
Abstract
Background
The family Hoplopleuridae contains at least 183 species of blood-sucking lice, which widely parasitize both mice and rats. Fragmented mitochondrial (mt) genomes have been reported in two rat lice (Hoplopleura kitti and H. akanezumi) from this family, but some minichromosomes were unidentified in their mt genomes.
Methods
We sequenced the mt genome of the rat louse Hoplopleura sp. with an Illumina platform and compared its mt genome organization with H. kitti and H. akanezumi.
Results
Fragmented mt genome of the rat louse Hoplopleura sp. contains 37 genes which are on 12 circular mt minichromosomes. Each mt minichromosome is 1.8–2.7 kb long and contains 1–5 genes and one large non-coding region. The gene content and arrangement of mt minichromosomes of Hoplopleura sp. (n = 3) and H. kitti (n = 3) are different from those in H. akanezumi (n = 3). Phylogenetic analyses based on the deduced amino acid sequences of the eight protein-coding genes showed that the Hoplopleura sp. was more closely related to H. akanezumi than to H. kitti, and then they formed a monophyletic group.
Conclusions
Comparison among the three rat lice revealed variation in the composition of mt minichromosomes within the genus Hoplopleura. Hoplopleura sp. is the first species from the family Hoplopleuridae for which a complete fragmented mt genome has been sequenced. The new data provide useful genetic markers for studying the population genetics, molecular systematics and phylogenetics of blood-sucking lice.

Similar content being viewed by others


Background
Blood-sucking lice are known vectors and transmit various disease agents and cause significant vector-borne diseases in humans, domestic and wild mammals [1]. The family Hoplopleuridae contains at least 183 described species of blood-sucking lice currently classified into eight genera [2]. Of the eight genera, Hoplopleura Enderlein, 1904 is the most species-rich (165 described species) found on rodents [3]. The Hoplopleura spp. are common ectoparasites of both mice and rats, causing pruritus, alopecia, dermal irritation and even anemia.
Metazoan mitochondrial (mt) genomes are usually circular DNA molecules (13–20 kb) with 36–37 genes that contain 12–13 protein-coding genes, two rRNA genes and 22 tRNA genes [4]. Some parasitic lice have an unusual, fragmented mt genome organization, but not all species of parasitic lice have been shown to have a fragmented genome. Fragmentation of the mt genome was first found in the human body louse, Pediculus humanus corporis (suborder Anoplura) [5]. Since then, 11 other blood-sucking lice, P. humanus capitis, P. schaeffi, Pthirus pubis, Haematopinus suis, H. apri, H. asini, H. akanezumi, H. kitti, Polyplax asiatica, P. spinulosa and Microthoracius praelongiceps (suborder Anoplura); eight avian feather lice, Bovicola bovis, B. ovis, B. caprae, Trichodectes canis, Columbicola columbae, C. macrourae, C. passerinae 1 and 2 (suborder Ischnocera), and the elephant louse, Haematomyzus elephantis (suborder Rhynchophthirina), have been found with fragmented mt genomes [6,7,8,9,10,11,12,13,14,15]. While seven feather lice, Colpocephalum griffoneae, Amyrsidea minuta and Heterodoxus macropus (suborder Amblycera), Ibidoecus bisignatus, Campanulotes compar, Bothriometopus macrocnemis and Falcolipeurus quadripustulatus (suborder Ischnocera), do not have fragmented mt genomes [15]. To date, the complete mt genomes of 12 blood-sucking lice have been sequenced and deposited in GenBank, but the complete mt genomes have been only reported for two rat lice (H. kitti and H. akanezumi) from this family Hoplopleuridae [7]. In addition, three genes (nad1, nad3 and nad5) or minichromosomes were unidentified in the mt genomes of two Hoplopleura spp. [7]. Interestingly, gene rearrangement has been reported in the fragmented mt genome of two Hoplopleura spp. [7]. Therefore, Hoplopleura mt genomes may represent one of the most frequently rearranged/fragmented mt genomes within the family Hoplopleuridae.
To understand the composition of mt minichromosomes in species of the same genus, Hoplopleura, we sequenced the complete mt genome of the rat louse Hoplopleura sp. and compared its mt genome organization with other two Hoplopleura species, and re-constructed its phylogenetic relationships within the suborder Anoplura using protein sequences derived from coding genes.
