
Abstract
Background
Fragmented mitochondrial (mt) genomes and extensive mt gene rearrangements have been frequently reported from parasitic lice (Insecta: Phthiraptera). However, relatively little is known about the mt genomes from the family Philopteridae, the most species-rich family within the suborder Ischnocera.
Methods
Herein, we use next-generation sequencing to decode the mt genome of Falcolipeurus suturalis and compare it with the mt genome of F. quadripustulatus. Phylogenetic relationships within the family Philopteridae were inferred from the concatenated 13 protein-coding genes of the two Falcolipeurus lice and members of the family Philopteridae using Bayesian inference (BI) and maximum likelihood (ML) methods.
Results
The complete mt genome of F. suturalis is a circular, double-stranded DNA molecule 16,659 bp in size that contains 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes, and three non-coding regions. The gene order of the F. suturalis mt genome is rearranged relative to that of F. quadripustulatus, and is radically different from both other louse species and the putative ancestral insect. Phylogenetic analyses revealed clear genetic distinctiveness between F. suturalis and F. quadripustulatus (Bayesian posterior probabilities = 1.0 and bootstrapping frequencies = 100), and that the genus Falcolipeurus is sister to the genus Ibidoecus (Bayesian posterior probabilities = 1.0 and bootstrapping frequencies = 100).
Conclusions
These datasets help to better understand gene rearrangements in lice and the phylogenetic position of Falcolipeurus and provide useful genetic markers for systematic studies of bird lice.
Graphical Abstract

Similar content being viewed by others

Background
The typical insect mitochondrial (mt) genome is a circular, double-stranded DNA molecule of about 12–20 kb in length that contains 37 genes: 13 protein-coding genes (PCGs), 22 transfer RNAs (tRNA), and two ribosomal RNAs (rRNA) [1, 2]. However, some lineages of parasitic lice (Insecta: Phthiraptera) are notable exceptions. For example, several groups show extensively fragmented mt genomes, where the genes are separated onto multiple circular chromosomes, including in the families Haematopinidae [3], Hoplopleuridae [4, 5], Menoponidae [6], Pediculidae [7,8,9], Polyplacidae [10], and Trichodectidae [11]. Parasitic lice are currently divided into four suborders, namely Ischnocera, Amblycera, Rhynchophthirina, and Anoplura, based on morphological structure.
Lice are permanent, obligate, and often host-specific ectoparasites commonly found on birds and mammals. The suborder Ischnocera (approximately 3120 species) is currently divided into two families, Philopteridae (approximately 2600 species) and Trichodectidae [12]. Complete mt genomes of only a limited number of philopterid species have been sequenced: Bothriometopus macrocnemis [13], Campanulotes bidentatus compar [14], Campanulotes compar [11], Coloceras sp. [15], Falcolipeurus quadripustulatus [11], Ibidoecus bisignatus [15], Columbina picui, Columbina cruziana, and Columbicola columbae [16]. These studies have found extensive gene rearrangements in philopterid mt genomes. A recent report has demonstrated that highly fragmented, mt minicircles are present in four species of the genus Columbicola [16], indicating that fragmented mt genomes are more prevalent in parasitic lice than previously hypothesized [11]. These studies demonstrate that our knowledge of the structure in mt genomes of bird lice is far from comprehensive. Additional data is needed to understand the pattern and mechanisms of genome fragmentation and rearrangement in bird lice.
Owing to maternal inheritance, relatively high evolution rate, conserved gene components, and low rate of recombination, the mt genome has been widely used as a genetic marker for comparative, evolutionary, and phylogenetic analysis at different taxonomic levels [17, 18]. In this study, we (i) characterize the complete mt genome sequences of F. suturalis from the tawny eagle, (ii) compare it with that of F. quadripustulatus from the vulture, and (iii) assess the phylogenetic position of Falcolipeurus within the Philopteridae.
