
Abstract
Background
Ticks (Arthropoda, Ixodida), after mosquitoes, are the second most prevalent vector of infectious diseases. They are responsible for spreading a multitude of pathogens and threatening the health and welfare of animals and human beings. However, given the history of tick-borne pathogen infections in the Inner Mongolia Autonomous Region of China, surprisingly, neither the genetic diversity nor the spatial distribution of haplotypes within ticks has been studied.
Methods
We characterized the haplotype distribution of Dermacentor nuttalli in four main pastoral areas of the Inner Mongolia Autonomous Region, by sampling 109 individuals (recovered from sheep) in April–August 2019. The 16S rRNA gene, cytochrome c oxidase subunit I (COI), and the internal transcribed spacer 2 region (ITS2) were amplified and sequenced from extracted DNA.
Results
Twenty-six haplotypes were identified using 16S rRNA sequences, 57 haplotypes were identified with COI sequences, and 75 haplotypes were identified with ITS2 sequences. Among the three genes, total haplotype diversity was greater than 0.7, while total nucleotide diversity was greater than 0.06. Neutrality tests revealed a significantly negative Tajima’s D result, while Fu's Fs was not significantly positive. Fixation index values (FST) indicated that the degree of genetic differentiation among some sampled populations was small, while for others it was moderate. Analysis of molecular variance (AMOVA) revealed that the variation within populations was greater than that among populations. The mismatch analysis of D. nuttalli exhibited double peaks.
Conclusion
The genetic diversity of D. nuttalli populations in our region can likely adapt to different geographical environments, thereby leading to genetic diversity, and creating genetic differentiation among different populations. However, genetic differentiation is cryptic and does not form a pedigree geographical structure.
Graphical Abstract

Similar content being viewed by others


Background
Dermacentor nuttalli (Acari: Ixodidae) is widely distributed across northern China, Russia and Mongolia [1], and is generally found in arid grasslands suitable for grazing cattle and sheep [2]. D. nuttalli is responsible for spreading a variety of diseases, including spotted fever, tick-borne Rickettsia, Crimean-Congo hemorrhagic fever and babesiosis [2,3,4,5], in addition to being an important vector of spirochetes and forest encephalitis [6, 7]. Humans may experience symptoms similar to viral infections after being bitten by ticks, including headaches, muscle pain, fevers, enlarged local lymph nodes or plaques, as well as severe liver and kidney damage and damage to the central nervous system [8, 9]. D. nuttalli is an important storage host and transmission medium of Rickettsia, resulting in the rapid spread of Rickettsia through bites and causing a variety of zoonotic diseases. As a result, D. nuttalli is quickly becoming one of the most dangerous tick species to public health in Inner Mongolia.
Yet, despite the increasing danger to the health and economy of the Inner Mongolia Autonomous Region, neither the genetic diversity nor the spatial distribution of haplotypes in D. nuttalli populations has been studied. In recent years, research into the genetic diversity within species and even between populations has become increasingly common [10,11,12,13,14]. Genetic diversity is affected by many biotic and abiotic factors, including natural selection, genetic variation and anthropogenic influences [15]. Ticks and pathogens have also been identified as influencing host evolution [16, 17]. Nevertheless, there remains a paucity of data for tick populations within China. Therefore, the goal of this study was to characterize the genetic diversity of various D. nuttalli populations to further our understanding not only of their diffusion, but also of their evolution, in order to protect the health of humans and animals. We hope these data will create a foundation for further study into the spread of tick-borne diseases in China and provide evidence for the origin and continued evolution of tick species.
We chose three gene regions for our study, as they are all easily amplified and commonly used in molecular systematics and population genetics [19,20,21]. While the 16S rRNA gene has a relatively slow rate of evolution [18], and the cytochrome c oxidase subunit I gene (COI) is a highly conserved region, they are both widely used in the study of intra- and interspecific genetic diversity, as well as in phylogenetic geography among different geographical populations [22, 23]. In comparison, the internal transcribed spacer 2 region (ITS2) evolves rapidly, resulting in rich polymorphism. As a result, it is commonly used to study historical population dynamics of species with close interspecific relationships [24,25,26,27,28]. In this study, we analyzed the haplotype distribution of the 16S rRNA, COI and ITS2 genes within individuals of D. nuttalli sampled from four main pastoral areas of the Inner Mongolia Autonomous Region.
