
Abstract
Background
The apolipoprotein E (APOE) ε4 allele is the strongest genetic risk factor for late onset Alzheimer’s disease, whilst the ε2 allele confers protection. Previous studies report differential DNA methylation of APOE between ε4 and ε2 carriers, but associations with epigenome-wide methylation have not previously been characterised.
Methods
Using the EPIC array, we investigated epigenome-wide differences in whole blood DNA methylation patterns between Alzheimer’s disease-free APOE ε4 (n = 2469) and ε2 (n = 1118) carriers from the two largest single-cohort DNA methylation samples profiled to date. Using a discovery, replication and meta-analysis study design, methylation differences were identified using epigenome-wide association analysis and differentially methylated region (DMR) approaches. Results were explored using pathway and methylation quantitative trait loci (meQTL) analyses.
Results
We obtained replicated evidence for DNA methylation differences in a ~ 169 kb region, which encompasses part of APOE and several upstream genes. Meta-analytic approaches identified DNA methylation differences outside of APOE: differentially methylated positions were identified in DHCR24, LDLR and ABCG1 (2.59 × 10−100 ≤ P ≤ 2.44 × 10−8) and DMRs were identified in SREBF2 and LDLR (1.63 × 10−4 ≤ P ≤ 3.01 × 10−2). Pathway and meQTL analyses implicated lipid-related processes and high-density lipoprotein cholesterol was identified as a partial mediator of the methylation differences in ABCG1 and DHCR24.
Conclusions
APOE ε4 vs. ε2 carrier status is associated with epigenome-wide methylation differences in the blood. The loci identified are located in trans as well as cis to APOE and implicate genes involved in lipid homeostasis.
Similar content being viewed by others
Background
The ε4 allele of the apolipoprotein E gene (APOE) is the strongest genetic risk factor for late-onset (> 65 years) Alzheimer’s disease (AD) [1,2,3]. Inheritance of one copy of this allele increases late-onset AD risk by two to fourfold, with two copies conferring an eight to twelvefold increase in risk compared to the ε3/ε3 genotype [4, 5]. The ε4 allele is also associated with a younger age-of-onset, with ε4 homozygotes having an average age-of-onset of 68 years compared to 84 years for ε3 homozygotes [4]. In contrast, the ε2 allele has been associated with a ~ 50% reduction in AD risk compared to the ε3/ε3 genotype [5].
The three APOE alleles (ε2/ε3/ε4) are defined by two APOE exon 4 single-nucleotide polymorphisms (SNPs) and encode functionally distinct ApoE isoforms. Isoform-dependent behaviours have been observed for many ApoE functions, including lipid metabolism, amyloid beta (Aβ) metabolism, tau phosphorylation, inflammation and synaptic plasticity, with ApoE4 and ApoE2 conferring effects consistent with increased and reduced AD risk, respectively [6, 7].
Despite the wealth of evidence linking ApoE to processes implicated in AD pathogenesis, understanding of the specific mechanism(s) by which genetic variation at this locus alters risk remains incomplete. APOE genotype acts in conjunction with other genetic and/or environmental factors to confer AD risk: the lifetime risk of dementia or mild cognitive impairment is 31%–40% for ε4/ε4 homozygotes [8] but the effects of APOE ε4 have been shown to be modified by ethnic background and sex [5, 9]. DNA methylation is associated with both genetic and environmental factors, and previous studies have identified associations with AD and neuropathological hallmarks of AD [10,11,12], AD risk factors (e.g. ageing [13], obesity [14] and lipid levels [15]), as well as modifiers of APOE genotype effects (e.g. sex [16] and ethnicity [17, 18]).
The two APOE haplotype-defining SNPs are located in a CpG island and have a direct effect on methylation by creating/destroying CpG sites [19]. The APOE ε2/ε3/ε4 haplotype is associated with methylation at other CpG sites within APOE [20, 21] but, to date, associations with methylation across the epigenome have not been assessed. We hypothesised that characterising these associations would yield insights into the biological context in which APOE acts, thus facilitating the search for mechanisms conferring risk/resilience for AD. Importantly, by studying individuals who are free from AD, we have the potential to identify pathogenic processes that precede the onset of irreversible neurodegeneration.
Methods
Participants
The participants were selected from the Generation Scotland: Scottish Family Health Study (GS:SFHS) cohort (~ 24,000 participants aged ≥ 18 years at recruitment), which has been described previously [22, 23]. The participants included in this study were of European (predominantly British) ancestry, following the exclusion of participants with likely recent Italian or African/Asian ancestry by principal components (PC) analysis [24]. Participants attended a baseline clinical appointment at which they were phenotyped for social, demographic, health and lifestyle factors, completed cognitive assessments and provided physical measurements and samples for DNA extraction. GS:SFHS obtained ethical approval from the NHS Tayside Committee on Medical Research Ethics, on behalf of the National Health Service (reference: 05/S1401/89) and has Research Tissue Bank Status (reference: 15/ES/0040).
Blood sample collection and DNA extraction
DNA was extracted from blood (9 ml) collected in EDTA tubes using the Nucleon BACC3 Genomic DNA Extraction Kit (Fisher Scientific), following the manufacturer’s instructions [25].
Genotyping of APOE
The APOE ε2/ε3/ε4 haplotypes are defined by two SNPs, rs429358 and rs7412, which were genotyped using TaqMan probes at the Clinical Research Facility, Edinburgh.
Measurement of cholesterol levels
Total and high-density lipoprotein (HDL) cholesterol were measured at the GS:SFHS baseline appointment and non-HDL cholesterol levels were calculated by subtracting HDL cholesterol from total cholesterol. The non-HDL cholesterol level reflects a combination of low-density lipoprotein (LDL) cholesterol and very low-density lipoprotein.
Genome-wide DNA methylation profiling for EWAS analyses
DNA methylation was profiled using the Infinium MethylationEPIC BeadChip (Illumina Inc.) in a discovery (n = 5190) and replication (n = 4583) sample, as described previously [26,27,28] (Supplementary Methods). The discovery and replication samples were normalised separately and converted to M values. The discovery data was corrected for relatedness (Supplementary Methods). Participants in the replication sample were unrelated (SNP-based relatedness< 0.05) to each other and/or discovery sample participants.
Poor performing probes, X/Y chromosome probes and participants with unreliable self-report data or potential XXY genotype were excluded (Supplementary Methods). The final discovery dataset comprised M values at 760,943 loci for 5087 participants; the replication dataset comprised M values at 758,332 loci for 4450 participants. All subsequent analyses of the DNA methylation data were carried out using R versions 3.6.0., 3.6.1., or 3.6.2 [29, 30].