Methods
Sample collection and DNA extraction
Adult samples of F. suturalis were taken from a tawny eagle Aquila rapax in the Beijing Wildlife Rescue Center, China. Lice were washed twice with sterile physiological saline solution (0.85%), and initial identification as F. suturalis made based on morphology and host species (Fig. 1) [19], and then stored in 95% (v/v) ethanol at −40 °C. Total genomic DNA was extracted from 60 individual lice (30 females and 30 males) using the DNeasy Tissue Kit (Promega, Madison, USA) following manufacturer instructions. Two pairs of primers [20], mtd6-mtd11 and 12SA-12SB, were used to amplify fragments of cox1 (600 bp) and rrnS (350 bp) genes, respectively, for use as assembly baits [4].

Female (a) and male (b) Falcolipeurus suturalis (dorsal side and ventral side). 1: head, 2: antenna, 3: thorax, 4: legs, 5: abdomen
Sequencing, assembling and verification
The quality of extracted genomic DNA was tested by agarose gel electrophoresis [21] and DNA concentration were quantified by Qubit 2.0 Fluorometer (Thermo Scientific). A genomic DNA library (350 bp inserts) was prepared and sequenced by Novogene Bioinformatics Technology Co., Ltd. (Tianjin, China) using Illumina HiSeq 2500 (250 bp paired-end reads). Raw reads were filtered with PRINSEQ [22]. Preliminary cox1 and rrnS sequences of F. suturalis were used as initial references for assembly in Geneious v 11.1.5 (minimum overlap identity = 99%, minimum overlap = 150 bp, maximal gap size = 5 bp) [23]. Genome size and circular organization were verified by long PCR using four pairs of specific primers (Additional file 1: Table S1; Additional file 2: Figure S1).
Annotation and sequence analysis
Protein-coding and rRNA genes were identified by alignment to homologous genes of previously sequenced mt genome of the vulture louse F. quadripustulatus [11] using the MAFFT v7.122 software [24]. tRNA genes were identified using ARWEN [25] and tRNAscan-SE [26], with manual adjustment. Annotated mt genomes were illustrated using the visualize module of MitoZ [27]. Nucleotide composition and amino acid sequences of each protein-coding genes and codon usage were analyzed using MEGA v6.0 [28]. Asymmetry of base composition was calculated as the following formula: AT-skew = (A−T)/(A+T), GC-skew = (G−C)/(G+C) [29].
Phylogenetic analysis
A total of 23 mt genomes from lice, including ten species from the Philopteridae, three species from the Trichodectidae, one elephant louse, Haematomyzus elephantis, and eight species of sucking lice, were used for phylogenetic analysis [3, 5, 7, 9,10,11,12,13,14,15,16, 20, 30,31,32], with one wallaby louse, Heterodoxus macropus (GenBank: AF270939), used as an outgroup [33]. Amino acid sequences of 12 protein-coding genes (except for nad2 because it is unidentified in the H. elephantis mt genome) were aligned individually using MAFFT [23]. Alignments of the individual genes were concatenated into a single dataset. Ambiguously aligned areas were removed by Gblocks 0.91b with the option of less stringent selection [34], and subjected to phylogenetic analyses under Bayesian inference (BI) and maximum likelihood (ML). BI was conducted with four independent Markov chains run for 5,000,000 metropolis-coupled MCMC generations, sampling trees every 500 generations in MrBayes v3.2.7 [35]. The initial 25% (2500) trees of each MCMC were discarded as burn-in and the majority-rule consensus tree used to calculate Bayesian posterior probabilities (Bpp). For ML analysis, the alignment was partitioned by gene, and bootstrapping frequencies (Bf) performed using the rapid bootstrapping option with 100 iterations. The MtART (all 12 genes) model was selected as the most suitable model of evolution by ProtTest v2.4 based on the Akaike information criterion (AIC) [36]. ML analyses were computed using RAxML v2.2.3 [37]. Phylograms were drawn using FigTree v.1.31.
