
Abstract
Background
Although the tick-borne pathogen Anaplasma phagocytophilum is currently described as a single species, studies using genetic markers can distinguish groups of variants associated with different hosts, pathogenicity, zoonotic potential and biotic and geographic niches. The objective of our study was to investigate the genetic diversity of A. phagocytophilum and Ixodes ricinus ticks attached to people.
Methods
In collaboration with a commercial diagnostic company, a total of 52 DNA samples were obtained from ticks that tested positive for A. phagocytophilum by quantitative PCR. The genetic profile of each sample was determined using the groEL and ankA genes. Identification of the tick species was confirmed by partial sequencing of the COI subunit and a portion of the TROSPA gene.
Results
All 52 ticks were identified as I. ricinus. Two protocols of nested PCR amplifying 1293- and 407-bp fragments of groEL of A. phagocytophilum yielded amplicons of the expected size for all 52 samples. Among all sequences, we identified 10 unique genetic variants of groEL belonging to ecotype I and ecotype II. The analysis targeting ankA was successful in 46 of 52 ticks. Among all sequences, we identified 21 unique genetic variants phylogenetically belonging to three clusters.
Conclusions
Our results indicate that ticks attached to people harbor distant genetic variants of A. phagocytophilum, some of which are not recognized as zoonotic. Further studies are needed to determine the risk of human infection by genetic variants other than those designated as zoonotic.
Graphical Abstract

Similar content being viewed by others


Background
Anaplasma phagocytophilum is a Gram-negative bacterium responsible for human granulocytic anaplasmosis (HGA) in humans, tick-borne fever (TBF) in ruminants, equine granulocytic anaplasmosis (EGA) in horses and granulocytic anaplasmosis in dogs and cats [1]. In the Palearctic region, the dominant genetic variants of A. phagocytophilum are transmitted by Ixodes ricinus and Ixodes persulcatus, while in the Nearctic region Ixodes scapularis and Ixodes pacificus are the known vectors of this bacterium [2, 3].
Genetic variants of A. phagocytophilum in North America and Europe differ in host preference and clinical symptoms, with TBF and EGA dominant in Europe and HGA dominant in North America [4, 5]. The genetic diversity of European strains of A. phagocytophilum of different origins has been demonstrated by phylogenetic analyses of genes such as groEL [6,7,8], ankA [4] and msp4 [9]. Different target genes led to different names of the genetic variants and consequently different terminology, such as ecotype (groEL), cluster (ankA and groEL) and genotype (msp4) [4, 10].
The heat shock operon groESL contains two genes encoding the chaperone proteins groES and groEL, respectively, as well as the intergenic region. A 5′ fragment of groEL has been widely used for genotyping A. phagocytophilum, especially those from Europe [11,12,13]. Currently, based on groEL genotyping studies, four ecotypes and eight clusters with different pathogenicity and geographical origin are distinguished [7, 8].
The variable ankA gene encodes the ankyrin repeat-containing protein AnkA (153–160 kDa). The ankA differentiates variants corresponding with the species of their animal hosts and exhibits higher sequence variability compared to that of 16S ribosomal DNA, groEL and msp4 [3]. Studies have found an association among genetic variants and vertebrate hosts, tick vectors and geographic locations such that regardless of the gene used for analysis, infected humans, whether in Europe or the Americas, appear to share related strains belonging to the same genetic group [5, 14].
The number of HGA cases reported in the USA has been steadily increasing since reporting of the disease was initiated, increasing from 348 in 2000 to 5762 cases in 2017 (https://www.cdc.gov/anaplasmosis/stats/index.html). In Europe, the total annual number of HGA cases has not exceeded 300 [2, 15]. Several HGA cases have also been reported in Canada, Russia, China, Taiwan, South Korea and Japan (for review, see [3]). According to the National Institute of Public Health in the Czech Republic, 53 cases of HGA were reported between 2007 and 2017; however, in the seroprevalence study from 2014, specific antibodies were detected in 34 of 314 individuals tested [16]. More recently, 12.6% of 103 patients with clinical symptoms persisting after antibiotic treatment of diagnosed Lyme disease were positive for A. phagocytophilum immunoglobulin G antibodies [17]. Similar rates of seroprevalence were reported in other European countries, including Norway, Sweden and Poland, where Anaplasma antibodies were detected in 11.0%, 12.0% and 11.8% of the general population, respectively [17]. The disease is likely to be greatly underdiagnosed due to its nonspecific flu-like symptoms, such as fever, headache and myalgias, which usually resolve without treatment [1]. If the immune system fails and the infection is left untreated, the disease can cause life-threatening symptoms, such as respiratory failure, severe gastrointestinal bleeding, renal failure and liver damage [18]. Although the number of surveillance reports of A. phagocytophilum in tick vectors is increasing worldwide, the epidemiological risk of infection by this pathogen is underestimated [19]. Detection of pathogens in blood-feeding ticks shows the risk of human exposure better than studies on foraging ticks [20]. In this context, the main objective of our study was to investigate the genetic diversity of A. phagocytophilum in ticks attached to people in the Czech Republic nationwide.