Methods
Sample collection
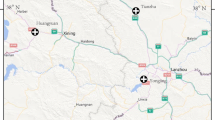
A total of 109 D. nuttalli individuals were sampled from Chengchuan Town, early Banner of Etoke Banner, Ordos City (EEDS), Siziwang Banner, Hohhot City (SZWQ), the Bayan WenduSumu area, Arukorqin Banner, Chifeng City (CF) and Xinbarhu right Banner, Hulun Buir City (HLBE), Inner Mongolia. All ticks were recovered from sheep using tweezers and rubber gloves between April and August 2019 (Table 1, Fig. 1) and were stored at −80 °C.

Collection site map. Samples of Dermacentor nuttalli in four regions of Inner Mongolia were collected. Each color corresponds to different collection regions in Inner Mongolia, and each graphic represents the approximate geographical coordinates of the location in the map for each collection site
DNA extraction
Each D. nuttalli individual was washed in three sterile water baths followed by one absolute ethanol bath, air dried and collected in a sterile EP tube. Each tick was then placed inside a 1.5 ml sterile micro-tube containing sterile micro-beads (Jingxin, Shanghai, China) with a 2:1 ratio of two small steel micro-beads (2 mm diameter) to one large steel micro-bead (4 mm diameter). Samples were cooled in liquid nitrogen for 2 minutes and crushed by a tissue grinder (Jing Xin, Shanghai, China) for two cycles (2 minutes, frequency of 60). The tubes were then briefly centrifuged at a speed of 12,000 rpm and then extracted using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) following the protocol for tissue extraction. The final elution volume was 70 μL, and the samples were stored at −20 °C [29].
Amplification and sequencing
To analyze the genetic structure of our the four populations, we amplified the 16S rRNA, COI and ITS2 gene regions. The primer templates for all three genes were downloaded from the NCBI database. The upstream and downstream primers of 16S rRNA gene were located in the v4 and v7 variable regions, respectively. The COI and ITS2 amplification products were approximately full length (Oligo7). The primer sets of the 16S rRNA, COI and ITS2 gene sequences were synthesized by Shanghai Sangon Biotechnology (Shanghai, China). The 40 μL PCR reaction mixture contained 20 μL Taq PCR Master Mix (Sangon), 1 μL of each primer, 16 μL ddH2O and 2 μL of DNA from each sample. Details about PCR primer pairs, size of the amplification (base pairs [bp]), and annealing temperatures are presented in Table 2. Negative control samples (sterile double-distilled water) were included in each PCR reaction. After electrophoresis, gel imaging was used to determine whether amplification was successful, and if the amplified band was the target band, it was retained. If the target band was bright and free, it was sent directly for sequencing, while other bands were excised, purified using a TIANGEN Gel DNA recovery Kit (TIANGEN, Beijing, China) and cloned using the pGM-T Easy Vector System.
Data analysis
Nucleotide sequences from the three amplified regions (16S rRNA, COI and ITS2) were spliced by SeqMan 7.1. Our sequences were compared to those previously uploaded to GenBank using the Basic Local Alignment Sequence Tool (BLAST) search engine. In order to ascertain the degree of genetic diversity among these four D. nuttalli populations, we estimated a number of parameters under a range of neutrality tests (including Tajima’s D, Fu and Li’s and Fu’s tests) in DnaSP 5.10, including the number of segregating sites, the average number of nucleotide differences, the number of haplotypes, haplotype diversity and the distribution pattern of DNA variation [30, 31]. The genetic differences among populations were calculated by the fixed index (FST). In order to determine the relationship between haplotypes [32], PopART [Population Analysis with Reticulate Trees] version 1.7 was used to construct TCS haplotype network maps. The phylogenetic tree was constructed for all haplotypes of the 16S rRNA, COI and ITS2 gene sequences of the four geographic populations of D. nuttalli in MEGA7.0 using the adjacency method. We downloaded the 16S rRNA, COI and ITS2 gene sequences of the three genes from different geographic populations in China (NCBI), and the higher homology of D. nuttalli with Haemaphysalis longicornis was chosen as the outgroup. The model was the Kimura two-parameter model, and the stability of the tree was evaluated by 1000 bootstrap repeats [33,34,35].
