
Abstract
Introduction
Antibodies against mutated citrullinated vimentin (AMCV) represent a useful diagnostic marker with correlation to disease activity in patients with rheumatoid arthritis (RA). Since seropositivity for citrullinated autoantibodies was predictive for response to B-cell depleting therapy (BCDT) with rituximab (RTX), we investigated whether differences in antibody fine reactivity and immunoglobulin (Ig) isotype kinetics among AMCV-positive patients could provide additional information about outcome.
Methods
A total of 50 AMCV IgG-positive RA patients (RTX responders (RRs) n = 37 and non-responders (NRRs) n = 13) were analyzed for reactivity against MCV epitopes and co-existent AMCV isotypes IgM and IgA. Antibody titers were determined by enzyme-linked immunosorbent assay at baseline and 24 weeks after the first cycle of RTX, and compared to kinetics of rheumatoid factor (RF) and antibodies against cyclic citrullinated peptide (ACCP).
Results
Recognized MCV epitopes by AMCV IgG of RRs and NRRs showed similar baseline patterns, with reducing reactivity in RRs and unchanged or even expanding reactivity in NRRs upon RTX treatment. At baseline, RRs were more frequently negative for AMCV subtypes, especially for IgA (68 %), compared to NRRs (31 %). Being AMCV IgA-negative at baseline indicated a good treatment response to RTX (negative predictive value = 0.86). Co-existence of AMCV IgA and IgG with stable titers upon treatment were associated with poorer responses to RTX. Furthermore, reductions of AMCV IgA levels upon RTX correlated with the improvement of 28-joint Disease Activity Score (DAS28). In comparison, subtypes of RF and ACCP were not of additional value for prediction of RTX response.
Conclusions
Restrictive IgG seropositivity against MCV with treatment-associated decline in fine reactivity and titers was predictive for response to RTX. Double-positivity for AMCV IgG and IgA was associated with failure to respond to BCDT, suggesting a pathogenetic and less sensitive IgA-producing B-cell subset in NRRs.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is one of the most common systemic autoimmune diseases worldwide, characterized by chronic and erosive arthritis, as well as by an increased mortality, mainly due to infections, cardiovascular events and malignant lymphoma [1]. Early diagnosis and treatment with synthetic and biological disease-modifying anti-rheumatic drugs (DMARDs) is crucial for remission of RA [2]. Since its authorization in 2006, biological treatment with rituximab (RTX) was approved in cases of inadequate response to tumor necrosis factor-alpha inhibitors (TNFi) [3]. Due to its beneficial safety profile and cost-efficacy, RTX has been currently recommended as the first-line biologic treatment in many countries worldwide [4]. Nevertheless, up to one third of RA patients still fail to respond to biologics, including RTX, leading to an individual as well as medical and economic burden [5]. Therefore, to facilitate a more personalized medicine, predictive biomarkers for response are urgently needed.
Response to a B-cell targeted therapy with RTX is usually evaluated by clinical and laboratory signs (cellular and humoral parameters) [6–8]. Due to the mode of action of RTX, follow-up investigations on cellular subsets and humoral factors were of special interest reflecting clinical response to RTX [9]. RA-associated autoantibodies, like rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPAs), were shown to be of predictive value for response to RTX [6, 10, 11]. In this context, RF showed treatment-related reductions, whereas antibodies against cyclic citrullinated peptide (ACCP) remained rather stable over the course of treatment [12, 13]. Of note, ACPA-seropositivity includes heterogeneous fine specificities against diverse citrullinated proteins [14]. Antibodies against MCV (AMCV) were shown to be highly sensitive and to correlate with disease activity of RA, probably due to the synovial appearance of the antigen during inflammation [15–18]. In particular, a concomitant presence of immunoglobulin (Ig) A with IgG AMCV was associated with a severe disease course, suggesting predictive properties of AMCV isotypes [19]. AMCV positivity was also postulated to predict (moderate) RTX response, but AMCV kinetics, especially of isotypes under B-cell depletion therapy (BCDT), have not been studied in greater detail so far [6]. Although seropositivity of autoantibodies seems to be a positive predictor for response to BCDT, some seropositive patients respond to biological treatment insufficiently.
An objective of this study was to differentiate subgroups of seropositive patients for response to RTX. For this purpose, we investigated the epitope recognition patterns against MCV and the AMCV isotypes in AMCV IgG-positive patients with RA, in relation to their therapeutic outcome to RTX. The aim was to determine a predictive and monitoring parameter for RTX treatment, and to gain further insights into the differential behavior of humoral autoimmune responses under such targeted therapies.