Methods
Tick identification
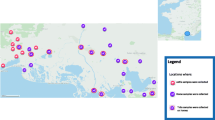
A total of 52 DNA samples of ticks that tested positive for A. phagocytophilum by quantitative PCR [21] and were attached to people at collection were acquired in collaboration with a commercial diagnostic company that offered pathogen detection in ticks sent by the general public (Fig. 1). Tick species, developmental stage and sex were determined by the specialist from the collaborating institution. Samples were collected in the period 2011–2020, and each sample was individually homogenized, following which nucleic acids were isolated by the ExiPrep Plus Viral DNA/RNA Kit using the Exiprep 16 Plus nucleic acid extraction system (Bioneer, Daejeon, Republic of Korea).

Map of the collection sites of the 46 samples of ticks that tested positive for Anaplasma phagocytophilum across the Czech Republic. The collection site for 6 ticks was not known. Numbers in parentheses represent the number of ticks from the same locality
The identification of the tick species was confirmed by partial sequencing of the cytochrome c oxidase I subunit gene (COI) and the intron part of the TROSPA gene, as described by [22]. The amplification of the COI was performed in a total reaction volume of 20.0 µl comprising the Phusion Green Hot Start II High-Fidelity PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions at an annealing temperature of 55 °C. Amplification of TROSPA was performed in a total reaction volume of 25.0 µl comprising 2× PCR BIO Taq Mix Red (PCR Biosystems Ltd., London, UK), following the manufacturer instructions at an annealing temperature of 65 °C. Amplicons were visualized, processed and sequenced as described in the following section. Sequence identity was determined by BLASTn analyses of the GenBank database at NCBI.
Amplification of groEL and ankA
To determine the groEL ecotype, nested PCR was performed targeting a 407-bp portion of the variable fragment of groEL, as described previously [7, 23]. Whenever possible, a longer fragment of groEL (1297 bp) was amplified following the nested PCR protocol [6] using slightly modified primers [24]. Subsequently, the samples were analyzed using two nested PCR assays aimed at the ankA gene (Additional file 1: Table S1). The first assay (herein called protocol 1) amplified an approximately 530-bp fragment of the ankA gene, and the second assay (protocol 2) amplified an approximately 500 bp. All PCRs were performed using the commercial master mix (2× PCR BIO Taq Mix Red; PCR Biosystems Ltd.) following the manufacturer’s instructions. A total volume of 25.0 μl was prepared for each reaction, comprising 12.5 μl of the master mix, 10.0 pmol of each primer, 2.0 μl of template DNA or 1.0 μl of PCR product from the first round in the case of nested PCR, with PCR water used for the remaining volume. All PCR products were visualized on an 1.5% agarose gel with the Midori Green Advance DNA System (Nippon Genetics Europe GmbH, Düren, Germany). Products of the expected size were purified using the Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech Ltd., New Taipei City, Taiwan) and sequenced at the Macrogen capillary sequencing services (Macrogen Europe, Amsterdam, the Netherlands) using the amplification primers.
Sequence and phylogenetic analysis
All sequences were assembled, edited and analyzed using the Geneious Prime® software version 2022.0.1 and compared to those available in the GenBank database by BLASTn analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi). For individual ticks, wherever possible, sequences from two separate assays that targeted overlapping regions of the ankA gene were assembled into a single sequence (approx. 800 bp). One sample that yielded mixed chromatogram signals was cloned using the pGEM®-T Easy Vector System (Promega, Madison, WI, USA). Cloned plasmid DNA was purified from the bacterial culture using the GenElute™ Plasmid Miniprep Kit (Sigma-Aldrich, St. Louis, MO, USA) and sequenced using the universal SP6 primer.