Results
Population genetic analysis
16S rRNA
With the 16S rRNA sequences, the final alignment consisted of 664 base pairs, with 453 variable sites. We detected 26 haplotypes (GenBank accession numbers MW486582–MW486607) that contained seven shared haplotypes (S1, S3, S4, S11, S14, S15, S18), with shared haplotypes varying in certain genetic differentiation among populations. The most frequent haplotype detected was S1, with 55 sequences (50.5% of all sequences). S1 was located in the center of the haplotype network diagram and was a shared haplotype of the four geographic populations, indicating that these populations were relatively stable and could adapt to different environments. The shared haplotype was the dominant haplotype among groups and likely could adapt to the environment more successfully than the remaining 19 unique haplotypes (Fig. 2a). Total average haplotype diversity (Hd) was 0.732 and varied among populations (ranging from 0.5 to 1), and total nucleotide diversity was 0.06091. Diversity values were calculated for each sampled locality. HLBE was the locality with the highest haplotype diversity (h = 0.946), while EEDS had the lowest diversity (h = 0.849). Regarding nucleotide diversity, the lowest diversity was found in SZWQ (pi = 0.00721), and the highest (pi = 0.10363) in CF (Table 3).

a TCS haplotype network of Dermacentor nuttalli based on 16S rRNA genes from four different populations in Inner Mongolia. b TCS haplotype network of Dermacentor nuttalli based on COI from four different populations in Inner Mongolia. c TCS haplotype network of Dermacentor nuttalli based on ITS2 from four different populations in Inner Mongolia
Negative neutrality results obtained for most of the locations analyzed indicated an excess of recently derived haplotypes. While Tajima's D values were significant, Fu’s Fs values were not significant, also indicating that the population had not experienced expansion recently. The FST value reflects the degree of genetic differentiation between two populations, representing the allelic variation between the populations, which was inversely proportional to gene flow. The FST value between CF and SZWQ was greater than 0.05, indicating that there was moderate genetic differentiation among populations, with a small degree of gene flow. The FST values among the other regions were less than 0.05, indicating that the genetic differentiation among these populations was very small, likely as a result of high gene flow (Table 4). The results of analysis of molecular variance (AMOVA) indicated that the population variation of D. nuttalli mainly arises from within the population, and the genetic differentiation between populations is very small (Table 5). The mismatch analysis chart exhibited double peaks, indicating that the four geographic populations did not experience rapid population expansion (Fig. 3a).

a Mismatch distribution analysis for the Dermacentor nuttalli groups based on 16S rRNA. b Mismatch distribution analysis for the Dermacentor nuttalli groups based on COI. c Mismatch distribution analysis for the Dermacentor nuttalli groups based on ITS2
Cytochrome c oxidase subunit I (COI)
In the COI sequences, the final alignment consisted of 1267 base pairs, with 992 variable sites. A total of 57 haplotypes (GenBank Accession Numbers MW507375-MW507431) contained 6 shared haplotypes (C2, C6, C12, C23, C28, C35). The most frequent haplotype detected was C12, with a total of 24 sequences (22% of all sequences), although C35 was located in the center of the network. Given that C35 possessed the greatest number of connections and was most closely related to other haplotypes, it is likely an ancient haplotype (Fig. 2b). Total average haplotype diversity (Hd) was 0.936, while total nucleotide diversity was 0.07402. EEDS was the locality with the highest haplotype diversity (h = 0.997), while SZWQ had the lowest diversity (h = 0.957). Regarding nucleotide diversity, the lowest diversity was found in EEDS (pi = 0.00698) and the highest (pi = 0.16498) in CF (Table 6).