Statistical analyses
A flow chart indicating all analyses is presented in Fig. 1.

Flow chart indicating the analyses carried out in this study. Yellow boxes indicate datasets used for the analysis, blue boxes describe the analysis performed and green boxes contain the results of the analysis. Arrows indicate the analyses for which the datasets were used, the order of the analyses and the results from each analysis
Epigenome-wide association studies
EWASs were implemented using limma [31]. CpG M values were the dependent variable and APOE ε4 vs. ε2 carrier status (a binary variable indicating APOE ε4 carriers with a “1” and APOE ε2 with a “0”; ε4/ε2 and ε3/ε3 participants were excluded) was the predictor-of-interest. Participants self-reporting AD (n = five) were excluded. Additional covariates were included as below:
Discovery sample
CpG site (pre-corrected for relatedness, estimated cell counts and processing batch) ~ APOE ε4 vs. ε2 + age + sex + smoking status + pack years + 20 methylation PCs
Replication sample
CpG site (M values) ~ APOE ε4 vs. ε2 + age + sex + smoking status + pack years + estimated cell counts (granulocytes, natural killer cells, B lymphocytes, CD4 + T lymphocytes and CD8 + T lymphocytes) + processing batch + 20 methylation PCs
The variables “smoking status”, “pack years” and the methylation PCs are explained in the Supplementary Methods.
An additional sensitivity analysis of the replication sample was performed in which the first 10 genetic PCs, calculated using GCTA [32], were included. The decision to include 10 PCs was based on inspection of a scree plot (Additional file 2: Fig. S1).
Limma was used to calculate empirical Bayes moderated t-statistics from which P values were obtained. The significance threshold in the discovery sample was P ≤ 3.6 × 10−8 [33]. Sites attaining significance in the discovery sample were assessed in the replication sample using a Bonferroni-corrected threshold of 0.05/no. sites assessed.
EWAS meta-analysis
Inverse variance-weighted fixed effects meta-analyses of 756,971 sites common to the discovery and replication EWAS results were performed using METAL [34]. Sites attaining a meta-analysis P ≤ 3.6 × 10−8 were considered significant.
Comparison of DNA methylation levels between APOE haplotypes
For the differentially methylated positions (DMPs) identified through the EWAS meta-analysis, pairwise differences in methylation levels between carriers of the APOE ε2/ε2, ε2/ε3, ε3/ε3, ε3/ε4 and ε4/ε4 haplotypes in the discovery sample were investigated, using the R package lsmeans [35]. P values were adjusted using a Bonferroni correction to account for the 10 within-CpG comparisons performed for each of the 20 CpGs assessed (i.e. an adjustment was performed for 200 tests). Corrected P ≤ 0.05 was considered statistically significant.
Identification of differentially methylated regions
DMRs associated with APOE ε4 vs. ε2 carrier status were identified using the dmrff.meta function from the dmrff R package [36]. Putative DMRs were defined as regions containing two to thirty sites separated by ≤ 500 bp with EWAS meta-analysis P ≤ .05 and methylation changes in a consistent direction. Following dmrff’s subregion selection step, DMRs with Bonferroni-adjusted P ≤ .05 were declared significant.
Gene ontology/KEGG pathway analyses
Gene ontology (GO) and KEGG pathway analyses were implemented using missMethyl’s gometh function [37]. The target list comprised probes that were suggestively associated with the phenotype-of-interest (P ≤ 1 × 10−5) in the meta-EWAS or that contributed to a significant DMR (adjusted P ≤ 0.05) and the gene universe included all analysed probes. Enrichment was assessed using a hypergeometric test, accounting for the bias arising from the variation in the number of probes-per-gene. Bonferroni-corrected significance thresholds of P ≤ 2.21 × 10−6 and P ≤ 1.48 × 10−4 were applied to account for the 22,578 GO terms and 337 KEGG pathways assessed.
Bootstrap mediation analysis
The roles of cholesterol levels (total cholesterol, HDL cholesterol and non-HDL cholesterol) in mediating any observed associations between APOE ε4 vs. ε2 carrier status and DNA methylation were assessed by bootstrap mediation analysis using the R package “mediation” [38]. The analyses were performed using 10,000 bootstrap samples in the discovery and replication samples separately and these results were then meta-analysed using inverse variance-weighted fixed effects meta-analyses to obtain meta-analyses P values and effect estimates. Significant mediation was declared when the meta-analysis P value met a Bonferroni-adjusted (to account for the assessment of 20 DMPs) significance threshold of P ≤ .05.
Genotyping and imputation
The genotyping and imputation of GS:SFHS to the Haplotype Reference Consortium reference panel release 1.1 [39] has been described previously [25, 40] (Supplementary Methods).
Identification of methylation quantitative trait loci
Methylation quantitative trait loci (meQTLs) were identified using the discovery sample. Following quality control, the data was normalised and corrected as described previously [41] (Supplementary Methods). Normalised and corrected data was available for 26 of the 31 CpGs-of-interest in this study. The resulting residuals were inverse rank normal transformed and entered as the dependent variable in simple linear model GWASs to identify meQTLs. GWASs were implemented using REGSCAN v0.5 [42]. SNPs that were associated with a CpG with P ≤ 1.92 × 10−9 (5 × 10−8/26) were declared to be meQTLs. SNPs located within one megabase up- or downstream of their associated CpG were defined as cis meQTLs; all other associated SNPs were defined as trans meQTLs. A look-up analysis of the GWAS catalog [43] (GWAS catalog v1.0.2., downloaded 07/09/20) was performed in which SNPs identified as meQTLs for the CpGs of interest were queried for their significant (P ≤ 5 × 10−8) disease or trait associations in the GWAS catalog.
Association analyses of APOE ε4 vs. ε2 carrier status
Association analyses were performed to assess whether meQTLs for the meta-analysis DMPs are associated with APOE ε4 vs. ε2 carrier status and, therefore, might contribute to the differences in methylation observed between APOE ε4 and ε2 carriers. Association tests used BOLT-LMM [44] to perform linear mixed models in participants with available APOE genotypes (ε2 n = 2613; ε4 n = 5401). BOLT-LMM adjusts for population structure and relatedness between individuals whilst assessing association. Sex was included as a covariate. Associations were considered significant when P ≤ 5 × 10−8.
Results
Sample demographics
The EWAS discovery sample comprised 1253 APOE ε4 and 596 APOE ε2 allele carriers and the replication sample comprised 1216 APOE ε4 and 522 APOE ε2 allele carriers. Twenty-seven ε2/ε2, 569 ε2/ε3, 2926 ε3/ε3, 1128 ε3/ε4 and 125 ε4/ε4 participants from the discovery sample were available for the pairwise analysis of genotypes. Key sample demographic information is presented in Additional file 1: Table S1.