The phylogeny was computed separately for groEL and ankA. For groEL, phylogenetic analysis was performed on 87 sequences from the GenBank representing all four described ecotypes [8] and eight clusters [7] together with 10 unique sequences from this study and two sequences of Anaplasma platys used as an outgroup. The ankA gene phylogenetic analysis was performed on 74 sequences from the GenBank representing different clusters together with 21 unique sequences from this study and sequences of Anaplasma marginale as an outgroup.
Due to an uneven length of sequences, the alignments were calculated in two steps by the MAFFT algorithm, using “Auto” strategy for sequences > 1000 nt and function –add for implementing sequences < 1000 nt to the alignment. The phylogenetic trees were inferred by the maximum likelihood method by IQTREEv. 1.6.5 [25]. The best-fit evolution model was selected based on the Bayesian information criterion (BIC) computed by implemented ModelFinder [26]. Branch supports were assessed by the ultrafast bootstrap (UFBoot) approximation [27] and by the SH-like approximate likelihood ratio test (SH-aLRT) [28]. Trees were visualized and edited in FigTree v1.4.1 and Inkscape 0.91. The map of localities was constructed in the Datawrapper online software (https://app.datawrapper.de/).
Results
Ticks were identified by morphology and sequencing of TROSPA and COI (data not shown). All 52 ticks were identified as I. ricinus and no other Ixodes species, including I. inopinatus, were found. Two protocols of nested PCR, amplifying 1293- and 407-bp fragments of groEL, respectively, yielded amplicons of the expected size for all 52 samples: 19 (6.3%) adults (14 females, 5 males, respectively) and 31 (10.3%) nymphs (Table 1); for two ticks, data on sex and stage development were not available. In total, eight short and 44 long sequences of groEL were obtained. Inspection of the chromatograms did not reveal any multiple peaks suggestive of mixed infections. Among all sequences, we identified 10 unique genetic variants with 97.9–99.9% sequence identity. The main variant (GT15) was detected in 39 individuals (75%), followed by four other variants, each detected in two ticks. The remaining five genetic variants were represented by a single individual (Fig. 1; Table 2). BLAST analysis was performed for each genetic variant. Representative sequences were deposited to the NCBI GenBank database under the accession numbers OP265397-OP265406.
The analysis targeting the ankA gene was successful in 46 of 52 ticks, with PCR yielding products of the expected size in 38 (455–530 bp) and 33 (470–574 bp) ticks using protocol 1 and 2, respectively (Fig. 1; Table 2). In 24 ticks, products from both assays were obtained, which enabled the assembly of longer sequence. Inspection of chromatograms of the ankA gene revealed multiple peaks, indicating mixed infection in one tick (GT213). Sequencing of five clones from this individual tick yielded two A. phagocytophilum variants with 98.7% identity (differences observed in 7 nt: 3 silent and 4 missense mutations). Among all sequences, we identified 21 unique genetic variants with 62.0–99.9% sequence identity. Representative sequences from the study were deposited to the NCBI GenBank database under the accession numbers OP265407-OP265426.
Phylogenetic analysis
In the present study, we followed the classification of A. phagocytophilum based on the partial groEL gene sequences, introduced by Jahfari et al. [8] and extended by Jaarsma et al. [7]. In the phylogenetic analysis, three well-supported clades were distinguished. The largest clade consists of two ecotypes, I and II (Fig. 2). The ecotype I is monophyletic and contains a wide range of isolates from the USA and Europe collected from humans, horses, dogs and domestic ruminants as well as wild boar and urban wildlife. Ecotype II consists of two monophyletic clusters with a high level of bootstrap support. However, ecotype II is not monophyletic, and clusters III and IV clearly differ in geographical distribution, host and vector species preference. Sequences obtained from specimens from cluster III originating from Europe were found in I. ricinus and deer. Cluster IV contains specimens from Asia, including isolates from a human, I. persulcatus and rodents (Additional file 2: Fig. S1). All sequences from our study fell into this largest clade (Fig. 2). Forty-five isolates were in two clusters belonging to ecotype I, and the remaining six isolates belong to cluster III within ecotype II (Fig. 2; Additional file 2: Fig. S1).