Methods
Sample collection and DNA extraction
Adult specimens of Hoplopleura sp. were collected from the Edward’s long-tailed rats Leopoldamys edwardsi in Chongqing, China. The specific identity of the examined wild rats was determined by PCR-based sequencing of the mitochondrial (mt) cox1 gene using an established method [16]. These rat lice were washed five times in physiological saline solution, identified preliminarily to the genus level (as Hoplopleura sp.) based on morphological features [2], and stored in 70% (v/v) ethanol at − 20 °C. Whole genomic DNA including nuclear and mt DNA was extracted from 50 single rat lice (25 females and 25 males) using the DNeasy Tissue Kit (Promega, Madison, USA) according to the manufacturer’s recommendations. The identity of these specimens was further confirmed by polymerase chain reaction (PCR) amplification and subsequent sequencing of the mt cox1 and rrnS genes using primer pairs L6625 (5′-CCG GAT CCT TYT GRT TYT TYG GNC AYC C-3′) and H7005 (5′-CCG GAT CCA CNA CRT ART ANG TRT CRT G-3′), and 12SA (5′-TAC TAT GTT ACG ACT TAT-3′) and 12SB (5′-AAA CTA GGA TTA GAT ACC C-3′), respectively.
Sequencing and assembling
The purity of the extracted whole genomic DNA was assessed by agarose-gel electrophoresis [17]. The DNA concentration was determined using a Quantus Fluorometer (Invitrogen, Carlsbad, USA). A paired-end genomic DNA library (350 bp inserts) was constructed for high throughput sequencing with Miseq PE300 (Illumina, San Diego, CA, USA) and collected raw reads were exported in the FASTQ format. The raw reads were filtered by removing adaptor reads, redundant reads and ‘N’-rich reads. Finally, 2 Gb clean data (256 bp pair-end reads) was produced for this rat louse. Contigs were de novo assembled from Illumina sequence reads using Geneious 11.1.5 [18] based on cox1 and rrnS relatively conserved sequences. The assembly parameters were minimum overlap identity 99% and minimum overlap 150 bp. The two ends of the contig overlapped, indicating circular organization of the minichromosome. We observed in previous studies that each mt minichromosome has a distinct coding region but a well-conserved non-coding region [10,11,12,13]. The conserved non-coding region sequences were identified between the cox1 and rrnS minichromosomes and were used as references to align the Illumina sequence dataset. BLAST was used for alignment. We assembled these minichromosomes individually in full length using the same method stated above for cox1 and rrnS minichromosome assembly.
Annotation
Sequences were aligned against the mt minichromosome sequences of the rat louse H. kitti [7] available using the MAFFT 7.122 software [19] to identify gene boundaries. Protein-coding genes and rRNA genes were identified with BLAST searches of the NCBI database. Amino acid sequences of each protein-coding genes were inferred using MEGA 6.0 [20]. tRNA genes were identified using ARWEN [21] and the program tRNAscan-SE [22] with manual adjustment.
Verification of mt minichromosomes
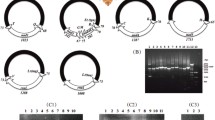
The size of each mt minichromosome of Hoplopleura sp. were verified by PCR using specific primers (Table 1). The forward primer and reverse primer in each pair were next to each other with a small gap in between (10–50 bp). PCR with these primers amplified each circular minichromosome in full length (Fig. 1). To obtain full-length sequences of the non-coding regions of the minichromosomes, these positive amplicons were also sequenced with high throughput sequencing as described above.

PCR verification of the 12 mt minichromosomes of the rat louse, Hoplopleura sp. Lane M: DL2000 DNA marker; Lane 1–12: I-cox1, rrnS, M-L1-rrnL-V, Q-nad1-G-nad3, D-Y-cox2-T, atp8-atp6-N, nad2, C-nad6-W-L2, R-nad4L-P-cox3-A, E-cytb-S1-S2, K -nad4 and H-nad5-F
Phylogenetic analysis
The phylogenetic relationships among representatives of the blood-sucking lice of suborder Anoplura were assessed based on concatenated amino acid sequences (Table 2), using one elephant louse, H. elephantis (GenBank: KF933032-41) as an outgroup [10]. Eight amino acid sequences (except for nad1, nad2, nad3, nad4 and nad5 because these genes were unidentified in some blood-sucking lice) were aligned individually using MAFFT 7.122 and were then concatenated to form a single dataset; ambiguously aligned regions were excluded using Gblocks 0.91b using default parameters [23]. The MtArt + I + G + F was selected as the most appropriate evolutionary model by ProtTest 2.4 based on the Akaike information criterion (AIC) [24]. Phylogenetic analyses were conducted with maximum likelihood (ML) using PhyML 3.0 with a BioNJ starting tree, and tree topology search was set from the subtree pruning and regrafting (SPR) method [25]. Bootstrap value was calculated using 100 bootstrap replicates. Phylograms were drawn using FigTree v.1.31.