Identification of differentially methylated positions and regions in APOE ε4 vs. ε2 carriers
An EWAS of APOE ε4 vs. ε2 carriers in the discovery sample identified eight significant DMPs, of which half were hypermethylated in APOE ε4 carriers. These DMPs had a mean absolute effect size of 0.070 (range 0.033–0.103) and P values ranging from 6.40 × 10−56 to 8.81 × 10−9. All eight sites were also significant (8.60 × 10−49 ≤ P ≤ 7.25 × 10−6) in the replication sample with a consistent direction of effect (mean absolute effect size = 0.102; range 0.049–0.170; Additional file 1: Table S2). The eight sites are located in a ~ 169 kb region on chromosome 19 (chr. 19: 45,242,346-45,411,802; GRCh37/hg19), which spans a region of the genome upstream of and including part of the APOE gene (chr19: 45,409,039–45,412,650; GRCh37/hg19). A sensitivity analysis of the discovery sample in which a methylation-based smoking score [45] was included as a covariate instead of the smoking covariates included in the original analysis (“smoking status” and “pack years”) produced highly similar results across all measured CpGs (correlation between effect sizes = 0.99, 95% confidence interval (CI) 0.99–0.99; P < 2.2 × 10−16; Additional file 1: Table S2). An additional sensitivity analysis in which the first 10 genetic PCs were included as additional covariates in the analysis of the replication sample also produced results that were highly correlated with those from the original replication sample analysis (r = 1.00, 95% CI 1.00–1.00, P < 2.2 × 10−16; Additional file 1: Table S2).
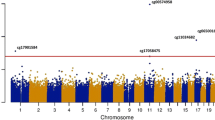
Inverse variance-weighted fixed effects meta-analysis of the discovery and replication samples identified 20 DMPs, with APOE ε4 carrier status associated with hypomethylation at 13 (65%) of these sites. Across all 20 DMPs, the mean absolute effect size was 0.052 (range 0.022–0.11) with P values ranging from 2.80 × 10−100 to 2.4 × 10−8 (Table 1; Fig. 2). Sixteen of these sites are located on chromosome 19q in a ~ 233 kb region (chr19: 45,221,584–45,454,752; GRCh37/hg19) encompassing APOE and several surrounding genes (Additional file 2: Fig. S2). Henceforth, the region containing APOE and neighbouring genes will be referred to as the “APOE locus”. The most significant DMP, cg13375295, is located ~ 4.5 kb upstream of poliovirus receptor-related 2 (PVRL2), a gene situated ~ 16.5 kb upstream of APOE. Four other DMPs (cg10762466, cg10178308, cg11643040 and cg06198803) are located either upstream or in the gene body of PVRL2. Two DMPs (cg06750524 and cg16471933) are located in APOE: cg06750524, the DMP with the largest effect size, in the intron between exons 2 and 3, and cg16471933 in exon 4, 139 bp 5′ of rs429358, one of the APOE ε4/ε2-defining SNPs. Although both the APOE DMPs are more highly methylated in APOE ε4 carriers; the DMPs in the surrounding region do not show a consistent direction of effect.

Manhattan plot showing the APOE ε4 vs. ε2 carrier EWAS and DMR meta-analyses results. Each point represents one of the 772,453 loci included in the EWAS meta-analysis, with the point’s position being determined by genomic position (x-axis) and significance in the EWAS meta-analysis (−log10 P value; y-axis). Sites attaining genome-wide significance (P ≤ 3.6 × 10−8) are indicated in red and those that are involved in a significant DMR (Bonferroni-correct P ≤ 0.05) are indicated in blue. The locations of DMRs are further indicated by vertical blue lines. The solid horizontal line is the threshold for genome-wide significance (P ≤ 3.6 × 10−8)
Four DMPs are located outside of chromosome 19q: cg17901584, 785 bp upstream of the 24-dehydrocholesterol reductase (DHCR24) gene on chromosome 1; cg19751789, 94 bp upstream of the low-density lipoprotein receptor (LDLR) gene on chromosome 19p; and two, cg16740586 and cg06500161, are located 668 bp apart in the same intron of multiple ATP binding cassette subfamily G member 1 (ABCG1) isoforms.
To further investigate the pattern of methylation observed at these 20 DMPs, pairwise comparisons were performed between carriers of the following APOE haplotypes: ε2/ε2, ε2/ε3, ε3/ε3, ε3/ε4 and ε4/ε4. These analyses revealed a range of allele-associated methylation patterns, which are depicted in Additional file 2: Fig. S3 and described in Additional file 1: Table S3. Carriers of the APOE ε2 allele (ε2/ε2 or ε2/ε3) differed from ε3/ε3 homozygotes at 14 of the DMPs, whilst carriers of the APOE ε4 allele (ε4/ε4 or ε3/ε4) differed from ε3/ε3 homozygotes at four DMPs. Dosage effects were observed at two DMPs for ε2 carriers (Additional file 2: Fig. S3A and S) and one DMP for ε4 carriers (Additional file 2: Fig. S3B), although the small numbers of participants who are homozygous for APOE ε2 (n = 27) and ε4 (n = 128) likely rendered our study underpowered to detect all dosage effects. For the two DMPs located within the APOE gene (cg06750524 and cg16471933), an increase in mean methylation levels was observed from ε2/ε2 homozygotes to ε3/ε3 homozygotes, with a further increase to the ε4/ε4 group (Additional file 2: Fig. S3B and E). At the four DMPs outside of the APOE locus, the methylation differences appear to be predominantly driven by the ε2 allele (Additional file 2: Fig. S3K, M, O and S).
Differentially methylated regions (DMRs) were identified using a meta-analysis approach, which identified six significant regions (Additional file 2: Fig. S4). Across all the DMRs, the mean absolute effect size was 0.182 (range 0.135–0.231) and Bonferroni-adjusted P values ranged from 1.63 × 10−4 to 3.01 × 10−2 (Table 2).Three of the DMRs are located at the APOE locus, two are in the first intron of sterol regulatory element binding transcription factor 2 (SREBF2) on chromosome 22, and the other is in the putative promoter of LDLR on chromosome 19p. All but one of the DMRs, which is located 190 bp upstream of the apolipoprotein C1 pseudogene 1 (APOC1P1) at the APOE locus, are hypomethylated in APOE ε4 carriers. Only one of the DMRs, located in an exon of a read-through transcript involving apolipoprotein C2 (APOC2) and apolipoprotein C4 (APOC4), contains CpGs that were identified as DMPs (cg13119609 and cg09555818).