Maximum likelihood phylogenetic tree based on the groEL gene ( groEL) of A. phagocytophilum (a). The bootstrap values (SH-aLRT/UFB) above the 80/95 threshold are displayed. Sequences of Anaplasma platys used as an outgroup are not displayed. The designation of the groEL clusters and ecotypes is given based on Jaarsma et al. [7] and Jahfari et al. [8], respectively. The highlighted clusters representing the ecotype I and ecotype II are displayed in detail in (b) where sequences acquired from the GenBank database are marked by their accession number and the host. Sequences obtained from this study are shown in red. SH-aLRT/UFB, SH-like approximate likelihood ratio test
The second well distinguishable clade consists of isolates of ecotype III with clusters V and VI. Cluster V is represented by sequences from rodents and Ixodes trianguliceps from Europe and Western Siberia, and cluster VI is composed of sequences from rodents and I. persulcatus from Asia. These two clusters are closely related to cluster VIII [7], representing isolates from Ixodes ventalloi exclusively. The last clade is represented by a small number of sequences in ecotype IV/ cluster VII. None of the genetic variants detected in our study belonged to ecotype III and IV.
To describe the results of phylogenetic analysis based on the ankA gene, we followed the classification used by Rar et al. [3]. In our analysis, three well-supported clades were formed. The first clade consists of six clusters originating from Europe, Asia and the USA (Fig. 3; Additional file 3: Fig. S2). The specimens originating from the USA are present in clusters 11 and 12. These clusters are sister groups to ankA cluster 1. European cluster 1 is the most diverse and contains all of the sequences from humans, small, medium and large mammals and I. ricinus. Thirty-nine of our sequences were placed in this cluster 1 (Table 2). The Asian group is referred to as cluster 8 and is represented by the sequences obtained from hybrids of I. persulcatus × Ixodes pavlovskyi. The ankA clusters 2 and 3, also containing A. phagocytophilum of the European origin, are mainly represented by specimens from wild and domestic ruminants and I. ricinus. Four of our sequences were classified in cluster 2.

Maximum likelihood phylogenetic tree based on the ankA gene (ankA) of A. phagocytophilum (a). The bootstrap values (SH-aLRT/UFB) above the 80/95 threshold are displayed. The sequence of A. marginale used as an outgroup is not displayed. The designation of ankA clusters is given according to Langenwalder et al. [29] and Rar et al. [3]. The highlighted clades representing clusters 1, 2, 4 and 7 are shown in detail in b). Sequences acquired from the GenBank database are marked by their accession number and the host. Sequences from this study are shown in red
The second, well supported clade consists of five clusters. Cluster 9 is represented by a single sequence from a tick collected from a woodchat shrike (Lanius senator) and it is related to clusters 4 and 7 with sequences from wild and domestic ruminants and I. ricinus from Europe (Fig. 3; Additional file 3: Fig. S2). Two of our sequences belong to cluster 4, and two sequences from a single cloned sample fall into cluster 7. The other branch of the second clade is formed by clusters 5 and 10 from Asia and Europe. Sequences belonging to these clusters have been detected in I. persulcatus, I. pavlovskyi, their natural hybrids and rodents (Fig. 3).
The third and smallest clade contains only a single cluster 6 with sequences from birds and Ixodes spp. from birds (Fig. 3; Additional file 3: Fig. S2).
Discussion
To our knowledge, this is the first elaborated study demonstrating the genetic diversity of A. phagocytophilum in ticks attached to humans based on groEL and ankA together. Little data are available on A. phagocytophilum detected in ticks feeding on humans, with current information coming from studies in Poland [30], Italy [31, 32], Romania [33] and Scotland [34]. In Europe, A. phagocytophilum is transmitted mainly by the tick I. ricinus. This tick has three feeding developmental stages, each with different host preferences, which in turn affects the range of pathogens ingested during each blood meal. Consequently, later developmental stages are more likely to carry the pathogens. The three-host life strategy of I. ricinus affects the transmission of A. phagocytophilum, which is shown in our study as well as in a study performed on questing ticks [31].