Results and discussion
Identity of the rat louse Hoplopleura sp.
Two blood-sucking louse species (H. kitti and P. insulsa) parasitize L. edwardsi (https://phthiraptera.info/category/mammal-wilson-reeder/mammals/rodentia/muridae/murinae/leopoldamys/leopoldamys-edwardsi). The Hoplopleura sp. has close morphological and morphometric similarities with H. kitti recovered from the same host (L. edwardsi). The mt cox1 and rrnS genes of Hoplopleura sp. shared 76% and 77.6% identity with previously published sequences of H. kitti (GenBank: KJ648943) from Berylmys bowersi and H. akanezumi (GenBank: KJ648928) from Apodemus chevrieri in China, respectively.
General features of the mt genome of the rat louse Hoplopleura sp.
We sequenced the Hoplopleura sp. genome and produced 3 Gb of Illumina short-read sequence data and obtained a total of 6,526,349 × 2 raw reads from adults of Hoplopleura sp. After quality filtration, 3,937,826 × 2 clean reads (2 Gb) were generated for assembly of the mt genome. We assembled these sequence-reads into contigs and identified 37 mt genes typical of bilateral animals (Fig. 2; Table 3). These genes are on 12 minichromosomes; each minichromosome is 1.8–2.7 kb in size and consists of a coding region and a non-coding region (NCR) in a circular organization (Table 3). The coding regions have 1–5 genes each and vary in size from 675 to 1760 bp (Table 3). All genes are transcribed in the same direction except for the nad1 gene. The nucleotide sequences of the mt minichromosomes of Hoplopleura sp. were deposited in the GenBank database under the accession numbers MT792483-MT792494.

The complete mitochondrial genome of rat louse, Hoplopleura sp. Each minichromosome has a coding region and a non-coding region (NCR, in black). The names and transcript orientation of genes are indicated in the coding region and the minichromosomes are placed in alphabetical order of protein-coding genes and rRNA genes. Abbreviations: atp6 and atp8, ATP synthase F0 subunits 6 and 8; cob, cytochrome b; cox1-3, cytochrome c oxidase subunits 1–3; nad1-6 and nad4L, NADH dehydrogenase subunits 1–6 and 4L; rrnS and rrnL, small and large subunits of ribosomal RNA. tRNA genes are indicated with their single-letter abbreviations of the corresponding amino acids
We sequenced the full-length non-coding regions of all of the 12 mt minichromosomes of the Hoplopleura sp., which range from 935 (H-nad5-F minichromosome) to 1305 bp (C-nad6-W-L2 minichromosome) (Table 3). The longest non-coding region of Hoplopleura sp. was shorter than the longest non-coding region of other sucking lice known, such as pig lice (2370 bp) [6] and horse lice (3276 bp) [13]. As in the human lice [12], rat lice [7] and pig lice [6], each coding region of Hoplopleura sp. is flanked by a conserved non-coding AT-rich motif (88 bp, 71.6%) upstream and a GC-rich motif (39 bp, 79.5%) downstream, indicating functional significance of these motifs in the mt genomes of blood-sucking lice.