GO analysis was carried out using the 19 Entrez IDs mapping to the 46 CpG sites with a meta-EWAS P ≤ 1 × 10−5 or that contributed to a significant DMR. This identified 78 significant GO terms (Table 3; Additional file 1: Table S4), the most significant of which was “cholesterol metabolic process” (P = 2.00 × 10−11). Significant enrichment for the KEGG pathways “cholesterol metabolism” (P = 5.93 × 10−10) and “steroid biosynthesis” (P = 1.22 × 10−4) was also observed.
Assessment of the role of cholesterol in mediating methylation differences between APOE ε4 and ε2 carriers
Given the well-established role of ApoE in cholesterol metabolism [6], bootstrap mediation analyses were performed to assess the role of cholesterol levels (total, HDL or non-HDL cholesterol) in mediating the association between APOE ε4 vs. ε2 carrier status and methylation at the 20 meta-analysis DMPs. Inverse variance-weighted fixed effects meta-analysis of the bootstrap mediation analyses in the discovery and replication samples identified HDL cholesterol as a significant mediator of the associations with the two ABCG1 DMPs cg06500161 (effect size = 0.006; effect size standard error = 0.001; P = 1.18 × 10−6) and cg16740586 (effect size = 0.004; effect size standard error = 0.001; P = 4.93 × 10−5), and the DHCR24 promoter DMP, cg17901584 (effect size = − 0.007; effect size standard error = 0.001; P = 6.04 × 10−6), for which it mediated 25.2%, 11.5%, and 18.2% of the relationship, respectively (Additional file 1: Table S5). For some sites, inspection of the P values indicated total and non-HDL cholesterol to be significant mediators but the proportion of the relationship between APOE ε4 vs. ε2 carrier status and methylation attributable to the mediator was negative (Additional file 1: Table S5). This indicates that, at these sites, the direction of the association between the cholesterol phenotypes and methylation is the opposite to the direction of the association between APOE ε4 vs. ε2 carrier status and methylation.
Assessment of meQTLs associated with loci that are differentially methylated between APOE ε4 and ε2 carriers
To explore the DMP and DMR CpGs further, meQTL analyses were performed. Whilst it was expected that meQTLs for the DMP and DMR CpGs would be identified at the APOE locus, the identification of meQTLs outside of this locus would be of particular interest. Should meQTLs outside of the APOE locus be found to be show non-random segregation with APOE ε4 vs. ε2 carrier status, these meQTL SNPs might contribute to the methylation differences observed in this study and APOE genotype effects more generally.
It was possible to assess meQTLs for 26 of the 31 CpGs of interest (from the DMP and DMR analyses); amongst these CpGs, 23 were associated with a meQTL. In total, 3727 significant CpG-SNP associations were identified for the 23 CpGs, involving 1654 unique SNPs (Fig. 3; Additional file 1: Table S6). Unsurprisingly, more than half of the meQTLs (n = 947) were located in a ~ 719 kb region (chr19: 45,004,645–45,723,446; GRCh37/hg19) spanning APOE. The APOE region meQTLs are associated with 16 CpGs, of which 14 are located at the APOE locus. None of these meQTLs is associated with all 16 CpGs: two are each associated with nine CpGs: rs7412, one of the APOE ε2/ε3/ε4-defining SNPs; and rs41290120, an intronic PVRL2 SNP that is in high linkage disequilibrium with rs7412 with D’ = 0.85 in the British population [46]. The two CpGs associated in trans with SNPs in the APOE region are cg16000331 in SREBF2 and cg19751789 in LDLR.

Circular plot indicating the locations of APOE ε4 vs. ε2 carrier-associated DMP and DMR CpGs. The first track shows a chromosome ideogram (hg19/GRCh37). The genomic locations of CpGs identified as being DMPs or in DMRs identified in APOE ε4 vs. ε2 carriers are indicated by blue lines on the second track and the meQTLs associated with these CpGs are indicated by the red lines on the third track. The connections between CpGs and meQTLs indicate regulatory relationships (cis interactions in red; trans interactions in blue). Gene symbols for genes located in each CpG/meQTL-harbouring region are indicated
Outside of the APOE locus, the remaining 707 meQTLs, which are associated with 10 CpGs, are located in 11 genomic regions (Fig. 3; Additional file 1: Table S7), with each region containing meQTLs associated with between one and eight CpGs-of-interest. To assess whether these meQTLs might contribute to APOE ε4 vs. ε2-associated methylation differences, their association with APOE ε4 vs. ε2 carrier status was assessed. No significant associations were observed, suggesting that the APOE ε4 vs. ε2-associated methylation differences are predominantly driven by genotype at the APOE locus.
To investigate potential trait/disease associations with variation in methylation levels at the CpGs-of-interest, the GWAS catalog was queried [43]. This identified 234/1654 meQTLs as having genome-wide significant associations with 316 traits (Additional file 1: Table S8). More than one third of the associations are with a lipid-related traits, including LDL, HDL and total cholesterol levels. As expected, many of the meQTL SNPs within the APOE locus have previously been associated with AD and related traits, such as “cerebrospinal fluid p-tau levels”, “cerebral amyloid deposition (PET imaging)” and “cognitive decline”. Interestingly, five SNPs located outside of the APOE locus have also been associated with traits related cognitive ability (“cognitive ability, years of educational attainment or schizophrenia (pleiotropy)”, “general cognitive ability”, “intelligence” and “self-reported math ability”). Four of these SNPs encompass the 3′ end of CCDC134 and most of the neighbouring SREBF2. Between them, these four SNPs are associated in cis with methylation at the four CpGs forming the two SREBF2 DMRs. The fifth SNP, which is located on chromosome 6 in the pseudogene CCDC162P, is associated with methylation at CpGs in SREBF2 and LDLR. Three meQTL SNPs have been associated with several age-related disorders (e.g. heart failure, stroke, and cancer) and endophenotypes of these disorders (including cholesterol levels, blood pressure and blood glucose) in a pleiotropic GWAS meta-analysis [47].
Discussion
We performed the first epigenome-wide comparison of DNA methylation between carriers of the APOE ε4 and ε2 haplotypes, which confer risk for and protection from AD, respectively. In large discovery and replication samples, we confirm the presence of APOE haplotype-associated methylation differences in APOE, demonstrate that differences in methylation at the APOE locus span a broad genomic locus encompassing several genes and find evidence for altered methylation at sites unlinked to the APOE locus. The observed methylation differences are located in a network of genes involved in lipid metabolism and homeostasis.