Despite the increasing number of studies on the genetic diversity of A. phagocytophilum, there is still insufficient data for a clear understanding of the geographic distribution, host preferences and pathogenicity to humans of the different genetic variants [24]. During the last decade, multilocus approaches gained attention as a promising tool that can help gain a better understanding of the epidemiology of A. phagocytophilum infections. In our study, we chose two genes and performed both analyses on the same samples. The association between ankA and groEL clusters was recently suggested by Rar et al. [3], who described concordance between specific clusters/ecotypes. In our study, 10 groEL and 21 ankA unique variants were detected, and subsequent phylogenies showed only a partial correlation between groEL ecotypes and ankA clusters. The groEL ecotype I has the broadest host range, and the vector I. ricinus has a wide distribution in the western Palearctic region and includes all genetically characterized strains detected from humans in Europe. Based on our results, groEL ecotype I genetic variants are represented in ankA clusters 1, 4 and 7, whereas groEL ecotype II corresponds to ankA clusters 2 and 4; these results support the hypothesis put forward by Rar et al. [3]. However, it should be noted that ankA cluster 4 consists of variants of A. phagocytophilum belonging to groEL ecotypes I and II. The ankA gene may be a virulence factor, and it has been suggested that it is involved in host adaptation underlying diversifying selection [2]. This may have direct implications for the observed genetic variability of this gene in A. phagocytophilum. The other explanation for the observed discrepancy is possible co-infection of different genetic variants of A. phagocytophilum in a tick sample. However, with the exception of one tick, double peaks indicative of co-infection were not observed in our study. Recent results characterizing tick-pathogen molecular interactions support the hypothesis that A. phagocytophilum has evolved common mechanisms to establish infections in tick vectors and vertebrate hosts, due to its adaptation to a variety of vector and reservoir species [7]. The mechanisms that A. phagocytophilum uses to infect and replicate in tick and vertebrate hosts are still largely unknown, and the interactions between different genetic variants of A. phagocytophilum are not fully explored [35]. In our study, we found ticks infected with A. phagocytophilum (groEL ecotype II/ankA clusters 2, 4 and 7) that are not known to be responsible for HGA in Europe. However, Kim et al. [36] reported a single clinical case, caused by A. phagocytophilum (in our study: ecotype II/cluster IV) from South Korea, where the pathogen is transmitted by ticks other than I. ricinus. Strains with zoonotic potential known from Europe represent only a small fraction of the A. phagocytophilum genetic variants and belong to ankA cluster 1/groEL ecotype I. These monophyletic groups are characterized by a wide host range, including domestic and wild animals, often with a synanthropic lifestyle or living closely to human settlements (e.g. foxes, hedgehogs, wild boars). The lack of reported clinical cases caused by A. phagocytophilum strains other than ecotype I/cluster 1 in Europe may therefore be related to their low prevalence in the environment, and their ability to infect humans needs to be confirmed.
Notwithstanding the noted variability of A. phagocytophilum, this tick-borne pathogen is considered to be a single species. This contrasts with a similar situation in B. burgdorferi sensu lato, which is subdivided into several operational taxonomic units (referred to as genospecies) with differences in geographic distribution, host species preferences, zoonotic potential and tissue tropism [37]. The introduction of a comprehensive and unified nomenclature and tools defining genetic lineages of A. phagocytophilum would allow easier communication of results and deeper understanding of the ecology and epidemiology of different genetic variants of this bacterium.
Availability of data and materials
Representative sequences of groEL and ankA were deposited to the NCBI GenBank database under accession numbers OP265397-OP265406 and OP265407-OP265426, respectively. They will not be released to the public database until 18 Sep 2023, or until the data or accession numbers appear in print, whichever is first.
References
Stuen S, Granquist EG, Silaghi C. Anaplasma phagocytophilum-a widespread multi-host pathogen with highly adaptive strategies. Front Cell Infect Microbiol. 2013;3:31.
Matei IA, Estrada-Peña A, Cutler SJ, Vayssier-Taussat M, Varela-Castro L, Potkonjak A, et al. A review on the eco-epidemiology and clinical management of human granulocytic anaplasmosis and its agent in Europe. Parasit Vectors. 2019;12:599.
Rar V, Tkachev S, Tikunova N. Genetic diversity of Anaplasma bacteria: twenty years later. Infect Genet Evol. 2021;91:104833.