Annotation
The boundaries between protein-coding genes of the mt genome of Hoplopleura sp. were determined by aligning its sequence and identifying translation initiation and termination codons with those of H. kitti and H. akanezumi [7]. Hoplopleura sp. mt genome encoded 13 protein-encoding genes, which had four initiation codons (ATT, ATG, TTG and GTG). Among them, both ATT (nad2, nad4L, nad5, cox3 and cytb) and ATG (nad3, nad4, nad6, atp6 and atp8) are the highest frequency of being used as initiation codons. Moreover, TTG (nad1 and cox2) and GTG (cox1) are used in the mt genome. This mt genome has three termination codons (TAA, TAG and T). Among them, TAG is most frequently used (five times altogether), by cox1, nad2, nad3, nad4L and cytb. TAA was second in frequency of recurrence (four times) as termination codons, cox2, atp6, atp8 and nad4, used it in the mt genome of Hoplopleura sp. Furthermore, cox3, nad1, nad5 and nad6 genes use T as termination codons. Incomplete terminations (TA and T) of protein-coding genes are commonly found in other mt genomes of blood-sucking lice, including H. suis [6], H. apri [6], H. asini [13], H. kitti [7], P. asiatica [8], P. spinulosa [8], P. schaeffi [9], M. praelongiceps [11] and P. pubis [12]. In the mt genome of Hoplopleura sp., the sizes of the rrnL and rrnS genes were 1125 bp and 675 bp, respectively. The 22 tRNA genes ranged from 59 to 71 bp in size. The secondary structure predictions in Hoplopleura sp. (not shown) were similar to those of H. kitti and H. akanezumi [7].
Variation in mt minichromosome composition among three rat lice
The complete mt genome sequences of Hoplopleura sp. fragmented into 12 circular minichromosomes. The incomplete mt genomes of H. kitti and H. akanezumi have 11 identified circular minichromosomes [7]. Eleven minichromosomes of the rat louse, Hoplopleura sp., have the same gene content and gene arrangement as their counterparts of the rat louse, H. kitti. Eight of these minichromosomes of the rat lice, Hoplopleura sp. and H. kitti, have the same gene content and gene arrangement as their counterparts of the rat louse, H. akanezumi [7]. However, the other two minichromosomes of the rat louse Hoplopleura sp. are not present in the rat louse H. akanezumi [7]. In Hoplopleura sp., one of the minichromosomes has four genes, D-Y-cox2-T (Fig. 2); however, in H. akanezumi this minichromosome has only three genes, D-Y-cox2. Similarly, another minichromosome of Hoplopleura sp. has five genes, R-nad4L-P-cox3-A (Fig. 2); however, in H. akanezumi this minichromosome has six genes, R-nad4L-P-cox3-A-T (Fig. 3). Interestingly, a chimeric minichromosome has been found in H. akanezumi which contains parts of the two rRNA genes, prrnL and prrnS, which are only 5% (51 bp) and 24% (172 bp) of the full-length rrnL and rrnS, respectively [7]. However, this chimeric minichromosome has not been identified in H. kitti and Hoplopleura sp.

The differences among all minichromosomes of three Hoplopleura lice. a 12 identified circular minichromosomes of Hoplopleura sp. b 11 identified circular minichromosomes of Hoplopleura akanezumi. c 11 identified circular minichromosomes of Hoplopleura kitti
Comparative mt genomic analyses of Hoplopleura sp. with H. kitti and H. akanezumi
A comparison of the nucleotide and the amino acid sequences of each protein-encoding gene (except for nad1, nad3 and nad5) of the three Hoplopleura species is given in Table 4. Pairwise comparisons of the nucleotide and amino acid sequences revealed identities of 50.6–77.2% and 37.5–90.2% among them, respectively. The greatest nucleotide variation was in the atp8 gene (49.4%), whereas the lowest differences (22.8%) were detected in the cox1 gene (Table 4). The difference across both concatenated nucleotide and amino acid sequences of the ten protein-coding genes was 37.5% and 36.8% between Hoplopleura sp. and H. kitti, 36.7% and 34.7% between Hoplopleura sp. and H. akanezumi, and 34.6% and 33.4% between H. kitti and H. akanezumi.
Phylogenetic relationships
In the present study, phylogenetic analysis of the concatenated amino acid sequence datasets for eight mt protein-coding genes (Fig. 4) showed that the family Hoplopleuridae (Hoplopleura sp., H. kitti and H. akanezumi) clustered to the exclusion of representatives of the families Polyplacidae (P. asiatica and P. spinulosa), Haematopinidae (H. apri, H. asini and H. suis), Pediculidae (P. humanus corporis, P. humanus capitis and P. schaeffi), Pthiridae (P. pubis), and the family Microthoraciidae (M. praelongiceps) clustered separately with strong nodal support (bootstrap = 100). Within the family Hoplopleuridae, Hoplopleura sp. and H. akanezumi clustered together with moderate support (bootstrap value = 73), to the exclusion of H. kitti, and then they formed a monophyletic group (bootstrap value = 100). The result was also strongly supported by RAxML analysis (bootstrap value = 100) (Additional file 1: Figure S1).