Methods
Patients
Our cohort was comprised of AMCV IgG-seropositive RA patients (n = 50) fulfilling the new ACR/EULAR classification criteria [20], who were recruited from the out-patient clinic of the Department of Rheumatology at the Charité-Universitätsmedizin Berlin, Germany (n = 37), and from the Federal Almazov Heart, Blood and Endocrinology Centre, St Petersburg, Russia (n = 13). Patients’ characteristics are summarized in Additional file 1. All patients were inadequate responders to conventional DMARDs, including methotrexate (MTX). RTX was applied in 30 patients as a second-line biologic after failure to TNFi-treatment. A total of 20 patients were biologically naïve and received RTX as a first-line biologic. Before RTX application, all patients had active diseases despite stable treatment with MTX (10 to 25 mg/week), as defined by 28-joint Disease Activity Score based on erythrocyte sedimentation rate (DAS28-ESR) >3.2. The first cycle of RTX (1,000 mg on days one and 15) was administered after standard premedication, and patients were followed up on for a period of six months. A good EULAR response to RTX treatment was evaluated by DAS28-ESR and defined as an improvement of ≥1.2 from baseline to end of follow-up period [21], identifying 37 (74 %) responders to RTX (RRs) and 13 (26-%) non-responders to RTX (NRRs). RRs showed a higher DAS28 at baseline compared to NRRs (6.23 versus 5.09). After the first RTX cycle, DAS28 was significantly reduced in RRs compared to NRRs (3.30 versus 4.72). However, according to EULAR response criteria [22], only a minority of RRs achieved either remission or low disease activity state at week 24 (see Additional file 1). After subsequent cycles of RTX, a further improvement in DAS28 was observed in RRs (data not shown). The study was approved by the local Ethics Committees at the Charité-Universitätsmedizin Berlin, Germany (vote EA1/193/10 april 26, 2012), and the Federal Almazov Heart, Blood and Endocrinology Centre, St. Petersburg, Russia (vote №124 may 21, 2012), and informed consent was given by all patients prior to serum sampling.
Methods
Anti-MCV reactivities in RRs and NRRs were investigated by an MCV epitope mapping of AMCV IgG, and by determination of AMCV isotype profiles in both groups at baseline and after 24 weeks of RTX-treatment.
From the patients in our cohort, the antibody reactivities of 34 AMCV IgG-positive RA patients (23 RRs compared with 11 NRRs) were tested against 88 epitopes of MCV using enzyme-linked immunosorbent assay (ELISA). Referring to the MCV sequence, 18-mer peptides with 12 overlapping amino acid residues to the adjacent peptide were generated (with the general structure Biotin-SGSG-PEPTIDE-Amide, JPT Peptide Technologies GmbH, Berlin, Germany). Mutations (from glycerine to citrulline and serine to histidine) and citrullinations (replacement of each arginine by citrulline) were inserted. The resulting peptides were applied in ascending order corresponding to the wells of a microtiter plate (A1-H1 (peptide 1–8), A2-H2 (peptide 9–16), A3-H3 (peptide 17–24), A4-H4 (peptide 25–32), A5-H5 (peptide 33–40), A6-H6 (peptide 41–48), A7-H7 (peptide 49–56), A8-H8 (peptide 57–64), A9-H9 (peptide 65–72), A10-H10 (peptide 73–80), A11-H11 (peptide 81–88)) (Additional file 2). Additional to the 88 peptides, biotinylated recombinant MCV, protein A and rheumatoid factor antigen were used as internal controls in a separate row of the microtiter plate (results not shown). Microtiter plates covering the entire peptide sequence were incubated with each patient’s serum sample in a dilution of 1:100 (Orgentec Diagnostika, Mainz; Germany and Medipan GmbH, Berlin/Dahlewitz, Germany) and 1:101 (Generic Assays GmbH, Berlin/Dahlewitz, Germany) in sample buffer (Phosphate-buffered saline [PBS] + 0.1% Tween-20 [NaCl, 1.37 mM; KCl 27 mM; Na2HPO4 100 mM; KH2PO4 18 mM; 0.1 % Tween; pH 7.2], all chemicals from Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) for 30 minutes. After a washing step, peroxidase conjugated anti-human IgG was added for 30 minutes. After a second washing step, the enzyme- triggered reaction (Dianova GmbH, Hamburg, Germany) was performed by an incubation with 3,3,5,5′-tetra-methyl benzidine (TMB, Life Technologies GmbH, Darmstadt, Germany) as peroxidase substrate for 15 minutes and terminated by H2SO4 (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) stop solution. Visualization and quantification of reaction products was made by absorbance detection at 450/620 nm. The cut-off level was set at 300 mOD, following receiver operating characteristic (ROC) analysis.