Methylation differences were identified using discovery, replication and meta-analysis EWASs and DMR analysis. Eight DMPs located on chromosome 19 in a ~ 169 kb region spanning from upstream of BCL3 to the APOE’s fourth exon showed replicated association. An additional twelve DMPs, eight of which are located in a ~ 233 kb region at the APOE locus, were identified by meta-analysing the discovery and replication samples. DMR analysis identified six regions of differential methylation, both within and outside of the APOE locus.
Within the APOE gene, two DMPs, cg06750524, in the second intron, and cg16471933, in the fourth exon, were identified. APOE ε4 carriers showed higher methylation levels at both. This observation directly replicates a previous study [21] and is in line with Foraker et al.’s observation of increased methylation of the APOE exon 4 CpG island in ε4 carriers [20]. Moreover, we have previously demonstrated [48] that the pattern of methylation in APOE in our sample is consistent with that described by Ma et al. [21]. Pairwise comparisons revealed differences in APOE methylation to be driven both by differences between ε2 carriers and ε3/ε3 homozygotes and ε4 carriers and ε3/ε3 homozygotes. One interpretation of this observation is that the spectrum of methylation at the APOE DMPs reflects the spectrum of AD risk conferred by different AD genotypes. It is clear, however, that additional, likely experimental, studies are required to assess the implications of the observed methylation pattern.
The differentially methylated CpGs at the APOE locus span a broad region that encompasses several genes containing AD-associated variants [49]. Long-ranging linkage disequilibrium in the region complicates the interpretation of association signals; however, conditional analysis and fine-mapping studies suggest the presence of multiple independent AD risk loci across the region [3, 49]. As such, the methylation differences observed in this study may be associated with variants that, whilst being in LD with the APOE ε2/ε4-defining SNPs, confer risk via different pathways to these SNPs. This notion is supported by the observation that SNPs that define an APOE ε4-independent AD-risk haplotype in PVRL2 [49] are highly significant meQTLs for the most significant DMP identified in this study.
Beyond the APOE locus, DMPs were identified in an ABCG1 intron, and upstream of DHCR24 and LDLR. Comparisons with ε3/ε3 homozygotes suggested the ε2 allele to be the primary driver of these differences, suggesting the possibility that altered methylation of genes involved in lipid metabolism might contribute to this allele’s protective effects. DMRs were identified in the gene body of SREBF2 and in the putative promoter region of LDLR. The CpGs involved in the DMPs and DMRs located outside of the APOE locus are associated with several meQTLs, with all of the CpGs except those involved in the LDLR DMR being associated with meQTLs in cis as well as in trans. Our findings did not, however, support a role for cis meQTLs for these CpGs driving associations with APOE ε4 vs. ε2 carrier status.
The genes outside of the APOE locus that harbour differentially methylated CpGs are implicated in lipid metabolism or homeostasis. ABCG1, which is highly expressed in the brain, encodes a cholesterol and phospholipid transporter and is involved in regulating the sterol biosynthetic pathway [50]. DHCR24, which encodes the cholesterol biosynthesis enzyme 3ß-hydroxysterol-∆24 reductase, also known as seladin-1, plays a neuroprotective role in AD-related stress conditions, including Aβ toxicity, oxidative stress and inflammation [51, 52]. The alteration of seladin-1 expression in mouse brain and human neuroblastoma cell cultures has been shown to affect β-secretase processing of amyloid precursor protein, with reduced seladin-1 being associated with an increased rate of Aβ production [53]. Future studies should assess whether methylation-associated differences in the brain expression of seladin-1 [6] might mediate the established associations between APOE ε4 vs. ε2 haplotype and Aβ production. The LDLR gene encodes the LDL receptor, one of the neuronal receptors capable of mediating the endocytosis of ApoE, thus maintaining brain cholesterol homeostasis. LDLR expression is regulated, in part, by SREBF2, a transcriptional regulator of sterol-regulated genes, which contains a SNP that is associated both with SREBF2 expression and CSF levels of the AD biomarkers Aβ and tau [54].
The link between APOE ε4 vs. ε2-associated methylation differences and lipid-related processes and pathways was further supported by GO and KEGG analyses, the identification of meQTLs for the differentially methylated CpGs, which were clustered in genomic regions that contain several lipid-related genes, and their GWAS-associated phenotypes. It would be of interest to investigate the mechanisms underlying the clustering of meQTLs in these genomic regions. Future studies might assess, for example, the extent to which meQTLs associated with the differentially methylated CpGs are enriched in these regions and whether they disproportionately affect certain sequence motifs. Previous EWASs have also identified associations between some of the APOE ε4 vs. ε2-associated CpGs and cholesterol levels: the DHCR24 (cg17901584), ABCG1 (cg06500161) and SREBF2 (cg16000331) DMPs have been associated with HDL cholesterol, total cholesterol and triglyceride levels [15, 55,56,57]. Comparisons with previous EWASs are, however, limited by the fact that the majority of previous EWASs used the 450 K array, which does not contain 10 of the APOE ε4 vs. ε2-associated CpGs.
As differences in lipid metabolism between carriers of the APOE ε4 and ε2 haplotypes are well-documented [6], we assessed whether variation in blood cholesterol levels might mediate the observed APOE ε4 vs. ε2-associated methylation differences. HDL cholesterol was found to be a partial mediator of the relationship between APOE ε4 vs. ε2 carrier status and methylation at three loci located outside of the APOE locus (two within ABCG1 and one in the promoter of DHCR24), thus suggesting one mechanism that might underlie these trans effects. Consistent with our observation that methylation differences at these loci appear to be predominantly driven by APOE ε2 carriers (when compared to APOE ε3/ε3 homozygotes), higher HDL cholesterol levels have been reported in carriers of APOE ε2 [58]. The effect of HDL cholesterol on methylation varied between the three loci, with APOE ε2 carriers showing increased methylation at the site located in the DHCR24 promoter and decreased methylation at the two ABCG1 sites. This suggests that increased HDL cholesterol levels do not exert a general effect on methylation but rather that methylation varies in a locus-specific manner in response to variation in HDL levels. It should be noted that an assumption of this analysis is that reverse causation does not exist between the outcome, methylation, and the mediator, cholesterol. Previous Mendelian randomisation studies have predominantly supported this premise [59, 60]; however, the ability to identify robust genetic instruments has limited both the number of methylation sites assessed and the ability to assess reverse causation. Limitations to the GS:SFHS cholesterol data should also be noted when interpreting these findings: triglyceride levels were not measured, preventing LDL cholesterol assessment, and blood samples were not taken at a consistent time of day or after fasting.