Huhn C, Winter C, Wolfsperger T, Wüppenhorst N, Strašek Smrdel K, Skuballa J, et al. Analysis of the population structure of Anaplasma phagocytophilum using multilocus sequence typing. PLoS ONE. 2014;9:e93725.
Liveris D, Aguero-Rosenfeld ME, Daniels TJ, Karpathy S, Paddock C, Adish S, et al. A new genetic approach to distinguish strains of Anaplasma phagocytophilum that appear not to cause human disease. Ticks Tick Borne Dis. 2021;12:101659.
Liz JS, Sumner JW, Pfister K, Brossard M. PCR detection and serological evidence of granulocytic ehrlichial infection in roe deer (Capreolus capreolus) and chamois (Rupicapra rupicapra). J Clin Microbiol. 2002;40:892–7.
Jaarsma RI, Sprong H, Takumi K, Kazimirova M, Silaghi C, Mysterud A, et al. Anaplasma phagocytophilum evolves in geographical and biotic niches of vertebrates and ticks. Parasit Vectors. 2019;12:328.
Jahfari S, Coipan EC, Fonville M, van Leeuwen AD, Hengeveld P, Heylen D, et al. Circulation of four Anaplasma phagocytophilum ecotypes in Europe. Parasit Vectors. 2014;7:365.
de la Fuente J, Gortazar C. Wild boars as hosts of human-pathogenic Anaplasma phagocytophilum variants. Emerg Infect Dis. 2012;18:2094–5.
Stigum VM, Jaarsma RI, Sprong H, Rolandsen CM, Mysterud A. Infection prevalence and ecotypes of Anaplasma phagocytophilum in moose Alces alces, red deer Cervus elaphus, roe deer Capreolus capreolus and Ixodes ricinus ticks from Norway. Parasit Vectors. 2019;12:1–8.
Petrovec M, Bidovec A, Sumner JW, Nicholson WL, Childs JE, Avsic-Zupanc T. Infection with Anaplasma phagocytophila in cervids from Slovenia: evidence of two genotypic lineages. Wien Klin Wochenschr. 2002;114:641–7.
Katargina O, Geller J, Alekseev A, Dubinina H, Efremova G, Mishaeva N, et al. Identification of Anaplasma phagocytophilum in tick populations in Estonia, the European part of Russia and Belarus. Clin Microbiol Infect. 2012;18:40–6.
Von Loewenich FD, Stumpf G, Baumgarten BU, Röllinghoff M, Dumler JS, Bogdan C. A case of equine granulocytic ehrlichiosis provides molecular evidence for the presence of pathogenic Anaplasma phagocytophilum (HGE agent) in Germany. Eur J Clin Microbiol Infect Dis. 2003;22:303–5.
Madison-Antenucci S, Kramer LD, Gebhardt LL. Emerging tick-borne diseases. Clin Microbiol Rev. 2020;33:e00083.
Bakken JS, Dumler JS. Human granulocytic anaplasmosis. Infect Dis Clin North Am. 2020;22:85–91.
Dvořáková Heroldová M, Dvořáčková M. Seroprevalence of Anaplasma phagocytophilum in patients with suspected Lyme borreliosis. Epidemiol Mikrobiol Imunol. 2014;63:297–302.
Sloupenska K, Dolezilkova J, Koubkova B, Hutyrova B, Racansky M, Horak P, et al. Seroprevalence of Antibodies against tick-borne pathogens in Czech patients with suspected post-treatment Lyme disease syndrome. Microorganisms. 2021;9:2217.
Baker A, Wang HH, Mogg M, Derouen Z, Borski J, Grant WE. Increasing incidence of anaplasmosis in the United States, 2012 through 2016. Vector Borne Zoonotic Dis. 2020;20:855–9.
Pilloux L, Baumgartner A, Jaton K, Lienhard R, Ackermann-Gäumann R, Beuret C, et al. Prevalence of Anaplasma phagocytophilum and Coxiella burnetii in Ixodes ricinus ticks in Switzerland: an underestimated epidemiologic risk. New Microbes New Infect. 2019;27:22–6.
Lernout T, De Regge N, Tersago K, Fonville M, Suin V, Sprong H. Prevalence of pathogens in ticks collected from humans through citizen science in Belgium. Parasit Vectors. 2019;12:1–11.