Results and discussion
Identity of the eagle louse F. suturalis
Seven louse species (F. suturalis, Degeeriella fulva, Degeeriella aquilarum, Nosopon chanabense, Colpocephalum impressum, Laemobothrion Laemobothrion vulturis, and Laemobothrion Laemobothrion maximum) can parasitize the tawny eagle A. rapax (http://phthiraptera.info/category/avian/aves/falconiformes/accipitridae/aquila/aquila-rapax).
F. suturalis can be identified by the following morphological characters: (1) body slender in deep dark brown colour; (2) head longer than wide with pointed anterior and slightly broad posterior; (3) dorsally, 3 dark brown spots on each side of preantennal area and 1 dark brown horizontal stripe near antennal area; (4) antennae sexually dimorphic, male antennae stout and long on segment 1, female antenna slender and short on all 5 segments; (5) thorax widened from top to bottom; (6) dorsal thorax with 1 pronotum and 2 pteronotums, pronotum larger than pteronotum, 1 stout short seta on posterior margin of each pteronotum; (7) ventral thorax, mesosternum with 1 pair dark brown spots near midlegs; (8) 3 pairs of legs slender, forelegs small, midlegs and hindlegs progressively larger; (9) abdomen longer than wide; (10) dorsal abdomen deep dark brown colour, 2nd segment slightly narrower than thoracic pteronotum and with 2 divided tergite plates; (11) tergites on dorsal abdominal segments 3 to 6 slightly narrow in middle on both male and female; (12) male with 2 divided tergite plates laterally and female with 1 tergite on each dorsal abdominal segment of 7 and 8; (13) ventral abdomen with 9 segments and in white colour, 1 pair of dark brown spots laterally on each segment of 2 to 9; (14) female with 1 small dark brown spot in inverted triangle shape in middle of ventral abdominal segments 8 and 9.
Genome organization
A total of 3.7 Gb of data (about 20-fold coverage) was obtained from the Illumina HiSeq 2500 platform, raw sequencing of 15,178,382 paired reads. Reads were cleaned (9,630,532 clean pairs) and assembled. The longest assembled contig was 16,659 bp in size and was the complete mt genome of F. suturalis (GenBank accession: MW696813). All 37 mt genes typical of metazoan mt genomes were present. Gene arrangement was distinct from that of F. quadripustulatus [11] and other philopterids [15]. Overall nucleotide composition was: A = 27.8%, T = 44.8%, C = 11.1%, G = 16.3%. All mt genes were encoded on the heavy strand, except tRNA-Arg. Three overlapping regions in the mt genome were observed: nad4L/nad1, tRNA-His/tRNA-Asp and tRNA-Asp/tRNA-Arg, ranging from 4 to 8 bp overlaps (Table 1). In additional, 22 intergenic regions were observed, ranging from 1 to 180 bp in size. The longest spacer was between tRNA-S2 and tRNA-G (Table 1).
Total A + T and G + C content of the complete mt genome was 73.0% and 27.0%, respectively, consistent with the nucleotide content of lice reported in previous studies [11, 15] (Table 2). Negative AT skew (−23.3) and positive GC skew (18.9) were found (Table 2), consistent with other louse mt genomes [11, 15]. All bird lice from the Philopteridae reported to date have strong strand asymmetry (GC skew between 6.3% and 38.1%) (Table 2).
