missense SNP. As KLRC4 influences the function of NK cells, the interaction between NK cells and DCs may also account for the eQTL effect on YBX3 in DCs [41]. YBX3 is included in a pDC module associated with BS, and genetic risk factors may cause changes in the expression pattern of YBX3 in BS patients. YBX3, encoded by YBX3, is an RNA-binding protein that has been reported to bind to various genes, regulating their expression, and one of the genes reported to be regulated by YBX3 is JAK1 [42], which has been implicated in the pathogenesis of BS from both genetic studies [43] and from clinical studies [12]. In addition, YBX3 controls amino acid levels by influencing mRNA abundance of SLC7A5 and SLC3A2 [42], and as immunometabolism has been implicated in the function of various immune cells including DCs [44], YBX3 may contribute to the pathogenesis of BS by altering the function of DCs and is a potential therapeutic target.
There are several limitations to this study. First, the number of patients was small, and 92% of the patients were receiving therapy for BS, making it difficult to discriminate the effect of treatment and the effect of the disease itself in certain instances. In addition, patients with high disease activity were excluded, and different changes may be seen in patients with highly active disease.
Conclusions
FCM analysis and transcriptome analysis of BS patients revealed an increase in Th17 cells in the peripheral blood and activation of the NFκB pathway in those cells. iWGCNA analysis revealed the association of BS with genes related to antigen-presenting cell activation, and YBX3, whose expression in pDC is influenced by a BS risk polymorphism, rs2617170, was identified as a potential key molecule, connecting genetic risk factors of BS with activation of antigen-presenting cells in BS. Furthermore, HLA-B51, an important genetic risk factor of BS, was associated with IL-17-associated genes in memory CD8+ T cells, suggesting a role for Tc17 cells in the pathogenesis of BS. In conclusion, our analysis suggested that Tc17 cells and YBX3 as potential therapeutic targets in BS.
Availability of data and materials
The RNA-seq data analyzed during the current study and the eQTL catalog are available at the National Bioscience Database Center (NBDC) (E-GEAD-397, E-GEAD-398, and E-GEAD-420) (https://biosciencedbc.jp/en/).
Abbreviations
- BS:
-
Behçet’s syndrome
- WGCNA:
-
Weighted gene co-expression network analysis
- pDC:
-
Plasmacytoid dendritic cell
- TNF-α:
-
Tumor necrosis factor-α
- FCM:
-
Flow cytometric analysis
- RNA-seq:
-
RNA sequencing
- DEG:
-
Differentially expressed genes
- PEER:
-
Probabilistic estimation of expression residuals
- HC:
-
Healthy controls
References
Hatemi G, Christensen R, Bang D, Bodaghi B, Celik A, Fortune F, et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis. 2018;77(6):808–18.
Bernabé E, Marcenes W, Mather J, Phillips C, Fortune F. Impact of Behçet’s syndrome on health-related quality of life: influence of the type and number of symptoms. Rheumatology (Oxford). 2010;49(11):2165–71.
Emmi G, Becatti M, Bettiol A, Hatemi G, Prisco D, Fiorillo C. Behçet’s syndrome as a model of thrombo-inflammation: the role of neutrophils. Front Immunol. 2019;10:1085.
Greco A, De Virgilio A, Ralli M, Ciofalo A, Mancini P, Attanasio G, et al. Behçet’s disease: new insights into pathophysiology, clinical features and treatment options. Autoimmun Rev. 2018;17(6):567–75.
Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat Genet. 2010;42(8):698–702.
Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013;45(2):202–7.
Hou S, Yang Z, Du L, Jiang Z, Shu Q, Chen Y, et al. Identification of a susceptibility locus in STAT4 for Behçet’s disease in Han Chinese in a genome-wide association study. Arthritis Rheum. 2012;64(12):4104–13.
Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behçet’s disease susceptibility. Nat Genet. 2017;49(3):438–43.
Su G, Zhong Z, Zhou Q, Du L, Ye Z, Li F, et al. Identification of novel risk loci for Behçet’s disease-related uveitis in a Chinese population in a genome-wide association study. Arthritis Rheum. 2022;74(4):671–81.
Yang Y, Tan H, Deng B, Yu H, Su G, Hu J, et al. Genetic polymorphisms of C-type lectin receptors in Behcet’s disease in a Chinese Han population. Sci Rep. 2017;7(1):5348.