Courtney JW, Kostelnik LM, Zeidner NS, Massung RF. Multiplex real-time PCR for detection of Anaplasma phagocytophilum and Borrelia burgdorferi. J Clin Microbiol. 2004;42:3164–8.
Noureddine R, Chauvin A, Plantard O. Lack of genetic structure among Eurasian populations of the tick Ixodesricinus contrasts with marked divergence from north-African populations. Int J Parasitol. 2011;41(2):183–92. https://doi.org/10.1016/j.ijpara.2010.08.010.
Alberti A, Zobba R, Chessa B, Addis MF, Sparagano O, Parpaglia MLP, et al. Equine and canine Anaplasma phagocytophilum strains isolated on the island of Sardinia (Italy) are phylogenetically related to pathogenic strains from the United States. Appl Environ Microbiol. 2005;71:6418–22.
Lesiczka PM, Hrazdilová K, Majerová K, Fonville M, Sprong H, Hönig V, et al. The role of peridomestic animals in the eco-epidemiology of Anaplasma phagocytophilum. Microb Ecol. 2021;82:602–12.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–95.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Langenwalder, D.B., Silaghi, C., Nieder, M. et al. Co-infection, reinfection and superinfection with Anaplasma phagocytophilum strains in a cattle herd based on ankA gene and multilocus sequence typing. Parasit Vectors. 2020;13:157. https://doi.org/10.1186/s13071-020-04032-2.
Grzeszczuk A, Stańczak J. High prevalence of Anaplasma phagocytophilum infection in ticks removed from human skin in north-eastern Poland. Ann Agric Environ Med. 2006;13:45–8.
Otranto D, Dantas-Torres F, Giannelli A, Latrofa M, Cascio A, Cazzin S, et al. Ticks infesting humans in Italy and associated pathogens. Parasit Vectors. 2014;7:328.
Audino T, Pautasso A, Bellavia V, Carta V, Ferrari A, Verna F, et al. Ticks infesting humans and associated pathogens: a cross-sectional study in a 3-year period (2017–2019) in northwest Italy. Parasit Vectors. 2021;14:1–10.
Matei IA, Kalmár Z, Lupşe M, D’Amico G, Ionică AM, Dumitrache MO, et al. The risk of exposure to rickettsial infections and human granulocytic anaplasmosis associated with Ixodes ricinus tick bites in humans in Romania: a multiannual study. Ticks Tick Borne Dis. 2017;8:375–8.
Hagedorn P, Imhoff M, Fischer C, Domingo C, Niedrig M. Human granulocytic anaplasmosis acquired in Scotland, 2013. Emerg Infect Dis. 2014;20:1079–81.
de la Fuente J, Estrada-Peña A, Cabezas-Cruz A, Kocan KM. Anaplasma phagocytophilum uses common strategies for infection of ticks and vertebrate hosts. Trends Microbiol. 2016;24:173–80.
Kim KH, Yi J, Oh WS, Kim NH, Choi SJ, Choe PG, Kim NJ, Lee JK, Oh MD. Human granulocytic anaplasmosis, South Korea, 2013. Emerg Infect Dis. 2014;20(10):1708–11. https://doi.org/10.3201/eid2010.131680.
Margos G. Population, genetics, taxonomy and phylogeny Borrelia burgdorrferi. Infect Genet Evol. 2012;2011:1545–63.
Acknowledgements
Not applicable.
Funding
This work was supported by the funding from the Grant Agency of the Czech Republic GACR 21-11661S to LZ and DM; KH was supported by the project National Institute of Virology and Bacteriology (Programme EXCELES, ID Project No. LX22NPO5103) - Funded by the European Union - Next Generation EU.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and data collection were performed by VH and PML. Data analyses were performed by PML. The first draft of the manuscript was written by PML, KH and LZ. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers used in the study.
Additional file 2: Figure S1.
Detailed maximum likelihood phylogenetic tree based on the groEL gene (groEL) of A. phagocytophilum.
Additional file 3: Figure S2.
Detailed Maximum likelihood phylogenetic tree based on the ankA gene (ankA) of A. phagocytophilum.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lesiczka, P.M., Hrazdilova, K., Hönig, V. et al. Distant genetic variants of Anaplasma phagocytophilum from Ixodes ricinus attached to people. Parasites Vectors 16, 80 (2023). https://doi.org/10.1186/s13071-023-05654-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-023-05654-y