As was the case with 16S rRNA, neutrality results revealed non-significant Tajima's D values and significant Fu's Fs values, confirming that the population had not experienced recent expansion. The FST value between CF and EEDS was the highest (FST = 0.15940), while the FST values for CF, SZWQ and HLBE were all greater than 0.05, indicating that there was moderate genetic differentiation among populations with a small degree of gene flow. The FST values among the three regions of SZWQ, EEDS and HLBE were all less than 0.05, indicating that the genetic differentiation among these populations was very small, likely as a result of high gene flow (Table 7). The results of AMOVA analysis and mismatch analysis were consistent with the 16S rRNA results (Table 8, Fig. 3b).
Internal transcribed spacer 2 (ITS2)
The final alignment of ITS2 sequences consisted of 1689 base pairs, with 1172 variable sites. Seventy-five haplotypes (GenBank accession numbers MW477806–MW477880) were obtained through ITS2 gene analysis, including three shared haplotypes. The most frequent haplotype detected was I1, represented by 17 sequences (14.8% of all sequences). I19 was in the center of the network map, while other shared haplotypes formed small cluster centers. The various clusters were surrounded, which may be related to the close geographic distance. There were no shared haplotypes in CF or HLBE, indicating that there was genetic differentiation between these two geographical populations (Fig. 2c). Total average haplotype diversity (Hd) was 0.966, while the total nucleotide diversity was 0.2619. EEDS was the locality with the highest haplotype diversity (h = 0.997), while CF had the lowest diversity (h = 0.931). Regarding nucleotide diversity, the lowest diversity was found in CF (pi = 0.21148) and the highest (pi = 0.28319) in SZWQ (Table 9).
Neutrality results were the same as that of 16S rRNA and COI. The FST values for CF and SZWQ, and CF and HLBE were all greater than 0.05, and the FST values of other regions were all less than 0.05, indicating that the genetic differentiation among these populations was very small, likely as a result of high gene flow (Table 10). The results of AMOVA analysis and mismatch analysis were consistent with 16S rRNA and COI results (Table 11, Fig. 3c).
Phylogenetic analysis
16S rRNA
We found that the inner populations of D. nuttalli were clustered into one group, and the four geographic populations were mixed, with no obvious geographical differentiation structure. This implies that the genetic evolution among populations was not closely related to the geographical structure. Genetic analysis found that D. nuttalli and D. silvarum were closely related, and others were distributed in the periphery (Fig. 4).

Phylogenetic tree based on the 16S rRNA gene of Dermacentor nuttalli
Cytochrome c oxidase subunit I (COI)
As in the case of 16S rRNA analysis, the haplotypes were gathered as a branch and mixed with each other. However, an additional phylogenetic tree revealed that D. nuttalli was related to D. silvarum and D. marginatus (Fig. 5).

Phylogenetic tree based on COI of Dermacentor nuttalli
Internal transcribed spacer 2 (ITS2)
The phylogenetic tree based on ITS2 gene sequences was identical to that created using the 16S rRNA sequences (Fig. 6).

Phylogenetic tree based on ITS2 of Dermacentor nuttalli
Discussion
This is the first report on the genetic structure of Dermacentor nuttalli, which is the dominant tick species in the Inner Mongolia Autonomous Region [36]. Haplotype diversity (Hd) and nucleotide diversity (Pi) are two important indicators to measure the diversity of a species population [37]. In this study, the genetic diversity of D. nuttalli populations was characterized among four different geographical populations in the Inner Mongolia Autonomous Region with three genes, revealing high haplotype diversity and high nucleotide diversity.