Immunoglobulin isotypes IgG, IgM and IgA against MCV were measured before and 24 weeks after the first application of RTX in an expanded cohort of 50 patients. Additionally, antibody subtypes of RF IgG, IgA, IgM and ACCP IgG were determined. Detection of all antibodies and subtypes was performed using ELISA. AMCV IgG, IgA and IgM were detected by ELISA (Orgentec Diagnostika, Mainz, Germany) with a cut-off level of 20 U/ml (ROC curve analysis for AMCV IgA test is shown in Additional file 3). RF IgG, IgM and IgA were determined by ELISA (Generic Assays GmbH, Dahlewitz/Berlin, Germany) with a cut-off level for RF IgG and IgA of 30 IU/ml, and 15 IU/ml for IgM. ACCP IgG (second-generation assay) was measured by ELISA (Medizym® anti-CCP, Medipan GmbH, Dahlewitz/Berlin, Germany) with a cut-off level of 30 U/ml. All tests were performed according to the manufacturers’ instructions.
Statistics
The comparison of antibody reactivities was performed by non-parametric tests with GraphPad Prism 4 (GraphPad Software, Inc., La Jolla, US). The Wilcoxon signed rank test was applied to compare the autoantibody titer difference from baseline to week 24 for RRs and NRRs. In order to compare the titers of RRs with NRRs for each autoantibody subtype, for differences at baseline and at week 24, we used the Mann–Whitney U test. Furthermore, the frequency distribution of seropositivity of antibody subtypes in RRs and NRRs was tested by the chi-squared and Fisher’s exact test. P values lower than 0.05 were considered to be significant. For correlation analysis, Pearson coefficients were calculated.
Results
Epitope recognition reactivities to mutated citrullinated vimentin peptides of anti-mutated citrullinated vimentin IgG in responders and non-responders to rituximab treatment
In order to clarify whether AMCV IgG-positive RRs (n = 23) and NRRs (n = 11) to RTX differ in their AMCV reactivities, we performed an epitope mapping using overlapping MCV peptides that were recognized by AMCV IgG of the patients. An illustration of the recognition pattern is shown for RRs (Fig. 1) and NRRs (Fig. 2) as they were targeted on the microtiter plate (x axis: wells A to H; y axis: wells 1 to 12; z axis: number of positive patients). At baseline, only minor differences could be detected between both groups. In NRRs, antibody reactivity against several sequences of the MCV protein with few predominant epitopes (A4, A5, C5 and D5) could be observed. After RTX treatment, differences between RRs and NRRs became more apparent: 69.6 % (16 out of 23) of the RRs and only 27.3 % (three out of 11) of the NRRs showed a reduction of their initial recognition pattern. Moreover, epitope recognition in 45.4 % (five out of 11) of NRRs remained unchanged, and expanded in 27.3 % ( three out of 11).

MCV epitope mapping in responders to RTX. The pattern of recognized MCV epitopes by IgG anti-MCV antibodies (AMCV) of RTX responders (n = 23) were reduced from baseline (a) to 24 weeks (b) after RTX treatment (x axis: wells A to H; y axis: wells 1 to 12; z axis: number of positive patients)

MCV epitope mapping in non-responders to RTX. The pattern of recognized MCV epitopes by IgG anti-MCV antibodies (AMCV) of RTX non-responders (n = 11) remained stable or even expanded from baseline (a) to 24 weeks (b) after RTX treatment (x axis: wells A to H; y axis: wells 1 to 12; z axis: number of positive patients)
Anti-mutated citrullinated vimentin isotype profiles at baseline in responders and non-responders to rituximab
In order to clarify whether the response outcome among seropositive patients is associated with Ig isotypes, we analyzed the qualitative distribution of IgM and IgA in AMCV IgG-positive patients and compared them with isotypes of RF and ACCP IgG. At baseline, a lower prevalence of AMCV subtypes was observed in RRs compared to NRRs, with significant differences for AMCV IgA (Table 1 and Fig. 3). In detail, one third (12 out of 37) of the RRs were positive for AMCV IgA titer, whereas 69 % (9/13) of the NRRs were seropositive at baseline (χ2 P = 0.0203). Being AMCV IgA-negative at baseline with an isolated IgG response could indicate a better response to RTX, (negative predictive value = 86.02 %). In return, baseline double-positivity of IgA and IgG was predictive for a poor response to RTX (positive predictive value = 42.86 %).

Baseline autoantibody profiles of responders and non-responders to RTX. Non-responders (blue) more frequently showed IgA and IgM antibodies against MCV (AMCV) than responders (red), whereas the antibody distribution of rheumatoid factor (RF) and antibodies against cyclic citrullinated peptide was nearly similar in both groups
Similar but less significant findings were observed for the qualitative difference of AMCV IgM in RRs and NRRs (Table 1). RRs were also less frequently AMCV IgM-positive at baseline compared to NRRs (43 % versus 69 %), but this difference did not reach statistical significance (χ2 P = 0.105). In contrast, IgG, IgM and IgA subclasses of RF, as well as ACCP IgG, showed nearly similar distributions in RRs and NRRs without apparent differences at baseline (Table 2 and Fig. 3).