Phylogenetic relationships among 13 species of the suborder Anoplura inferred by maximum likelihood of deduced amino acid sequences of eight mitochondrial proteins using PhyML. The elephant louse, Haematomyzus elephantis, was used as the outgroup. Bootstrap values are indicated at nodes
The work of Johnson et al. [26] created robustness and stability in higher systematics within the order Phthiraptera based on analyses of 1107 single-copy orthologous genes from sequenced genomes of 46 species of lice [26]. Their result has indicated that the genera Hoplopleura and Haematopinus were more closely related than to the genus Pediculus with a strong bootstrap value [26]. However, mt genomic phylogenetic relationships deviated from phylogenies derived from the nuclear genome. Shao et al. [11] performed a phylogenetic analysis with mt genomes, indicating that the genera Haematopinus and Pediculus were more closely related than to the genus Hoplopleura with a strong bootstrap value. Our result also showed the genera Haematopinus and Pediculus were more closely related than to the genus Hoplopleura, but with a weak bootstrap value (bootstrap value = 55) (Fig. 3). Although the number of sucking lice mt genome sequences is increasing, so far, mt genomes of many lineages of sucking lice are underrepresented or not represented. Insufficient taxon sampling for the suborder Anoplura mt genomes might be the cause of the discordance between the mt and nuclear phylogenies.
Many studies have indicated that the mt genome sequence is a valuable genetic marker for phylogenetic studies at various taxonomic levels of different organisms [27, 28], including lice [14, 15]. The fragmentation of the mt genome may have arisen independently in multiple louse clades. Therefore, the mt genome sequences of rat louse Hoplopleura sp. could promote to reassess the systematic relationships of lice within the suborder Anoplura using mt genomic datasets. No species from the other genera (Ancistroplax, Ferrisella, Haematopinoides, Paradoxophthirus, Pterophthirus, Schizophthirus and Typhlomyophthirus) within the family Hoplopleuridae was included in our analyses. Therefore, more expanded taxa sampling is necessary for future phylogenetic studies of the family Hoplopleuridae using mt genomic datasets.
Conclusions
Comparison among the three rat lice revealed variation in the composition of mt minichromosomes among species of the genus Hoplopleura. Hoplopleura sp. is the first species from the family Hoplopleuridae for which a complete fragmented mt genome has been sequenced. The new data provide useful genetic markers for studying the population genetics, molecular systematics and phylogenetics of blood-sucking lice.
Availability of data and materials
The fragmented mitochondrial genome sequences of Hoplopleura sp. from the Edward’s long-tailed rats have been deposited in the GenBank database under the accession numbers MT792483–MT792494.
Abbreviations
- mt:
-
mitochondrial
- rDNA:
-
ribosomal DNA
- nad1:
-
NADH dehydrogenase subunit 1
- nad3:
-
NADH dehydrogenase subunit 3
- nad5:
-
NADH dehydrogenase subunit 5
- cox1:
-
cytochrome c oxidase subunit 1
- rrnS:
-
small subunit of rRNA
- rrnL:
-
large subunit of rRNA
- tRNA:
-
transfer RNA
- nad2:
-
NADH dehydrogenase subunit 2
- nad4:
-
NADH dehydrogenase subunit 4
- nad4L:
-
NADH dehydrogenase subunit 4L
- cox3:
-
cytochrome c oxidase subunit 3
- cytb:
-
cytochrome b
- nad6:
-
NADH dehydrogenase subunit 6
- atp6:
-
ATP synthase F0 subunit 6
- atp8:
-
ATP synthase F0 subunit 8
- cox2:
-
cytochrome c oxidase subunit 2
References
Tsai YL, Chang CC, Chuang ST, Chomel BB. Bartonella species and their ectoparasites: selective host adaptation or strain selection between the vector and the mammalian host? Comp Immunol Microbiol Infect Dis. 2011;34:299–314.
Kim KC, Ludwig HW. The family classification of the Anoplura. Syst Entomol. 1978;3:249–84.
Durden LA, Musser GG. The sucking lice (Insecta, Anoplura) of the world: a taxonomic checklist with records of mammalian hosts and geographical distributions. Bull Am Mus Nat Hist. 1994;218:1–90.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80.
Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse Pediculus humanus. Genome Res. 2009;19:904–12.
Jiang H, Barker SC, Shao R. Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol Evol. 2013;5:1298–308.