Puccetti A, Fiore P, Pelosi A, Tinazzi E, Patuzzo G, Argentino G, et al. Gene expression profiling in Behcet’s disease indicates an autoimmune component in the pathogenesis of the disease and opens new avenues for targeted therapy. J Immunol Res. 2018;2018:4246965.
Tulunay A, Dozmorov M, Ture-Ozdemir F, Yilmaz V, Eksioglu-Demiralp E, Alibaz-Oner F, et al. Activation of the JAK/STAT pathway in Behcet’s disease. Genes Immun. 2015;16(2):170–5.
Kim SM, Park MJ, Park S, Cheng JY, Lee ES. Differential expression of novel genes and signalling pathways of senescent CD8+ T cell subsets in Behçet’s disease. Clin Exp Rheumatol. 2020;38 Suppl 127(5):17–25.
Ota M, Nagafuchi Y, Hatano H, Ishigaki K, Terao C, Takeshima Y, et al. Dynamic landscape of immune cell specific gene regulation in immunemediated diseases. Cell. 2021.184(11):3006-21.
International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078–80.
Lawton G, Bhakta BB, Chamberlain MA, Tennant A. The Behcet’s disease activity index. Rheumatology (Oxford, England). 2004;43(1):73-8.
Oliver J, Nair N, Orozco G, Smith S, Hyrich KL, Morgan A, et al. Transcriptome-wide study of TNF-inhibitor therapy in rheumatoid arthritis reveals early signature of successful treatment. Arthritis Res Ther. 2021;23(1):80.
Sumitomo S, Nagafuchi Y, Tsuchida Y, Tsuchiya H, Ota M, Ishigaki K, et al. Transcriptome analysis of peripheral blood from patients with rheumatoid arthritis: a systematic review. Inflamm Regen. 2018;38:21.
Plank K, Dorn C, Krause SW. The effect of erythrocyte lysing reagents on enumeration of leukocyte subpopulations compared with a no-lyse-no-wash protocol. Int J Lab Hematol. 2021;43(5):939–47.
Dobin A, Davis C, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England). 2013;29(1):15–21.
Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England). 2015;31(2):166–9.
Sun J, Nishiyama T, Shimizu K, Kadota K. TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics. 2013;14:219.
Risso D, Ngai J, Speed T, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol. 2014;32(9):896–902.
Robinson M, McCarthy D, Smyth G. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England). 2010;26(1):139–40.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17.
Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7(3):500–7.
Yu G, Wang L, Han Y, He Q. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–7.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7.
Mielke LA, Liao Y, Clemens EB, Firth MA, Duckworth B, Huang Q, et al. TCF-1 limits the formation of Tc17 cells via repression of the MAF-RORγt axis. J Exp Med. 2019;216(7):1682–99.
Liu J, Chang HW, Huang ZM, Nakamura M, Sekhon S, Ahn R, et al. Single-cell RNA sequencing of psoriatic skin identifies pathogenic Tc17 cell subsets and reveals distinctions between CD8 + T cells in autoimmunity and cancer. J Allergy Clin Immunol. 2020;147(6):31702–4.
Nakano H, Kirino Y, Takeno M, Higashitani K, Nagai H, Yoshimi R, et al. GWAS-identified CCR1 and IL10 loci contribute to M1 macrophage-predominant inflammation in Behçet’s disease. Arthritis Res Ther. 2018;20(1):124.
Lückel C, Picard FSR, Huber M. Tc17 biology and function: novel concepts. Eur J Immunol. 2020;50(9):1257–67.
Ekinci NS, Alpsoy E, Karakas AA, Yilmaz SB, Yegin O. IL-17A has an important role in the acute attacks of Behçet’s disease. J Invest Dermatol. 2010;130(8):2136–8.
Jadideslam G, Kahroba H, Ansarin K, Sakhinia E, Abhar A, Alipour S, et al. Interleukin-17 mRNA expression and serum levels in Behçet’s disease. Cytokine. 2020;127:154994.
Hamzaoui K, Borhani Haghighi A, Ghorbel IB, Houman H. RORC and Foxp3 axis in cerebrospinal fluid of patients with neuro-Behçet’s disease. J Neuroimmunol. 2011;233(1-2):249–53.
Chen G, Hardy K, Pagler E, Ma L, Lee S, Gerondakis S, et al. The NF-κB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J Immunol. 2011;187(9):4483–91.