The cross-sectional nature of this study precludes the observed methylation differences being interpreted as conferring risk, protection or compensation. Comparison of methylation at these loci in APOE ε4 and ε2 carriers with AD would be useful in addressing this question; however, the optimum study design would involve the longitudinal assessment of the trajectory of ε4 vs. ε2-associated methylation differences in AD-free individuals in midlife who either do or do not later develop AD. These analyses are currently not feasible due to the small sizes of existing AD patient blood-based DNA methylation samples and insufficient follow-up time of large population-based samples.
Studies assessing the association of neuropathological hallmarks (neuritic plaque burden and/or neurofibrillary tangles) of AD with DNA methylation in the brain have not identified the loci identified in the present study [10, 12, 61]. Although the phenotypes assessed differ, the existence of APOE haplotype-associated differences in Aβ metabolism and tau phosphorylation [6] suggest that some degree of overlap might be expected. The neuropathological hallmarks of AD are, however, complex phenotypes and APOE haplotype will be one of many contributing factors (De Jager et al. [10] reported that APOE ε4 could account for 13.9% of the variance in NP burden observed in their participants). In addition, the smaller samples assessed by De Jager et al. [10], Lunnon et al. [12] and Smith et al. [61] may have been inadequately powered to detect any methylation differences driven by APOE haplotype. Differences in age and methylation profiling platform are also likely to limit comparability: the participants assessed in these studies were much older (mean age > 75 years) than those assessed in our study (mean age ~ 50 years) and array differences mean that only two thirds of our DMP/DMR probes were assessed. Two important corollaries of the age difference are that brain-based studies are more likely to (i) suffer from survivor bias and (ii) be better suited to investigating end-of-disease processes. It is also important to note that APOE is involved in multiple processes, with APOE ε4 conferring risk for AD, at least in part, via mechanisms that are not related to Aβ or tau pathology. A recent study has indicated that APOE ε4-associated breakdown of the blood-brain barrier in the hippocampus and medial temporal lobe contributes to APOE ε4-associated cognitive decline independently of Aβ or tau [62].
The blood provides an easily accessible tissue that can be repeatedly sampled to characterise pre-morbid markers of risk. The extent to which it can provide mechanistic insights into diseases that are considered predominantly brain-based, however, is a perennial subject of debate. Cis meQTL effects tend to be highly correlated (r = 0.78) between the blood and the brain [63], supporting the use of the blood to study the effects of genetic risk factors for brain-based diseases. It is also important to note the increasing recognition of the role of peripheral processes in conferring risk for AD [64]. As the blood provides a conduit by which many circulating factors (e.g. plasma proteins and microbial metabolites) reach the brain and affect brain ageing [65], assessing DNA methylation in the blood is likely to be informative regarding systemic factors contributing to AD pathogenesis. Although APOE is synthesised separately in the blood and the brain and neither APOE nor cholesterol can cross the blood-brain barrier [66, 67], there is cross-talk between brain and blood cholesterol via oxysterols [67], levels of which vary by APOE ε2/ε3/ε4 haplotype [68]. Peripheral hypercholesterolemia has been associated with increased oxysterol levels in the brain, which have been implicated in with production and accumulation of Aβ, increased neuroinflammation and neuronal death [67].
The association between APOE genotype and AD varies between populations [5], with studies in populations of Hispanic and African ancestry often reporting attenuated effect sizes for the ε4 allele compared to studies involving European and Asian participants [69, 70]. Moreover, Rajabli et al. [70] have shown that genetic variation local to APOE is likely to confer protection from the effects of the ε4 allele in individuals of African ancestry. As the participants in the present study were of European ancestry, it should be noted that these findings are likely to be European-specific and future studies should assess their generalisability and relevance to AD pathogenesis in other populations.
Conclusions
This is the first study to characterise epigenome-wide DNA methylation differences between carriers of APOE ε4 and ε2. In AD-free individuals, we identified several methylation differences both at the APOE locus and in the rest of the genome, which converge on lipid-related pathways. Strengths of the study include the large samples available for EWAS analysis, the epigenome-wide approach, the use of a well-phenotyped cohort with genotype data and the avoidance of reverse causation by studying AD-free participants. Future studies should investigate the causal relationship between APOE genotype, DNA methylation and lipid-related processes and their role in AD pathogenesis.
Availability of data and materials
According to the terms of consent for GS:SFHS, individual-level data (‘omics and phenotypes) cannot be made publically available. The data that support the findings of this study are, however, available upon reasonable request and with permission of the GS Access Committee (access@generationscotland.org).
Abbreviations
- Aβ:
-
Amyloid beta
- AD:
-
Alzheimer’s disease
- CI:
-
Confidence interval
- DMP:
-
Differentially methylated position
- DMR:
-
Differentially methylated region
- EWAS:
-
Epigenome-wide association study
- GS:SFHS:
-
Generation Scotland: Scottish Family Health Study
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- PC:
-
Principal component
- SNP:
-
Single-nucleotide polymorphism
References
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–13.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–56.
Safieh M, Korczyn AD, Michaelson DM. ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019;17(1):64.
Tzioras M, Davies C, Newman A, Jackson R, Spires-Jones T. Invited review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2019;45(4):327–46.
Qian J, Wolters FJ, Beiser A, Haan M, Ikram MA, Karlawish J, et al. APOE-related risk of mild cognitive impairment and dementia for prevention trials: an analysis of four cohorts. PLoS Med. 2017;14(3):e1002254.
Babenko VN, Afonnikov DA, Ignatieva EV, Klimov AV, Gusev FE, Rogaev EI. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci. 2018;19(Suppl 1):16.
De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17(9):1156–63.
Gasparoni G, Bultmann S, Lutsik P, Kraus TFJ, Sordon S, Vlcek J, et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin. 2018;11(1):41.
Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17(9):1164–70.
Slieker RC, Relton CL, Gaunt TR, Slagboom PE, Heijmans BT. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin. 2018;11(1):25.
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541(7635):81–6.
Hedman AK, Mendelson MM, Marioni RE, Gustafsson S, Joehanes R, Irvin MR, et al. Epigenetic patterns in blood associated with lipid traits predict incident coronary heart disease events and are enriched for results from genome-wide association studies. Circ Cardiovasc Genet. 2017;10(1):e001487.
Singmann P, Shem-Tov D, Wahl S, Grallert H, Fiorito G, Shin SY, et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics Chromatin. 2015;8:43.
Galanter JM, Gignoux CR, Oh SS, Torgerson D, Pino-Yanes M, Thakur N, et al. Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures. Elife. 2017;6:e20532.
Yuan V, Price EM, Del Gobbo G, Mostafavi S, Cox B, Binder AM, et al. Accurate ethnicity prediction from placental DNA methylation data. Epigenetics Chromatin. 2019;12(1):51.