Our results indicate that only a few shared haplotypes were present across our four sampled populations, but that these shared haplotypes were also highly abundant. Shared haplotypes can persist for long periods in a population and can adapt to different environments [38]. While most of the haplotypes detected in our study were exclusive, indicating genetic differentiation among populations, the rich haplotype diversity and high genetic diversity indicate that D. nuttalli may have the ability to adapt to different environments within its large geographical range in Inner Mongolia. Our haplotype network diagram revealed highly connected shared haplotypes, indicating that not only was there frequent gene communication, but that there was also no effect of geographical isolation.
The neutral test results of Tajima's D and Fu's Fs can be used to analyze the historical dynamics of the population. If both are significantly negative, it means that the population has experienced rapid population expansion in history. Otherwise, there is no population expansion, and the population may differentiate. Therefore, all three genes exhibited genetic differentiation with no population expansion. The mismatch analysis maps are both bimodal, with no peak, which is consistent with the results of the neutrality test. FST values represent the genetic differentiation index of populations, which can measure the genetic differentiation among different populations [39]. In our study, the varying degrees of genetic differentiation among our four populations are likely determined by the different evolution rates of the three genes, but overall, the degree of genetic differentiation was small, with high gene flow present among populations. The phylogenetic tree and haplotype network illustrated that the haplotypes were not distributed according to the corresponding geographical units, and that the haplotypes of each geographical population were mixed. Therefore, D. nuttalli populations within Inner Mongolia did not conform to geographical isolation, and the reason for the formation of this structure was the combination of multiple factors.
In a study in Eastern Siberia, the genetic specificity and phylogeny of D. nuttalli across four sites characterized with 16S rRNA and ITS2 obtained only 11 haplotypes, which is significantly fewer than in our study [2]. While the documented population variation in Eastern Siberia was smaller, with lower diversity of haplotypes, the phylogenetic analysis was consistent with ours, in that the closest relative of D. nuttalli was D. silvarum and the next closest related species was D. marginatus. In this study, only four areas with D. nuttalli from Inner Mongolia were selected, which did not cover the distribution area of D. nuttalli in China. Further studies should increase the geographic sampling range to lay a foundation for controlling the further spread of tick-borne diseases and provide more information with which to study the origin and evolution of ticks.
Conclusion
The 16S rRNA gene, COI and ITS2 were used to study the genetic diversity of D. nuttalli sampled from four geographic localities in Inner Mongolia. The genetic diversity of D. nuttalli populations in the Inner Mongolia Autonomous Region reflects adaptation to different geographical environments, and the adaptability of different populations resulted in genetic diversity that led to genetic differentiation among them. However, the genetic differentiation among populations was cryptic and did not form a pedigree geographical structure. The genetic diversity was related not only to gene mutation, but to natural selection and other environmental factors [40]. It was also likely related to the migratory ability, strong adaptability, diversity of hosts, environment and climate, habitat fragments and human activities. The data in this study documented the genetic diversity of ticks in Inner Mongolia and provided enhanced scientific understanding of the evolution of ticks.
Availability of data and materials
All datasets have been included with this article and our sequences have been deposited within GenBank (accession numbers MW486582–MW486607 for 16S rRNA, accession numbers MW507375–MW507431 for COI and accession numbers MW477806–MW477880 for ITS2).
Abbreviations
- COI:
-
Cytochrome c oxidase subunit I
- ITS2:
-
Internal transcribed spacer 2
- PCR:
-
Polymerase chain reaction
- D. nuttalli :
-
Dermacentor nuttalli
- D. silvarum :
-
Dermacentor silvarum
- D. marginatus :
-
Dermacentor marginatus
- AMOVA:
-
Analysis of molecular variance
References
Wang F, Wang D, Guo G, Hu Y, Wei J, Liu J. Species delimitation of the Dermacentor ticks based on phylogenetic clustering and niche modeling. PeerJ. 2019;7:e6911.
Kulakova NV, Khasnatinov MA, Sidorova EA, Adel’shin RV, Belikov SI. Molecular identification and phylogeny of Dermacentor nuttalli (Acari: Ixodidae). Parasitol Res. 2014;113:1787–93.