Taken together, in our cohort RRs to RTX were characterized by an isolated IgG AMCV response at baseline. In return, co-existent AMCV IgA were able to indicate a poorer response to RTX.
Anti-mutated citrullinated vimentin isotype titer courses in responders and non-responders upon rituximab treatment
For quantitative analysis, we determined the mean titer courses of AMCV isotypes upon RTX treatment in RRs and NRRs and compared them with RF subtypes and ACCP IgG (Tables 1 and 2, Fig. 4). RRs showed lower mean titers of all AMCV subtypes at baseline, with a further substantial reduction upon treatment. In contrast, NRRs exhibited higher AMCV titers at baseline, which were less sensitive to change despite treatment (Fig. 4). Most notably, the mean baseline AMCV IgA titer of RRs decreased by nearly 67 % (from 90.14 to 29.84 U/ml; P <0.0001), whereas the titer of NRRs was twice as high at baseline and rather remained stable, with a non-significant increase of 19.76 % during the follow-up period (from 182.51 to 218.57; P = 0.17) (Fig. 5). To evaluate a clear difference of AMCV IgA variations between RRs and NRRs without bias of seronegative patients, we considered the titer changes of AMCV IgA-positive RRs and NRRs. The tendency became more obvious: a similar baseline titer in RRs and NRRs (261.55 versus 259.99 U/ml) decreased significantly in RRs by 69 % (to 81.56 U/ml; P = 0.0005), but increased slightly in NRRs by 20 % (to 312.25 U/ml; P = 0.2). As a result, the change of AMCV IgA in RRs was significantly different compared to NRRs at the end of the follow-up period (P = 0.007).

Autoantibody isotype-related mean titer change of responders (RRs) and non-responders (NRRs) upon RTX -treatment. Mean titer course of IgM and especially IgA antibodies against MCV (AMCV) were significantly different in RRs and NRRs to RTX, with higher reductions in RRs after six months. Changes in RF isotypes and antibodies against CCP (ACCP) showed no significant difference between groups

Individual IgA anti-MCV titer courses of responders and non-responders upon RTX treatment with mean titer change (red). Responders showed a greater tendency to reduce their initial titer from baseline to week 24 upon RTX treatment compared to non-responders, who rather remained stable
With respect to AMCV IgM, a titer decrease was observed in both RRs and NRRs, but with an increased decline in RRs (Table 1). In detail, AMCV IgM titers decreased significantly by 73.95 % in RRs (P <0.0001) and by 62.5 % in NRRs (P = 0.03), with a substantial difference between both groups at week 24 (P = 0.0003) (Fig. 4). More seropositive RRs normalized their titer levels for AMCV IgM and AMCV IgA under the cut-off level than NRRs after treatment. The number of patients who normalized their AMCV IgA levels was less frequent compared to AMCV IgM.
Such autoantibody kinetics were not observed for RF subtypes and ACCP IgG (Table 2). Although the mean baseline titers of RF IgG, IgM and IgA (with the exception of ACCP IgG in NRRs) decreased significantly with treatment in RRs and NRRs (P <0.0001), the difference did not reach statistical significance at week 24 (Mann–Whitney U P >0.05) (Fig. 4).
IgA anti-mutated citrullinated vimentin titers in relation to Disease Activity Score 28 treatment response
AMCV IgA titer reductions reflected improved treatment response according to the DAS28 and the EULAR response criteria (Table 3). In detail, RRs who achieved remission at week 24 had the greatest percentage reductions in AMCV IgA titers, RRs with low and moderate disease activity presented moderate decreases and RRs with high disease activity revealed only a minor decline in AMCV IgA titers (Fig. 6). In comparison, NRRs with moderate disease activity at week 24 had rather unchanged AMCV IgA titers over time and NRRs with high disease activity even showed an increase of AMCV IgA titers (Fig. 6). Interestingly, the absolute titer exerted no influence. Thus, only the relative change of AMCV IgA titers from baseline to the end of the follow-up period provides information about the response characteristic.