Annotation
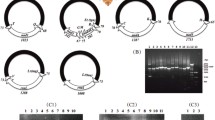
As the mt genomes of parasitic lice can contain non-standard initiation codons [1, 5, 13], their identification can be challenging. In F. suturalis, all protein-coding genes used ATNs (ATA, ATG, ATT) or TTG as their initiation codons. Four genes (cox1, atp6, cox3, and cox2) use ATA, five (nad6, nad2, nad5, atp8, and nad1) use ATG, three (nad3, nad4L, and nad4) use ATT, and one (cytb) uses TTG (Table 1). All protein-coding genes had standard (TAA or TAG) or partial (TA or T) termination codons (Table 1). Eight genes (cox1, atp6, nad6, cox3, nad2, cox2, atp8, and nad4L) use TAA, three genes (nad4, nad3, and cytb) use TAG, nad1 uses TA, and nad5 genes use T (Table 1). Incomplete termination codons (TA or T) were found for nad1 and nad5 genes, which has been found in other bird lice, including B. macrocnemis (nad1) and F. quadripustulatus (nad5, nad6 and nad1). In the F. suturalis mt genome, rrnL was located between rrnS and tRNA-Ala, while rrnS was between tRNA-Asn and rrnL (Fig. 2; Table 1). The lengths of rrnS and rrnL were 726 bp and 1075 bp, respectively. tRNA genes varied from 61 to 71 bp in length (Table 1). All 22 tRNA genes can be folded into the canonical cloverleaf structure (Fig. 3), consistent with previous studies [31, 32]. Apart from the coding regions, we identified three non-coding regions. Non-coding region (NC1) (829 bp), located between nad4 and tRNA-LeuUUR, has the highest A + T content of 75.4% while non-coding region (NC2) (722 bp; A + T = 74.4%) is located between cox2 and nad5 and non-coding region (NC3) (425 bp; A + T = 75.1%) is located between tRNA-Gly and nad3 (Table 1).

The mt genome of Falcolipeurus suturalis. All genes are on the same DNA strand and are transcribed clockwise, except for tRNA-Arg (R). Protein-coding and rRNA genes are indicated with the standard nomenclature. tRNA genes are indicated with the one-letter code of their corresponding amino acids. There are two tRNA genes for leucine: L1 for codons CUN and L2 for UUR; and two tRNA genes for serine: S1 for codons AGN and S2 for UCN. “NCR1” refers to the first non-coding region. “NCR2” refers to the second non-coding region. “NCR3” refers to the third non-coding region

22 tRNA secondary structures from Falcolipeurus suturalis
Comparative analyses between F. suturalis and F. quadripustulatus
The entire mt genome of F. suturalis is 537 bp longer than that of F. quadripustulatus [11]. A comparison of nucleotide and amino acid sequences in each protein-coding gene of the two Falcolipeurus species is given in Table 3. Nucleotide sequence differences across the entire mt genome was 31.4%. The magnitude of nucleotide sequence variation in each gene between F. suturalis and F. quadripustulatus ranged from 13.2 to 27.5%. The greatest variation was observed in atp8 (27.5%), and the least difference was found in cytb (13.2%) (Table 3). For rrnL and rrnS, sequence difference is 28.4% and 14.6%, respectively, between F. suturalis and F. quadripustulatus (Table 3). Amino acid sequences inferred from individual mt protein genes of F. suturalis were also compared with those of F. quadripustulatus. Amino acid sequence differences ranged from 4.5 to 41.2%, with cox1 being the most conserved protein, and atp8 the least conserved (Table 3). Our results are consistent with other species-level comparisons in lice. For example, amino acid divergence in the 13 protein-coding genes of C. picui and C. cruziana was 5.5–50% [16], while between C. bidentatus compar and C. compar it was 0–37.3% [11, 14]. Taken together, the molecular evidence presented here supports that F. suturalis and F. quadripustulatus represent distinct louse species.
Gene rearrangement
The mt genome arrangements of both Falcolipeurus species differ substantially from those of other species in the Philopteridae and from the putative ancestral insect (Fig. 4). Compared with the putative ancestral insect, no mt gene arrangements are shared with the mt genomes of Falcolipeurus species, as all genes are moved and/or inverted relative to their ancestral positions (Fig. 4). Of the 13 protein-coding genes, four (nad5, nad4, nad4L, and nad1) are inverted in both Falcolipeurus species relative to the putative ancestral insect. Only two gene blocks are shared between Bothriometopus and the putative ancestral insect: tRNA-Gly–nad3 and atp8–atp6 [13], while one gene block is shared between Campanulotes species and the ancestral insect: atp8-atp6 [11, 14, 33]. Three gene blocks, tRNA-Val–cox3, tRNA-Tyr–cox2, and tRNA-LeuCUN–nad4L, are shared between Falcolipeurus and Ibidoecus [11]. Two gene blocks, nad4–tRNA-LeuUUR and tRNA-Gly–nad3, are shared between Falcolipeurus and Campanulotes. In the Philopteridae, only one gene block, tRNA-Ile–cox1, is shared across all Philopterids, excepting I. bisignatus, Columbina, and Columbicola. Such a lack of conserved gene arrangements in the mt genome of bird lice complicates the accurate reconstruction and identification of rearrangement events across their history [13].