Voorhees MA, Padilla SL, Jamsransuren D, Koehler JW, Delp KL, Adiyadorj D, et al. Crimean-Congo Hemorrhagic Fever Virus, Mongolia, 2013–2014. Emerg Infect Dis. 2018;24:2202–9.
McFee RB. Tick borne illness - Rocky mountain spotted fever. Dis Mon. 2018;64:185–94.
Battsetseg B, Lucero S, Xuan X, Claveria F, Byambaa B, Battur B. Detection of equine Babesia spp. gene fragments in Dermacentor nuttalli Olenev 1929 infesting Mongolian horses, and their amplification in egg and larval progenies. J Vet Med Sci. 2002(64):727–30.
Wang YC, Li S, Wang Z, Zhang L, Cai Y, Liu Q. Prevalence and identification of Borrelia burgdorferi sensu lato genospecies in ticks from Northeastern China. Vector Borne Zoonotic Dis. 2019;19:309–15.
Kholodilov I, Belova O, Burenkova L, Korotkov Y, Romanova L, Morozova L, et al. Ixodid ticks and tick-borne encephalitis virus prevalence in the South Asian part of Russia (Republic of Tuva). Ticks Tick Borne Dis. 2019;10:959–69.
Sekeyová Z, Danchenko M, Filipčík P, Fournier PE. Rickettsial infections of the central nervous system. PLoS Negl Trop Dis. 2019;13:e0007469.
Sanchez-Vicente S, Tagliafierro T, Coleman JL, Benach JL, Tokarz R. Polymicrobial nature of tick-borne diseases. mBio. 2019;10:e02055-e2119.
Wang GZ, Chen SS, Chao TL, Ji ZB, Hou L, Qin ZJ, et al. Analysis of genetic diversity of Chinese dairy goats via microsatellite markers. J Anim Sci. 2017;95:2304–13.
Ma SJ, Sa KJ, Hong TK, Lee JK. Genetic diversity and population structure analysis in Perilla crop and their weedy types from northern and southern areas of China based on simple sequence repeat (SSRs). Genes Genomics. 2019;41:267–81.
Nagamitsu T, Yasuda M, Saito-Morooka F, Inoue MN, Nishiyama M, Goka K, et al. genetic structure and potential environmental determinants of local genetic diversity in Japanese honeybees (Apis cerana japonica). PLoS ONE. 2016;11:e0167233.
Balik-Meisner M, Truong L, Scholl EH, Tanguay RL, Reif DM. Population genetic diversity in zebrafish lines. Mamm Genome. 2018;29:90–100.
Li YH, Ahmed MZ, Li SJ, Lv N, Shi PQ, Chen XS, et al. Plant-mediated horizontal transmission of Rickettsia endosymbiont between different whitefly species. FEMS Microbiol Ecol. 2017;93:1.
Feng XJ. Genetic Diversity of Hamaphysalis longicornis Population from Four Geographic Regions in China. Hebei Normal University. 2016.
Silva JC, Cornillot E, McCracken C, Usmani-Brown S, Dwivedi A, Ifeonu OO, et al. Genome-wide diversity and gene expression profiling of Babesia microti isolates identify polymorphic genes that mediate host-pathogen interactions. Sci Rep. 2016;6:35284.
Jaimes-Dueñez J, Triana-Chávez O, Mejía-Jaramillo AM. Genetic, host and environmental factors associated with a high prevalence of Anaplasma marginale. Ticks Tick Borne Dis. 2018;9:1286–95.
Araya-Anchetta A, Busch JD, Scoles GA, Wagner DM. Thirty years of tick population genetics: a comprehensive review. Infect Genet Evol. 2015;29:164–79.
Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis. 2014;5:195–207.
Wang T, Zhang S, Pei T, Yu Z, Liu J. Tick mitochondrial genomes: structural characteristics and phylogenetic implications. Parasit Vectors. 2019;12:451.
Williams-Newkirk AJ, Burroughs M, Changayil SS, Dasch GA. The mitochondrial genome of the lone star tick (Amblyomma americanum). Ticks Tick Borne Dis. 2015;6:793–801.