Mean IgA anti-MCV titer course for responders and non-responders to RTX corresponding to their achieved Disease Activity Score 28 (DAS28) after RTX treatment. The achieved DAS28 and treatment response at week 24 according to the EULAR response criteria were reflected by the extent of anti-MCV IgA titer reductions. The better the DAS28 responses were, the greater the IgA anti-MCV reductions were. Responders in remission showed the greatest IgA anti-MCV reductions, whereas non-responders with high disease activity slightly increased their initial levels. EULAR, European League against Rheumatism; HDA, high disease activity; LDA, low disease activity; MDA, moderate disease activity
In line with these findings, a moderate correlation of AMCV IgA and DAS28 could be described (Fig. 7). In order to determine a purely quantitative relation between AMCV IgA and DAS28 without bias of two different time points, we correlated the difference from baseline to the end of the follow-up period in titers of AMCV IgA-positive patients (n = 21) in relation to their change in DAS28. A correlation coefficient of r = 0.49 was found (P = 0.023). The greater the difference and reduction of AMCV IgA titers were, the greater the difference and improvement in DAS28 were. This was in agreement with the above-mentioned AMCV IgA reductions in relation to their DAS28 response. In contrast, no significant correlation was observed for the other autoantibody subtypes and DAS28 values (data not shown).

Correlation of IgA anti-MCV and Disease Activity Score 28 (DAS28) in IgA anti-MCV-positive patients. The difference (delta) of anti-MCV IgA titers (delta anti-MCV IgA) from baseline to 24 weeks after RTX treatment correlated moderately with a greater improvement of DAS28 (delta DAS28) in anti-MCV IgA-positive patients (n = 21) (r = 0.49)
Coincidence of autoantibody subtypes in IgA antibody against mutated citrullinated vimentin-positive patients
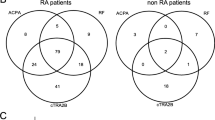
Interestingly, in our AMCV IgG-positive cohort, almost all patients were also positive for ACCP IgG (n = 46 out of 50) but negative for RF IgG (n = 47 out of 50) (Tables 1 and 2). A separation into AMCV IgA-positive (n = 21) versus AMCV IgA-negative (n = 29) patients revealed a higher association of the other AMCV subtypes, with consequent higher mean titers of IgM and IgG in the AMCV IgA-positive group (Table 4). In detail, AMCV IgG titers were twofold and IgM titers threefold higher in the AMCV IgA-positive group compared to the negative group (P = 0.012). Furthermore, only the subtype IgA of RF showed a significant coincidence with AMCV IgA. ACCP IgG and RF IgM were not significantly associated with AMCV IgA (P <0.05) (Fig. 8).

Coincidence of autoantibody subtypes with anti-MCV (AMCV) IgA-positive and -negative patients at baseline. AMCV IgA-positive patients (purple) showed a greater overlap with RA-associated autoantibodies than AMCV IgA-negative patients (green), with significant coincidence of AMCV IgA, IgM and RF IgA
Discussion
The objective of this study was to identify a predictive biomarker among RA patients undergoing BCDT with RTX. Since seropositivity for RA-associated autoantibodies are accepted indicators for response to RTX, we investigated whether differences in fine reactivity and Ig isotype kinetics among AMCV-positive patients could provide additional information. Since the pathogenetic role of these immunoglobulin isotypes is also of interest, we compared the profile of AMCV antibodies to established RF subtypes and ACCP IgG.
Analysis of AMCV IgG-positive patients revealed differences with respect to MCV epitope recognition patterns, serological baseline status and follow-up changes in relation to response outcome. Patients with response to RTX were characterized by a restrictive antibody response of AMCV IgG, decreasing mean titers and reduction of initially recognized epitope pattern upon treatment. In contrast, double-seropositivity for AMCV IgG and IgA (and IgM, but to a lesser degree), stable antibody mean titers and an unchanged or even expanded epitope recognition pattern during the follow-up period were associated with non-response to RTX. These findings were confirmed by a moderate correlation of AMCV IgA reductions with improved DAS28 response.
In general, non-response to RTX could be due to resistant B-cell repertoire or/and pharmacokinetic differences in individual cases, like different tissue penetration of RTX to lymphoid niches [23]. In this context, the results of our study provide evidence for a RTX-sensitive B-cell subpopulation in RRs with a reduced epitope recognition pattern and decreasing IgG titers under treatment. In contrast, an unchanged or even expanded reactivity after B-cell depletion could be explained by active IgG-producing B-cell subsets in NRRs, which are less sensitive to RTX. Furthermore, a higher coincidence of AMCV isotypes, including IgA, in NRRs could be related to refractory IgA-producing B-cell subsets.
A close link of RTX response to CD20-positive memory B-cell compartment has been shown by previous studies. In most of these investigations, NRRs had an incomplete depletion and higher repopulation numbers of memory B-cells [24–26]. These memory B-cells are involved in the formation of secondary germinal centers and differentiate into CD20-negative plasmablasts and plasma cells, contributing to active inflammatory responses with rise in antibody levels [27]. Both isotypes IgA and IgG are mainly produced by tissue-resident memory B-cells and plasma cells [28, 29]. Since the distribution and architecture of these compartments can vary between individuals, RTX can cause distinct depletion of these B-cells with individually varying IgA or IgG kinetics [30]. In this context, stable AMCV IgA titers in both RRs and NRRs could be explained by lower sensitivity of AMCV IgA-producing B-cells towards RTX treatment. As shown also by vaccination studies, BCDT affects mainly short-lived CD20-positive B-cells, explaining especially the decline of IgM titers, which was also observed in our cohort [31].