Gene rearrangements of mitochondrial genomes of bird lice within the family Philopteridae
Usually, gene arrangement in mt genomes is very conserved between congeneric lice [11, 14]. Gene rearrangements between F. suturalis and F. quadripustulatus were identified consisting of at least one translocation (Fig. 5). The nad3 gene is located between cox2 and tRNA-Thr in F. quadripustulatus, but between tRNA-Gly and tRNA-LeuCUN in F. suturalis (Fig. 5). This gene rearrangement between the mt genomes of two Falcolipeurus species indicated that the rate of rearrangements of mt genes may vary substantially among closely related groups of lice [38]. It is interesting that two congeneric Falcolipeurus species differ by a rearrangement. Although this pattern has been found in Columbicola [16] and multiple Anoplura species [11], it is the first time that it has been found in a louse without fragmented genomes.

Gene rearrangement in two Falcolipeurus species
One tRNA gene (tRNA-Gly) was lacking, while the duplication of three genes (tRNA-Thr, tRNA-Tyr, and cox2) was found in the F. quadripustulatus mt genome [11]. However, all 37 genes have been identified in the F. suturalis mt genome. Gene duplications have also been reported in the mt genomes of several families in the class Insecta, such as the Reduviidae and Thripidae [39,40,41]. In addition, tRNA loss has been also found in the mt genome of several families of the class Insecta [11, 42].
Phylogenetic relationships
Phylogenetic analysis shows the genetic distinctiveness between F. suturalis and F. quadripustulatus (Bpp = 1.0; Bf = 100). The branch leading to the two Falcolipeurus species is much longer than the branch of two Columbina species. The genus Falcolipeurus is more closely related to the genus Ibidoecus than to other genera (Bpp = 1.0, Fig. 6; Bf = 100, Fig. 7), which was consistent with that of a previous study [11]. Ten species of the Philopteridae were included in this study. The Philopteridae was paraphyletic with strong support in Bayesian analysis (Bpp > 0.9, Fig. 6) and weak support in ML analysis (Bf > 17, Fig. 7).

Phylogenetic relationships among 10 species of the family Philopteridae inferred by Bayesian inference from deduced amino acid sequences of 12 protein-coding genes. One wallaby louse, Heterodoxus macropus as an outgroup. Bayesian posterior probabilities (Bpp) were indicated at nodes

Phylogenetic relationships among 10 species of the family Philopteridae inferred by Maximum likelihood from deduced amino acid sequences of 12 protein-coding genes. One wallaby louse, Heterodoxus macropus as an outgroup. Bootstrapping frequencies (Bf) were indicated at nodes
DNA sequencing provides the opportunity to further evaluate phylogenetic relationships in the Philopteridae. Phylogenetic relationships in the Philopteridae have been investigated with analyses of the nuclear gene sequences. For example, Cruickshank et al. analyzed elongation factor-1 alpha (EF-1α) sequences of 127 species from the four louse suborders, showing the Philopteridae to be paraphyletic [43]. Yoshizawa and Johnson analyzed mt 12S and 16S rDNA sequences of 18 species also showed the family to be paraphyletic [44]. However, Johnson et al. analyzed 1107 single-copy orthologous genes of 46 species and showed that the Philopteridae was monophyletic [45]. De Moya et al. analyzed 2370 orthologous genes and also showed that Philopteridae was monophyletic [46].