Kobayashi S, Nagafuchi Y, Okubo M, Sugimori Y, Hatano H, Yamada S, et al. Dysregulation of the gene signature of effector regulatory T cells in the early phase of systemic sclerosis. Rheumatology (Oxford). 2022. https://doi.org/10.1093/rheumatology/keac031.
McGonagle D, Aydin SZ, Gül A, Mahr A, Direskeneli H. ‘MHC-I-opathy’-unified concept for spondyloarthritis and Behçet disease. Nat Rev Rheumatol. 2015;11(12):731–40.
Giza M, Koftori D, Chen L, Bowness P. Is Behçet’s disease a ’class 1-opathy’? The role of HLA-B*51 in the pathogenesis of Behçet’s disease. Clin Exp Immunol. 2018;191(1):11–8.
Arnold CE, Gordon P, Barker RN, Wilson HM. The activation status of human macrophages presenting antigen determines the efficiency of Th17 responses. Immunobiology. 2015;220(1):10–9.
Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol. 2002;2(12):957–64.
Cooke A, Schwarzl T, Huppertz I, Kramer G, Mantas P, Alleaume AM, et al. The RNA-binding protein YBX3 controls amino acid levels by regulating SLC mRNA abundance. Cell Rep. 2019;27(11):3097–106.
Hou S, Qi J, Zhang Q, Liao D, Li Q, Hu K, et al. Genetic variants in the JAK1 gene confer higher risk of Behcet’s disease with ocular involvement in Han Chinese. Hum Genet. 2013;132(9):1049–58.
Al-Khami AA, Rodriguez PC, Ochoa AC. Energy metabolic pathways control the fate and function of myeloid immune cells. J Leukoc Biol. 2017;102(2):369–80.
Acknowledgements
We would like to express our gratitude to all the study participants and the members of the Department of Allergy and Rheumatology for their cooperation in this study. The supercomputing resource, SHIROKANE, was provided by the Human Genome Center, The University of Tokyo.
Funding
This study was supported by Chugai Pharmaceutical Co., Ltd., Tokyo, Japan, and the Ministry of Health, Labour and Welfare, Ministry of Education, Culture, Sports, Science and Technology KAKENHI Grant-in-Aid for Scientific Research (C) (20K08771) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Contributions
K.F., K.Y., and T.O. conceived and designed the study. M. Okubo, H.Y., H.S., S.K., Y.S., J.M., H. Shirai., and Y.I. collected the samples and the patients’ clinical data. M. Okubo, S.S., Y. Tsuchida, Y.N., Y. Takeshima, H.H., and M. Ota performed the analysis. M. Okubo and Y. Tsuchida wrote the original draft. S.S., H. Shoda, M. Ota, and K.F. revised the draft. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of the University of Tokyo (G10095), and written consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
T.O., M.O., Y.N., and Y. Takeshima belong to the Social Cooperation Program, Department of Functional Genomics and Immunological Diseases, supported by Chugai Pharmaceutical. K.F. receives consulting honoraria and research support from Chugai Pharmaceutical.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Antibodies used for flow cytometry.
Additional file 2.
Definition of PBMC subsets.
Additional file 3.
Clinical symptoms of the BS patients during the entire disease course.
Additional file 4.
Th17 cells are increased in the peripheral blood of BS patients.
Additional file 5.
Genes upregulated in BS patients in each cell subset.
Additional file 6.
Genes downregulated in BS patients in each cell subset.
Additional file 7.
MA plot of each cell subset.
Additional file 8.
Heatmap of DEG in Th17 cells.
Additional file 9.
Pathway Analysis of DEG between BS Patients.
Additional file 10.
Top 20 members of modules with significant correlation with clinical parameters.
Additional file 11.
Modules and their relationship to clinical parameters.
Additional file 12
eQTL effect of rs1518111 on IL10.
Additional file 13
eQTL effect of rs4683184 on CCR1, CCR2, CCR3, and CCR5.
Additional file 14
eQTL effect of rs2617170 on KLRK1, KLRC4, and KLRC1.
Additional file 15.
Members of “pDC_15” associated with the diagnosis of BS.
Additional file 16.
Members of “MCD8_08”, the module with strongest positive correlation with HLA-B51 positivity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Okubo, M., Sumitomo, S., Tsuchida, Y. et al. Transcriptome analysis of immune cells from Behçet’s syndrome patients: the importance of IL-17-producing cells and antigen-presenting cells in the pathogenesis of Behçet’s syndrome. Arthritis Res Ther 24, 186 (2022). https://doi.org/10.1186/s13075-022-02867-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-022-02867-x