Dong WG, Song S, Guo XG, Jin DC, Yang QQ, Barker SC, et al. Fragmented mitochondrial genomes are present in both major clades of the blood-sucking lice (suborder Anoplura): evidence from two Hoplopleura rodent lice (family Hoplopleuridae). BMC Genomics. 2014;15:751.
Dong WG, Song S, Jin DC, Guo XG, Shao R. Fragmented mitochondrial genomes of the rat lice, Polyplax asiatica and Polyplax spinulosa: intra-genus variation in fragmentation pattern and a possible link between the extent of fragmentation and the length of life cycle. BMC Genomics. 2014;15:44.
Herd KE, Barker SC, Shao R. The mitochondrial genome of the chimpanzee louse, Pediculus schaeffi: insights into the process of mitochondrial genome fragmentation in the blood-sucking lice of great apes. BMC Genomics. 2015;16:661.
Shao R, Barker SC, Li H, Song S, Poudel S, Su Y. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci Rep. 2015;5:17389.
Shao R, Li H, Barker SC, Song S. The mitochondrial genome of the guanaco louse, Microthoracius praelongiceps: insights into the ancestral mitochondrial karyotype of sucking lice (Anoplura, Insecta). Genome Biol Evol. 2017;9:431–45.
Shao R, Zhu XQ, Barker SC, Herd K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol Evol. 2012;4:1088–101.
Song SD, Barker SC, Shao R. Variation in mitochondrial minichromosome composition between blood-sucking lice of the genus Haematopinus that infest horses and pigs. Parasit Vectors. 2014;7:144.
Sweet AD, Johnson KP, Cameron SL. Mitochondrial genomes of Columbicola feather lice are highly fragmented, indicating repeated evolution of minicircle-type genomes in parasitic lice. PeerJ. 2020;8:e8759.
Song F, Li H, Liu GH, Wang W, James P, Colwell DD, et al. Mitochondrial genome fragmentation unites the parasitic lice of eutherian mammals. Syst Biol. 2019;68:430–40.
Robins JH, Hingston M, Matisoo-Smith E, Ross HA. Identifying Rattus species using mitochondrial DNA. Mol Ecol Notes. 2007;7:717–29.
Almal S, Jeon S, Agarwal M, Patel S, Patel S, Bhak Y, et al. Sequencing and analysis of the whole genome of Indian Gujarati male. Genomics. 2019;111:196–204.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Laslett D, Canbäck B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5.
Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W57.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Johnson KP, Nguyen NP, Sweet AD, Boyd BM, Warnow T, Allen JM. Simultaneous radiation of bird and mammal lice following the K-Pg boundary. Biol Lett. 2018;14:20180141.
Li R, Wang Y, Shu X, Meng L, Li B. Complete mitochondrial genomes of three Oxya grasshoppers (Orthoptera) and their implications for phylogenetic reconstruction. Genomics. 2020;112:289–96.
Kim JS, Kim MJ, Jeong JS, Kim I. Complete mitochondrial genome of Saturnia jonasii (Lepidoptera: Saturniidae): genomic comparisons and phylogenetic inference among Bombycoidea. Genomics. 2018;110:274–82.
Acknowledgements
Not applicable.
Funding
This study was supported by the Planned Programme of Hunan Province Science and Technology Innovation (Grant No. 2018RS3085) and the Training Programme for Excellent Young Innovators of Changsha (Grant No. KQ1905013).
Author information
Authors and Affiliations
Contributions
YTF and GHL conceived and designed the study, and critically revised the manuscript. YTF performed the experiments. YTF and GHL analyzed the data. DYD and GHL drafted the manuscript. YN helped in study design, study implementation, and manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures involving animals in the present study were approved and this study was approved by the Animal Ethics Committee of Hunan Agricultural University (No. 43321503).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Phylogenetic relationships among 13 species of the suborder Anoplura inferred from maximum likelihood of deduced amino acid sequences of 8 mitochondrial proteins using RAxML. One elephant louse, Haematomyzus elephantis was used as the outgroup. Bootstrap values were indicated at nodes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, YT., Nie, Y., Duan, DY. et al. Variation of mitochondrial minichromosome composition in Hoplopleura lice (Phthiraptera: Hoplopleuridae) from rats. Parasites Vectors 13, 506 (2020). https://doi.org/10.1186/s13071-020-04381-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04381-y