Mt genome sequences are effective molecular markers for the study of phylogenetic and systematic relationships at various taxonomic ranks across the phylum Arthropoda, including for ectoparasites [44,45,46,47,48]. Recently, a study has also indicted that mt genes can provide reliable reconstructions of evolutionary relationships in parasitic lice [6]. In the present study, the monophyly of the Philopteridae was rejected by mt genomic phylogenetic analyses. Song et al. inferred the high-level phylogeny of parasitic lice with the mt genome sequences of 25 species of parasitic lice, and showed that the Philopteridae was paraphyletic [11]. To date, the phylogenetic position of the Philopteridae within the Phthiraptera has not been confidently determined. Although mt genomic data has proven to be useful as genetic markers to explore the phylogenetic relationships among major lineages of parasitic lice [9, 11], mt genome sequences of many lineages are under- or not represented. Analyses of mt genome sequences in the current and previous studies has indicated that the Philopteridae was paraphyletic. However, many species of this family were not included across these studies [11]. Additional complete mt genomes of bird louse species representing multiple families should be included in future analysis to help resolve the phylogenetic position of the Philopteridae within the Phthiraptera.
Conclusions
The current study presents the entire mt genome of F. suturalis and compared it with the mt genome of F. quadripustulatus. Its gene order is rearranged relative to other Falcolipeurus species, and represents a new pattern within the Phthiraptera. These novel datasets will help to better understand the gene rearrangements and phylogenetic position of Falcolipeurus and provide useful genetic markers for systematics and phylogenetic studies of bird lice.
Availability of data and materials
The complete mitochondrial genome sequences of Falcolipeurus suturalis have been deposited in the GenBank database under the accession number MW696813.
Abbreviations
- mt:
-
Mitochondrial
- rDNA:
-
Ribosomal DNA
- BI:
-
Bayesian inference
- nad2:
-
NADH dehydrogenase subunit 2
- cox1:
-
Cytochrome c oxidase subunit 1
- cox2:
-
Cytochrome c oxidase subunit 2
- atp6:
-
ATP synthase F0 subunit 6
- cox3:
-
Cytochrome c oxidase subunit 3
- nad3:
-
NADH dehydrogenase subunit 3
- nad5:
-
NADH dehydrogenase subunit 5
- nad4:
-
NADH dehydrogenase subunit 4
- nad4L:
-
NADH dehydrogenase subunit 4L
- nad6:
-
NADH dehydrogenase subunit 6
- cytb:
-
Cytochrome b
- atp8:
-
ATP synthase F0 subunit 8
- nad1:
-
NADH dehydrogenase subunit 1
- tRNA:
-
Transfer RNA
- rrnL:
-
Large subunit of rRNA
- rrnS:
-
Small subunit of rRNA
- BI:
-
Bayesian inference
- ML:
-
Maximum likelihood
- AIC:
-
Akaike information criterion
- Bpp:
-
Bayesian posterior probabilities
- Bf:
-
Bootstrapping frequencies
References
Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992;141:173–216.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80.
Song SD, Barker SC, Shao R. Variation in mitochondrial minichromosome composition between blood-sucking lice of the genus Haematopinus that infest horses and pigs. Parasit Vectors. 2014;7:144.
Dong WG, Song S, Guo XG, Jin DC, Yang Q, Barker SC, et al. Fragmented mitochondrial genomes are present in both major clades of the blood-sucking lice (suborder Anoplura): evidence from two Hoplopleura rodent lice (family Hoplopleuridae). BMC Genomics. 2014;15:751.
Fu YT, Nie Y, Duan DY, Liu GH. Variation of mitochondrial minichromosome composition in Hoplopleura lice (Phthiraptera: Hoplopleuridae) from rats. Parasit Vectors. 2020;13:506.
Sweet AD, Johnson KP, Cao Y, de Moya RS, Skinner RK, Tan M, et al. Structure, gene order, and nucleotide composition of mitochondrial genomes in parasitic lice from Amblycera. Gene. 2021;768:145312.
Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse Pediculus humanus. Genome Res. 2009;19:904–12.
Herd KE, Barker SC, Shao R. The mitochondrial genome of the chimpanzee louse, Pediculus schaeffi: insights into the process of mitochondrial genome fragmentation in the blood-sucking lice of great apes. BMC Genomics. 2015;16:661.
Fu YT, Dong Y, Wang W, Nie Y, Liu GH, Shao R. Fragmented mitochondrial genomes evolved in opposite directions between closely related macaque louse Pedicinus obtusus and colobus louse Pedicinus badii. Genomics. 2020;112:4924–33.
Dong WG, Song S, Jin DC, Guo XG, Shao R. Fragmented mitochondrial genomes of the rat lice, Polyplax asiatica and Polyplax spinulosa: intra-genus variation in fragmentation pattern and a possible link between the extent of fragmentation and the length of life cycle. BMC Genomics. 2014;15:44.
Song F, Li H, Liu GH, Wang W, James P, Colwell DD, et al. Mitochondrial genome fragmentation unites the parasitic lice of eutherian mammals. Syst Biol. 2019;68:430–40.
Price RD, Hellenthal RA, Palma RL. World checklist of chewing lice with host associations and keys to families and genera. In: Price RD, Hellenthal RA, Palma RL, Johnson KP, Clayton DH, editors. The chewing lice: world checklist and biological overview, vol. 24. Illinois: Illinois Natural History Survey Special Publication; 2003. p. 1–448.
Cameron SL, Johnson KP, Whiting MF. The mt genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J Mol Evol. 2007;65:589–604.
Covacin C, Shao R, Cameron S, Barker SC. Extraordinary number of gene rearrangements in the mt genomes of lice (Phthiraptera: Insecta). Insect Mol Biol. 2006;15:63–8.
Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genomics. 2011;12:394.
Sweet AD, Johnson KP, Cameron SL. Mitochondrial genomes of Columbicola feather lice are highly fragmented, indicating repeated evolution of minicircle-type genomes in parasitic lice. PeerJ. 2020;8:e8759.
Li R, Wang Y, Shu X, Meng L, Li B. Complete mitochondrial genomes of three Oxya grasshoppers (Orthoptera) and their implications for phylogenetic reconstruction. Genomics. 2020;112:289–96.
Kim JS, Kim MJ, Jeong JS, Kim I. Complete mitochondrial genome of Saturnia jonasii (Lepidoptera: Saturniidae): genomic comparisons and phylogenetic inference among Bombycoidea. Genomics. 2018;110:274–82.
Tandan BK. Mallophaga from birds of the Indian subregion. Part VI Falcolipeurus Bedford. In: Proceedings of the royal entomological society of London. Series B, Taxonomy; 2009.
Shao R, Li H, Barker SC, Song S. The mitochondrial genome of the guanaco louse, Microthoracius praelongiceps: insights into the ancestral mitochondrial karyotype of sucking lice (Anoplura, Insecta). Genome Biol Evol. 2017;9:431–45.
Almal S, Jeon S, Agarwal M, Patel S, Patel S, Bhak Y, et al. Sequencing and analysis of the whole genome of Indian Gujarati male. Genomics. 2019;111:196–204.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Laslett D, Canbäck B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5.
Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–7.
Meng G, Li Y, Yang C, Liu S. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019;47:e63.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353–8.
Jiang H, Barker SC, Shao R. Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol Evol. 2013;5:1298–308.
Shao R, Zhu XQ, Barker SC, Herd K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol Evol. 2012;4:1088–101.
Shao R, Barker SC, Li H, Song S, Poudel S, Su Y. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci Rep. 2015;5:17389.
Shao R, Campbell NJ, Barker SC. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol Biol Evol. 2001;18:858–65.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Shao R, Campbell NJ, Schmidt ER, Barker SC. Increased rate of gene rearrangement in the mitochondrial genomes of three orders of hemipteroid insects. Mol Biol Evol. 2001;18:1828–32.