Lv J, Wu S, Zhang Y, Chen Y, Feng C, Yuan X, et al. Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasit Vectors. 2014;7:93.
Zhang DX, Hewitt GM. Assessment of the universality and utility of a set of conserved mitochondrial COI primers in insects. Insect Mol Biol. 1997;6:143–50.
Navajas M, Lagnel J, Gutierrez J, Boursot P. Species-wide homogeneity of nuclear ribosomal ITS2 sequences in the spider mite Tetranychus urticae contrasts with extensive mitochondrial COI polymorphism. Heredity (Edinb). 1998;80:742–52.
Alvarez I, Wendel JF. Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol. 2003;29:417–34.
Hillis DM, Dixon MT. Ribosomal DNA: molecular evolution and phylogenetic inference. Q Rev Biol. 1991;66:411–53.
Marinho MA, Junqueira AC, Azeredo-Espin AM. Evaluation of the internal transcribed spacer 2 (ITS2) as a molecular marker for phylogenetic inference using sequence and secondary structure information in blow flies (Diptera: Calliphoridae). Genetica. 2011;139:1189–207.
Song ZK, Wang XZ, Liang GQ. Molecular evolution and phylogenetic utility of the internal transcribed spacer 2 (ITS2) in Calyptratae (Diptera: Brachycera). J Mol Evol. 2008;67:448–64.
Halos L, Jamal T, Vial L, Maillard R, Suau A, Le Menach A, et al. Determination of an efficient and reliable method for DNA extraction from ticks. Vet Res. 2004;35:709–13.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Rozas J. DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol. 2009;537:337–50.
Meirmans PG, Hedrick PW. Assessing population structure: F(ST) and related measures. Mol Ecol Resour. 2011;11:5–18.
Mendell NL, Reynolds ES, Blanton LS, Hermance ME, Londoño AF, Hart CE, et al. Detection of rickettsiae, borreliae, and ehrlichiae in ticks collected from Walker County, Texas, 2017–2018. Insects. 2019;10:315.
Li K, Stanojević M, Stamenković G, Ilić B, Paunović M, Lu M, et al. Insight into diversity of bacteria belonging to the order Rickettsiales in 9 arthropods species collected in Serbia. Sci Rep. 2019;9:18680.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Zhang YK, Zhang XY, Liu JZ. Ticks (Acari: Ixodoidea) in China: geographical distribution, host diversity, and specificity. Arch Insect Biochem Physiol. 2019;102:e21544.
Vrijenhoek RC. Genetic diversity and fitness in small populations. Conserv Genetics. 1994;1:37–53.
Hackett SJ, Griffiths CS, Bates JM, Klein NK. Re: A commentary on the use of sequence data for phylogeny reconstruction. Mol Phylogenet Evol. 1995;4:350–6.
Weight S. Evolution and the genetics of population variability within and among natural population. Chicago: University of Chicago Press; 1978. p. 10–5.
Slatkin M. Gene flow and the geographic structure of natural populations. Science. 1987;236:787–92.
Acknowledgements
We are very grateful to the Inner Mongolia Center for Disease Control and Prevention for providing tick samples. We are very grateful to the Molecular Biology Research Center of Inner Mongolia Medical University for providing experimental facilities and conditions.
Funding
This work received financial support from the PhD Research Startup Foundation of Inner Mongolia Medical University (Grant No. ykd2017bp009) and the Science and Technology Million Project of Inner Mongolia Medical University (Grant No. YKD2017KJBW002).
Author information
Authors and Affiliations
Contributions
ZG, LW and HC performed laboratory analysis, analyzed the data and wrote the first draft. LM and JFY revised the manuscript. XYS participated in sample collection. SYF directed the experiment and helped to revise the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate.
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gui, Z., Wu, L., Cai, H. et al. Genetic diversity analysis of Dermacentor nuttalli within Inner Mongolia, China. Parasites Vectors 14, 131 (2021). https://doi.org/10.1186/s13071-021-04625-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04625-5