The association of non-response to RTX with the appearance of IgA anti-MCV points to the significance of this Ig subset in RA. At early stages of RA investigations, the presence of IgA RF was shown to be associated with increased disease activity and severity [32]. Furthermore, IgA RF also preceded disease onset and was linked to extraarticular manifestations and resistance to treatment with TNFi [33, 34]. Of note, overall serum IgA and IgG levels have been described as rather stable, even after repeated cycles of RTX in RA patients [35]. Finally, co-expression of IgA and IgG for AMCV has been associated with disease severity by another prior study [19], which supports the relevance of an interaction between the mucosal and systemic humoral responses. Mei et al. have identified the source of serum IgA from circulating plasmablasts and suggested their origin to be from mucosal memory B-cells [36]. Due to loss of CD20 receptors, these mucosal plasmablasts were not depleted by RTX [37], and subsequently, levels of IgA remained stable. As known from vaccination studies, IgA responses were mainly induced by oral but not systemic exposure to antigen within the mucosa-associated lymphoid tissues [38]. Thus, the appearance of IgA autoantibodies suggests primarily mucosal triggers for such autoimmune responses. This connection of mucosal immune reactions and autoimmune disorders is also supported by known oral or gastrointestinal triggers in RA [39, 40], including nutrition-dependent increase in antibody titers [41]. In summary, it can be hypothesized that tissue-resident memory B-cells could represent the main source for such plasmablasts, causing treatment resistance to RTX.
Our study is limited by the relatively small sample size and inclusion of patients from two cohorts, of whom one part received RTX as a first-line biologic. Thus, different pretreatment with biologicals and concomitant medication could have influenced our results. Furthermore, by preselecting for AMCV IgG-positive patients, we could have missed different subtype kinetics in other serologic subgroups. Although this study design limits the comparability to existing studies, we were able to perform detailed analyses of the autoantibody kinetics in RTX-treated patients. In summary, our results confirm a correlation of AMCV subtypes with disease activity, and provide evidence for their potential prediction and monitoring value.
Conclusions
Restrictive baseline seropositivity for anti-MCV IgG was identified as a predictor for response to RTX. Co-expression of IgA was associated with treatment failure to RTX. Reduction of AMCV IgA titers under RTX showed a correlation with EULAR response in RA. Furthermore, rigid IgA isotype under such targeted therapies could be related to refractory B-cell subsets, giving evidence of greater heterogeneity within the so called seropositive RA cohort.
Abbreviations
- ACCP:
-
antibodies against cyclic citrullinated peptide/protein
- ACPA:
-
anti-cyclic citrullinated protein antibodies
- AMCV:
-
antibodies against mutated citrullinated vimentin
- BCDT:
-
B-cell depletion therapy
- DAS28:
-
Disease Activity Score in 28 joints
- DMARDs:
-
disease-modifying anti-rheumatic drugs
- ESR:
-
erythrocyte sedimentation rate
- EULAR:
-
European League against Rheumatism
- HDA:
-
high disease activity
- IgG/ IgM/ IgA:
-
Immunoglobulin G/ Immunoglobulin M/ Immunoglobulin A
- LDA:
-
low disease activity
- MDA:
-
moderate disease activity
- MTX:
-
methotrexate
- NRRs:
-
non-responders to RTX
- RA:
-
rheumatoid arthritis
- RF:
-
rheumatoid factor
- ROC:
-
receiver operating curve
- RRs:
-
responders to RTX
- RTX:
-
rituximab
- SD:
-
standard deviation
- TNFi:
-
Tumor necrosis factor- alpha inhibitor
References
Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev. 2005;4:130–6.
Bykerk VP, Keystone EC, Kuriya B, Larche M, Thorne JC, Haraoui B. Achieving remission in clinical practice: lessons from clinical trial data. Clin Exp Rheumatol. 2013;31:621–32.
Smolen JS, Keystone EC, Emery P, Breedveld FC, Betteridge N, Burmester GR, et al. Consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:143–50.
Smolen JS, Landewe R, Breedveld FC, Buch M, Burmester G, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73:492–509.
Singh JA, Cameron DR. Summary of AHRQ’s comparative effectiveness review of drug therapy for rheumatoid arthritis (RA) in adults--an update. J Manag Care Pharm. 2012;18:S1–18.