Kocher A, Kamilari M, Lhuillier E, Coissac E, Péneau J, Chave J, et al. Shotgun assembly of the assassin bug Brontostoma colossus mitochondrial genome (Heteroptera, Reduviidae). Gene. 2014;552:184–94.
Sun Z, Liu Y, Wilson JJ, Chen Z, Song F, Cai W, et al. Mitochondrial genome of Phalantus geniculatus (Hemiptera: Reduviidae): trnT duplication and phylogenetic implications. Int J Biol Macromol. 2019;129:110–5.
Jiang P, Li H, Song F, Cai Y, Wang J, Liu J, et al. Duplication and remolding of tRNA genes in the mitochondrial genome of Reduvius tenebrosus (Hemiptera: Reduviidae). Int J Mol Sci. 2016;17:951.
Tyagi K, Chakraborty R, Cameron SL, Sweet AD, Chandra K, Kumar V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci Rep. 2020;10:695.
Cruickshank RH, Johnson KP, Smith VS, Adams RJ, Clayton DH, Page RD. Phylogenetic analysis of partial sequences of elongation factor 1 alpha identifies major groups of lice (Insecta: Phthiraptera). Mol Phylogenet Evol. 2001;19:202–15.
Yoshizawa K, Johnson KP. Phylogenetic position of Phthiraptera (Insecta: Paraneoptera) and elevated rate of evolution in mitochondrial 12S and 16S rDNA. Mol Phylogenet Evol. 2003;29:102–14.
Johnson KP, Dietrich CH, Friedrich F, Beutel RG, Wipfler B, Peters RS, et al. Phylogenomics and the evolution of hemipteroid insects. Proc Natl Acad Sci USA. 2018;115:12775–80.
de Moya RS, Yoshizawa K, Walden KKO, Sweet AD, Dietrich CH, Johnson KP. Phylogenomics of parasitic and non-parasitic lice (Insecta: Psocodea): combining sequence data and exploring compositional bias solutions in next generation datasets. Syst Biol. 2020:syaao75.
Liu GH, Chen F, Chen YZ, Song HQ, Lin RQ, Zhou DH, et al. Complete mitochondrial genome sequence data provides genetic evidence that the brown dog tick Rhipicephalus sanguineus (Acari: Ixodidae) represents a species complex. Int J Biol Sci. 2013;9:361–9.
Gu XB, Liu GH, Song HQ, Liu TY, Yang GY, Zhu XQ. The complete mitochondrial genome of the scab mite Psoroptes cuniculi (Arthropoda: Arachnida) provides insights into Acari phylogeny. Parasit Vectors. 2014;7:340.
Acknowledgements
Not applicable.
Funding
This study was supported by the Planned Programme of Hunan Province Science and Technology Innovation (Grant No. 2018RS3085) and the Training Programme for Excellent Young Innovators of Changsha (Grant No. KH2002001).
Author information
Authors and Affiliations
Contributions
GHL and YT conceived and designed the study, and critically revised the manuscript. YN performed the experiments. YN, YTF, and GHL analyzed the data. YN and YTF drafted the manuscript. YZ, YPD, and WW helped in study design, study implementation, and manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures involving animals in the present study were approved and this study was approved by the Animal Ethics Committee of Hunan Agricultural University (No. 43321503).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers used for assembly validation.
Additional file 2: Figure S1.
PCR amplicons from the mitochondrial genome of Falcolipeurus suturalis. Amplicons generated with the F. suturalis primers. M: DL8000 DNA marker, 1: Validation_01, 2: Validation_02, 3: Validation_03, 4: Validation_04, 5: Validation_05, 6: Negative control.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nie, Y., Fu, YT., Zhang, Y. et al. Highly rearranged mitochondrial genome in Falcolipeurus lice (Phthiraptera: Philopteridae) from endangered eagles. Parasites Vectors 14, 269 (2021). https://doi.org/10.1186/s13071-021-04776-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04776-5