Yu CE, Cudaback E, Foraker J, Thomson Z, Leong L, Lutz F, et al. Epigenetic signature and enhancer activity of the human APOE gene. Hum Mol Genet. 2013;22(24):5036–47.
Foraker J, Millard SP, Leong L, Thomson Z, Chen S, Keene CD, et al. The APOE gene is differentially methylated in Alzheimer’s disease. J Alzheimers Dis. 2015;48(3):745–55.
Ma Y, Smith CE, Lai CQ, Irvin MR, Parnell LD, Lee YC, et al. Genetic variants modify the effect of age on APOE methylation in the Genetics of Lipid Lowering Drugs and Diet Network study. Aging Cell. 2015;14(1):49–59.
Smith BH, Campbell A, Linksted P, Fitzpatrick B, Jackson C, Kerr SM, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int J Epidemiol. 2013;42(3):689–700.
Smith BH, Campbell H, Blackwood D, Connell J, Connor M, Deary IJ, et al. Generation Scotland: the Scottish Family Health Study; a new resource for researching genes and heritability. BMC Med Genet. 2006;7:74.
Amador C, Huffman J, Trochet H, Campbell A, Porteous D, Generation S, et al. Recent genomic heritage in Scotland. BMC Genomics. 2015;16:437.
Kerr SM, Campbell A, Murphy L, Hayward C, Jackson C, Wain LV, et al. Pedigree and genotyping quality analyses of over 10,000 DNA samples from the Generation Scotland: Scottish Family Health Study. BMC Med Genet. 2013;14:38.
Barbu MC, Shen X, Walker RM, Howard DM, Evans KL, Whalley HC, Porteous DJ, Morris SW, Deary IJ, Zeng Y, Marioni RE, Clarke TK, McIntosh AM. Epigenetic prediction of major depressive disorder. Mol Psychiatry. 2020.
Madden RA, McCartney DL, Walker RM, Hillary RF, Bermingham ML, Rawlik K, Morris SW, Campbell A, Porteous DJ, Deary IJ, Evans KL, Hafferty J, McIntosh AM, Marioni RE. Birth weight associations with DNA methylation differences in an adult population.Epigenetics. 2020:1–14.
Bermingham ML, Walker RM, Marioni RE, Morris SW, Rawlik K, Zeng Y, et al. Identification of novel differentially methylated sites with potential as clinical predictors of impaired respiratory function and COPD. EBioMedicine. 2019;43:576–86.
Team RC. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2019.
Team RC. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2020.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82.
Saffari A, Silver MJ, Zavattari P, Moi L, Columbano A, Meaburn EL, et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol. 2018;42(1):20–33.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–1.
Lenth RV. Least-squares means: the R package lsmeans. J Stat Softw. 2016;69(1):1–33.
Suderman M, Staley JR, French R, Arathimos R, Simpkin A, Tilling K. dmrff: identifying differentially methylated regions efficiently with power and control. bioRxiv. 2018:508556.
Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32(2):286–8.
Tingley D, Yamamoto T, Hirose K, Keele L, Imai K. mediation: R package for causal mediation analysis. Journal of Statistical Software. 2014;59(5):1–38.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–83.
Nagy R, Boutin TS, Marten J, Huffman JE, Kerr SM, Campbell A, et al. Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017;9(1):23.
Zeng Y, Amador C, Xia C, Marioni R, Sproul D, Walker RM, et al. Parent of origin genetic effects on methylation in humans are common and influence complex trait variation. Nat Commun. 2019;10(1):1383.
Haller T, Kals M, Esko T, Magi R, Fischer K. RegScan: a GWAS tool for quick estimation of allele effects on continuous traits and their combinations. Brief Bioinform. 2015;16(1):39–44.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D12.
Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjalmsson BJ, Finucane HK, Salem RM, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47(3):284–90.
Maas SCE, Vidaki A, Wilson R, Teumer A, Liu F, van Meurs JBJ, et al. Validated inference of smoking habits from blood with a finite DNA methylation marker set. Eur J Epidemiol. 2019;34(11):1055–74.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–7.
He L, Kernogitski Y, Kulminskaya I, Loika Y, Arbeev KG, Loiko E, et al. Pleiotropic meta-analyses of longitudinal studies discover novel genetic variants associated with age-related diseases. Front Genet. 2016;7:179.
Mur J, McCartney DL, Walker RM, Campbell A, Bermingham ML, Morris SW, Porteous DJ, McIntosh AM, Deary IJ, Evans KL, Marioni RE. DNA methylation in APOE: The relationship with Alzheimer's and with cardiovascular health. Alzheimers Dement. 2020;6(1):e12026.
Zhou X, Chen Y, Mok KY, Kwok TCY, Mok VCT, Guo Q, et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat Commun. 2019;10(1):3310.
Burgess BL, Parkinson PF, Racke MM, Hirsch-Reinshagen V, Fan J, Wong C, et al. ABCG1 influences the brain cholesterol biosynthetic pathway but does not affect amyloid precursor protein or apolipoprotein E metabolism in vivo. J Lipid Res. 2008;49(6):1254–67.
Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T, Behl C, et al. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J Neurosci. 2000;20(19):7345–52.
Martiskainen H, Paldanius KMA, Natunen T, Takalo M, Marttinen M, Leskela S, et al. DHCR24 exerts neuroprotection upon inflammation-induced neuronal death. J Neuroinflammation. 2017;14(1):215.
Crameri A, Biondi E, Kuehnle K, Lutjohann D, Thelen KM, Perga S, et al. The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J. 2006;25(2):432–43.
Picard C, Julien C, Frappier J, Miron J, Theroux L, Dea D, et al. Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol Aging. 2018;66:180 e1–9.
Braun KVE, Dhana K, de Vries PS, Voortman T, van Meurs JBJ, Uitterlinden AG, et al. Epigenome-wide association study (EWAS) on lipids: the Rotterdam Study. Clin Epigenetics. 2017;9:15.
Pfeiffer L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S, et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet. 2015;8(2):334–42.
Sayols-Baixeras S, Subirana I, Lluis-Ganella C, Civeira F, Roquer J, Do AN, et al. Identification and validation of seven new loci showing differential DNA methylation related to serum lipid profile: an epigenome-wide approach. The REGICOR study. Hum Mol Genet. 2016;25(20):4556–65.
Tan CE, Tai ES, Tan CS, Chia KS, Lee J, Chew SK, et al. APOE polymorphism and lipid profile in three ethnic groups in the Singapore population. Atherosclerosis. 2003;170(2):253–60.