Ferraccioli G, Tolusso B, Bobbio-Pallavicini F, Gremese E, Ravagnani V, Benucci M, et al. Biomarkers of good EULAR response to the B cell depletion therapy in all seropositive rheumatoid arthritis patients: clues for the pathogenesis. PLoS One. 2012;7, e40362.
Soliman MM, Hyrich KL, Lunt M, Watson KD, Symmons DP, Ashcroft DM, et al. Effectiveness of rituximab in patients with rheumatoid arthritis: observational study from the British Society for Rheumatology Biologics Register. J Rheumatol. 2012;39:240–6.
Daien CI, Morel J. Predictive factors of response to biological disease modifying antirheumatic drugs: towards personalized medicine. Mediators Inflamm. 2014;2014:386148.
Buch MH, Smolen JS, Betteridge N, Breedveld FC, Burmester G, Dorner T, et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70:909–20.
Quartuccio L, Fabris M, Salvin S, Atzeni F, Saracco M, Benucci M, et al. Rheumatoid factor positivity rather than anti-CCP positivity, a lower disability and a lower number of anti-TNF agents failed are associated with response to rituximab in rheumatoid arthritis. Rheumatology. 2009;48:1557–9.
Benucci M, Manfredi M, Puttini PS, Atzeni F. Predictive factors of response to rituximab therapy in rheumatoid arthritis: What do we know today? Autoimmun Rev. 2010;9:801–3.
Toubi E, Kessel A, Slobodin G, Boulman N, Pavlotzky E, Zisman D, et al. Changes in macrophage function after rituximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:818–20.
Vancsa A, Szabo Z, Szamosi S, Bodnar N, Vegh E, Gergely L, et al. Longterm effects of rituximab on B cell counts and autoantibody production in rheumatoid arthritis: use of high-sensitivity flow cytometry for more sensitive assessment of B cell depletion. J Rheumatol. 2013;40:565–71.
Willemze A, Bohringer S, Knevel R, Levarht EW, Stoeken-Rijsbergen G, Houwing-Duistermaat JJ, et al. The ACPA recognition profile and subgrouping of ACPA-positive RA patients. Ann Rheum Dis. 2012;71:268–74.
Van Steendam K, Tilleman K, De Ceuleneer M, De Keyser F, Elewaut D, Deforce D. Citrullinated vimentin as an important antigen in immune complexes from synovial fluid of rheumatoid arthritis patients with antibodies against citrullinated proteins. Arthritis Res Ther. 2010;12:R132.
Qin X, Deng Y, Xu J, Li TJ, Li S, Zhao JM. Meta-analysis: diagnostic value of serum anti-mutated citrullinated vimentin antibodies in patients with rheumatoid arthritis. Rheumatol Int. 2011;31:785–94.
Ioan-Facsinay A, Willemze A, Robinson DB, Peschken CA, Markland J, van der Woude D, et al. Marked differences in fine specificity and isotype usage of the anti-citrullinated protein antibody in health and disease. Arthritis Rheum. 2008;58:3000–8.
Zahran WE, Mahmoud MI, Shalaby KA, Abbas MH. Unique correlation between mutated citrullinated vimentine IgG autoantibodies and markers of systemic inflammation in rheumatoid arthritis patients. Indian J Clin Biochem. 2013;28:272–6.
Svard A, Kastbom A, Soderlin MK, Reckner-Olsson A, Skogh T. A comparison between IgG- and IgA-class antibodies to cyclic citrullinated peptides and to modified citrullinated vimentin in early rheumatoid arthritis and very early arthritis. J Rheumatol. 2011;38:1265–72.
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham 3rd CO, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81.
Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Rheum Dis Clin North Am. 2009;35:745–57. vii–viii.
van Riel PL. The development of the disease activity score (DAS) and the disease activity score using 28 joint counts (DAS28). Clin Exp Rheumatol. 2014;32 5 Suppl 85:S-65–74.
Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15:S3.
Nakou M, Katsikas G, Sidiropoulos P, Bertsias G, Papadimitraki E, Raptopoulou A, et al. Rituximab therapy reduces activated B cells in both the peripheral blood and bone marrow of patients with rheumatoid arthritis: depletion of memory B cells correlates with clinical response. Arthritis Res Ther. 2009;11:R131.
Sellam J, Rouanet S, Hendel-Chavez H, Abbed K, Sibilia J, Tebib J, et al. Blood memory B cells are disturbed and predict the response to rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2011;63:3692–701.
Trouvin AP, Jacquot S, Grigioni S, Curis E, Dedreux I, Roucheux A, et al. Usefulness of monitoring of B cell depletion in rituximab-treated rheumatoid arthritis patients in order to predict clinical relapse: a prospective observational study. Clin Exp Immunol. 2015;180:11–8.
McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat Rev Immunol. 2012;12:24–34.