Dekkers KF, van Iterson M, Slieker RC, Moed MH, Bonder MJ, van Galen M, et al. Blood lipids influence DNA methylation in circulating cells. Genome Biol. 2016;17(1):138.
Sayols-Baixeras S, Tiwari HK, Aslibekyan SW. Disentangling associations between DNA methylation and blood lipids: a Mendelian randomization approach. BMC Proc. 2018;12(Suppl 9):23.
Smith RG, Pishva E, Shireby G, Smith AR, Roubroeks JAY, Hannon E, et al. Meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights 220 differentially methylated loci across cortex. bioRxiv. 2020:2020.02.28.957894.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9(1):2282.
Morris G, Berk M, Maes M, Puri BK. Could Alzheimer’s disease originate in the periphery and if so how so? Mol Neurobiol. 2019;56(1):406–34.
Pluvinage JV, Wyss-Coray T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat Rev Neurosci. 2020;21:93–102.
Liu M, Kuhel DG, Shen L, Hui DY, Woods SC. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(9):R903–8.
Gamba P, Testa G, Gargiulo S, Staurenghi E, Poli G, Leonarduzzi G. Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front Aging Neurosci. 2015;7:119.
Jenner AM, Lim WL, Ng MP, Wenk MR, Shui G, Sharman MJ, et al. The effect of APOE genotype on brain levels of oxysterols in young and old human APOE epsilon2, epsilon3 and epsilon4 knock-in mice. Neuroscience. 2010;169(1):109–15.
Blue EE, Horimoto A, Mukherjee S, Wijsman EM, Thornton TA. Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement. 2019;15(12):1524–32.
Rajabli F, Feliciano BE, Celis K, Hamilton-Nelson KL, Whitehead PL, Adams LD, et al. Ancestral origin of ApoE epsilon4 Alzheimer disease risk in Puerto Rican and African American populations. Plos Genet. 2018;14(12):e1007791.
Acknowledgements
We are grateful to all the families who took part, the general practitioners and the Scottish School of Primary Care for their help in recruiting them and the whole Generation Scotland team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, healthcare assistants and nurses.
Funding
This work was supported by a Wellcome Trust Strategic Award “STratifying Resilience and Depression Longitudinally” (STRADL) [104036/Z/14/Z] to AMM, KLE, CSH, DJP and others and an MRC Mental Health Data Pathfinder Grant [MC_PC_17209] to AMM and DJP. REM is supported by an Alzheimer’s Research UK major project grant [ARUK-PG2017B-10]. KV is funded by the Wellcome Trust Translational Neuroscience PhD Programme at the University of Edinburgh [108890/Z/15/Z]. ADB would like to acknowledge funding from the Wellcome PhD training fellowship for clinicians [204979/Z/16/Z], the Edinburgh Clinical Academic Track (ECAT) programme. Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates [CZD/16/6] and the Scottish Funding Council [HR03006]. Genotyping of the GS:SFHS samples was carried out by the Genetics Core Laboratory at the Clinical Research Facility, Edinburgh, Scotland, and was funded by the UK’s Medical Research Council and the Wellcome Trust [104036/Z/14/Z]. DNA methylation profiling of the GS:SFHS samples was funded by the Wellcome Trust Strategic Award [10436/Z/14/Z] with additional funding from a 2018 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation [27404].
Author information
Authors and Affiliations
Contributions
Conception and design: RMW, KLE, REM; data analysis: RMW, KV, MLB, ADB, CH. Drafting the article: RMW and KLE; data preparation: RMW, MLB, SWM, KR, AC, ADB, YZ, CA; data collection: AMM, KLE, CSH, DJP. Revision of the article: RMW, KV, MLB, ADB, YZ, CA, AC, CSH, DJP, REM, KLE; all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study is part of GS:SFHS, which obtained ethical approval from the NHS Tayside Committee on Medical Research Ethics, on behalf of the National Health Service (reference: 05/S1401/89). All participants provided broad and enduring written informed consent for biomedical research. GS:SFHS has Research Tissue Bank Status (reference: 15/ES/0040), providing generic ethical approval for a wide range of uses within medical research. All experimental methods were in accordance with the Helsinki declaration.
Consent for publication
Not applicable
Competing interests
AMM has received grant support from Pfizer, Eli Lilly, Janssen and The Sackler Trust. These sources are not connected to the current investigation. AMM has also received speaker fees from Janssen and Illumina. The remaining authors declare that they have no competing interests. The funding bodies did not play any role in the design of the study, the collection, analysis or interpretation of the data or in the writing of the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1:
Sample demographic information for the discovery and replication samples and the individual APOE genotype groups within the discovery sample. Table S2. Differentially methylated positions (DMPs) showing replicated association with APOE ε4 vs. ε2 carrier status and sensitivity analyses results. Table S3. Pairwise comparison of DNA methylation levels between APOE genotypes for each of the 20 CpGs identified as being a DMP in the meta-analysis of the discovery and replication samples. Table S4. Gene ontology (GO) terms showing significant enrichment (P ≤ 2.21 × 10−6) for the CpG sites identified as showing suggestive (P ≤ 1 × 10−5) association with APOE ε4 vs. ε2 carrier status through DMP analysis or which contributed to a significant DMR. Table S5. Assessment of total, HDL or non-HDL cholesterol as potential mediators of APOE ε4 vs. ε2 carrier status on methylation. Table S6. meQTLs for the APOE ε4 vs. ε2-associated DMPs and DMR sites. Table S7. meQTLs for APOE ε4 vs. ε2-associated DMPs and DMR sites grouped by genomic region. Table S8. GWAS catalogue entries for the meQTLs associated with the APOE ε4 vs. ε2-associated DMPs and DMR sites.
Additional file 2: Fig. S1.
Scree plot showing the eigenvalues of the first 50 genetic principal components for the Generation Scotland: Scottish Family Health Study. Fig. S2. The genomic region encompassing APOE, which contains 16 of the APOE ε4 vs. ε2-associated DMPs identified by meta-analysis (chr19: 45,221,584 – 45,454,752; GRCh37/hg19). Fig. S3. Bar charts showing the mean methylation levels (M-values) in the discovery sample for the 20 meta-DMPs split by APOE genotype (ε2/ε2, ε2/ε3, ε3/ε3, ε3/ε4, and ε4/ε4). Fig. S4. The genomic regions containing the six identified DMRs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Walker, R.M., Vaher, K., Bermingham, M.L. et al. Identification of epigenome-wide DNA methylation differences between carriers of APOE ε4 and APOE ε2 alleles. Genome Med 13, 1 (2021). https://doi.org/10.1186/s13073-020-00808-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-020-00808-4