Yoshida T, Mei H, Dorner T, Hiepe F, Radbruch A, Fillatreau S, et al. Memory B and memory plasma cells. Immunol Rev. 2010;237:117–39.
Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15:149–59.
DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas KM, et al. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. 2008;180:361–71.
Westra J, van Assen S, Wilting KR, Land J, Horst G, de Haan A, et al. Rituximab impairs immunoglobulin (Ig)M and IgG (subclass) responses after influenza vaccination in rheumatoid arthritis patients. Clin Exp Immunol. 2014;178:40–7.
Teitsson I. IgA rheumatoid factor as predictor of disease activity. Scand J Rheumatol Suppl. 1988;75:233–7.
Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–9.
Bobbio-Pallavicini F, Caporali R, Alpini C, Avalle S, Epis OM, Klersy C, et al. High IgA rheumatoid factor levels are associated with poor clinical response to tumour necrosis factor alpha inhibitors in rheumatoid arthritis. Ann Rheum Dis. 2007;66:302–7.
De La Torre I, Leandro MJ, Valor L, Becerra E, Edwards JC, Cambridge G. Total serum immunoglobulin levels in patients with RA after multiple B-cell depletion cycles based on rituximab: relationship with B-cell kinetics. Rheumatology. 2012;51:833–40.
Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, et al. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood. 2009;113:2461–9.
Mei HE, Frolich D, Giesecke C, Loddenkemper C, Reiter K, Schmidt S, et al. Steady-state generation of mucosal IgA+ plasmablasts is not abrogated by B-cell depletion therapy with rituximab. Blood. 2010;116:5181–90.
Hasegawa H, van Reit E, Kida H. Mucosal immunization and adjuvants. Curr Top Microbiol Immunol. 2015;386:371–80.
Koziel J, Mydel P, Potempa J. The link between periodontal disease and rheumatoid arthritis: an updated review. Curr Rheumatol Rep. 2014;16:408.
Taneja V. Arthritis susceptibility and the gut microbiome. FEBS Lett. 2014;588:4244–9.
Hvatum M, Kanerud L, Hallgren R, Brandtzaeg P. The gut-joint axis: cross reactive food antibodies in rheumatoid arthritis. Gut. 2006;55:1240–7.
Acknowledgements
The research leading to these results was funded within the European Union’s Research and Innovation FP7 HEALTH programme under the grant agreement FP7-HEALTH-F2-2012-305549, SFB 650 and by a scientific grant from Roche Pharma. We would like to thank Barbara Lehmann, Labor-Berlin GmbH, for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Holger Bang is an employee of Orgentec Diagnostika GmbH, Mainz, Germany. Esther Wittenborn is an employee of Roche Pharma. Eugen Feist received honoraria from Orgentec GmbH and grant support form Roche Pharma. Karl Egerer received grand support from Orgentec GmbH. Gerd-R Burmester and Thomas Dörner received honoraria form Roche Pharma. No non-financial conflicts of interest exist for any of the authors.
Authors’ contributions
LL carried out immunoassays, performed statistical analysis and drafted and revised the manuscript. LS participated in its coordination, contributed with patient data and helped to draft the manuscript. HB participated in its coordination, contributed with patient data and drafted and revised the manuscript. TD participated in coordination, contributed with patient data and helped to draft the manuscript. ALM participated in its coordination, contributed with patient data and helped to draft the manuscript. SVL carried out immunoassays and helped to draft the manuscript. EII participated in its coordination, contributed with patient data and helped to draft the manuscript. LMG participated in its coordination, performed statistical analysis and helped to draft the manuscript. HB provided antigenic peptides, carried out immunoassays and helped to draft the manuscript. EW participated in its design and helped to draft the manuscript. KE carried out immunoassays and helped to draft the manuscript. GRB participated in its design and helped to draft and revise the manuscript. EF conceived of the study, participated in its coordination and helped to draft and revise the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Patients’ characteristics. (DOCX 20 kb)
Additional file 2: Table S2.
Sequence of selected mutated citrullinated vimentin (MCV) with epitopes from 1 to 88. The entire protein sequence of MCV was cut into 88 peptides consisting of 17 amino acids, with 12 overlapping amino acids to the neighbourhood peptide, with following amino acid code: X = citrulline; X = mutated glycine = citrulline; H = mutated serine = histidine. (DOCX 18 kb)
Additional file 3: Figure S1.
ROC curve analysis for AMCV IgA assay. Cut-off level at 20 U/ml resulted in a specificity of 100 % and sensitivity of 42 %. (DOCX 23 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lindenberg, L., Spengler, L., Bang, H. et al. Restrictive IgG antibody response against mutated citrullinated vimentin predicts response to rituximab in patients with rheumatoid arthritis. Arthritis Res Ther 17, 206 (2015). https://doi.org/10.1186/s13075-015-0717-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-015-0717-z