
Abstract
Background
Ticks and tick-borne pathogens (TTBP) are a major constraint to livestock production in Pakistan; despite a high prevalence of TTBPs, knowledge on the capacity of Pakistani ticks to carry pathogens and endosymbionts is limited. Furthermore, mixed infections with multiple microorganisms further complicate and limit the detection potential of traditional diagnostic methods. The present study investigated the tick-borne microorganisms in bovine ticks in Pakistan, employing a high-throughput microfluidic real-time PCR based technique.
Methods
Ticks were collected from clinically healthy cattle (n = 116) and water buffaloes (n = 88) from 30 villages across six districts located in five agro-ecological zones (AEZs) of Pakistan from September to November 2017. The microfluidic real-time PCR was used to test the genomic DNA of individual ticks for the presence of 27 bacterial and eight parasitic microorganisms. Phylogenetic methods were used to assess the genetic relationship of DNA sequences determined herein.
Results
PCR detected DNA of at least one microorganism in each of 221 ticks tested (94.4%, 221/234). DNA-based detection inferred that single pathogens/endosymbionts were the most common (43.4%, 96/221) followed by double (38.9%, 86/221), triple (14.5%, 32/221), quadruple (2.3%, 5/221) and quintuple (0.9%, 2/221) mixed infections. Piroplasms (Babesia/Theileria spp.) were the most prevalent (31.6%, 74/234), followed by Ehrlichia spp. (20%, 47/234) and Anaplasma marginale (7.7%, 18/234). Anaplasma phagocytophilum, A. ovis, A. centrale, Babesia ovis, Borrelia spp., Rickettsia spp., R. massiliae, Bartonella spp. and Hepatozoon spp. were also detected. Endosymbionts such as Francisella-like (91.5%, 214/234) and Coxiella-like (1.3%, 3/234) organisms were also detected in ticks. The highest diversity of microorganisms was detected in Hyalomma anatolicum ticks (test-positive for 14/14 microorganisms), followed by Rhipicephalus microplus (4/14), Hy. hussaini (3/14) and Rh. annulatus (2/14). Ticks collected from cattle carried significantly more frequently piroplasms (41.2%, 54/131; P < 0.05) than those from buffaloes (19.4%, 20/103). However, the overall prevalence of microorganisms did not vary significantly among ticks from the two host species as well as across different AEZs.
Conclusions
To our knowledge, this is the first study to investigate a wide range of tick-borne microorganisms in bovine ticks using a high-throughput diagnostic method from different AEZs in Pakistan. These findings will aid in establishing the distribution patterns and the control of tick-borne pathogens of bovines in Pakistan.

Similar content being viewed by others

Background
Ticks (Acari: Ixodida) are obligate, blood-sucking ectoparasites of vertebrates and are distributed worldwide [1]. They pose a major health and production threat to global animal industries [2,3,4] by affecting their hosts directly by causing irritation, inflammation, anemia, skin/hide damage, toxicosis and paralysis, or indirectly by transmitting a diverse range of pathogens, leading to tick-borne diseases (TBD). It is estimated that 80% of the world’s cattle population, mainly in the tropics and subtropics, is at risk of ticks and tick-borne disease (TTBD) [5]. Furthermore, it is believed that current ongoing climatic and seasonal changes are contributing to the (re)emergence and spread of TTBDs in animals and humans [6].
TTBDs are one of the major health and production constraints for livestock in Pakistan [7] which is the mainstay of Pakistani farmers’ income. Almost 90% of livestock species are kept by small-scale farmers having less than 10 animals, mostly in rural areas [8]. Bovine population is comprised of water buffalo (Bubalus bubalis; n = 40 million) and cattle (Bos indicus and Bos taurus; n = 47.8 million) [9]. A recent study on the perceptions of farmers and veterinary health professional on major bovine health, production and welfare constraints in Pakistan revealed that TTBDs are one of the important challenges for cattle and buffalo productivity [Ghafar et al., 2019a, unpublished]. The main bovine ticks reported from Pakistan are Hyalomma spp. and Rhipicephalus spp. [10,11,12,13,14,15]. These ticks are responsible for transmitting three important TBDs in cattle and buffaloes, including anaplasmosis (caused by Anaplasma centrale and A. marginale), babesiosis (caused by Babesia bigemina and B. bovis) and tropical theileriosis (caused by Theileria annulata) [7]. Some of tick-borne pathogens (TBP) transmitted by Hyalomma anatolicum are of zoonotic importance (e.g. Crimean Congo haemorrhagic fever) [16].
The occurrence and prevalence of tick-borne pathogens (TBPs) in bovines have been reported from different parts of Pakistan [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. However, most of these studies have utilized conventional diagnostic methods for the detection of TBPs in bovines. Although economical and useful for diagnosing clinical cases, these methods have lower sensitivity and specificity [34]. Furthermore, previous studies were confined to the surrounding areas of major metropolitan cities without considering diverse climatic conditions and bovine production systems in Pakistan which are important factors in the design of epidemiological studies [7]. Recently, several studies have utilized molecular methods for the detection of pathogens in bovines [30, 31] and their ticks [18]. However, these studies only targeted the main TBPs pathogens (e.g. Anaplasma spp., Babesia spp. and Theileria spp.). As ticks usually can carry and transmit multiple pathogens, several non-pathogenic commensals and mutualistic microorganisms, known as endosymbionts, concurrently [35], it is important to explore these non-pathogenic organisms as well because they may interact with pathogens and evolve over time to become pathogenic to humans and/or animals (e.g. Coxiella burnetii) [35,36,37].
In the last decade, various TBPs using molecular methods have been reported from bovines in Pakistan [17, 18, 30, 31]. However, these methods are time-consuming as they can detect only a few pathogens at a time and require large volumes of DNA for the detection of multiple pathogens. Furthermore, very little is known about the occurrence of endosymbionts of ticks from Pakistan which might also play a role in tick species survival and disease ecology in bovines [17]. To address these issues, a novel microfluidic-based high-throughput method has been developed and used for epidemiological and surveillance studies in Europe and South Asia [37,38,39]. This method uses a small volume of the nucleic acid to perform parallel real-time PCRs on 48.48 or 96.96 chips and can process up to 2304 or 9216 individual reactions, respectively [37, 40].
This study aimed to investigate molecular epidemiology and the prevalence of microorganisms and their co-infections in bovine ticks from five AEZs of Pakistan using a novel microfluidic-based high-throughput method.
Methods
Tick collection, identification and DNA extraction
Based on physiography, climate, land use and soil type, Pakistan is divided into 10 AEZs [41]. However, the fodder availability and climatic conditions mainly govern the type of livestock species kept across different AEZs. The bovine population is mainly distributed in two provinces (Punjab and Sindh) of Pakistan. Ticks (n = 774) were collected from clinically healthy cattle (n = 242) and water buffaloes (n = 200) from 30 villages located in six districts of Punjab and Sindh from September to November 2017. These districts are located in five different AEZs and include Bahawalpur (sandy desert), Okara (northern irrigated plain), Jhelum and Layyah (arid; two districts were selected to cover diversity within this zone) districts in Punjab and Sukkur (southern irrigated plain) and Thatta (Indus delta) districts in Sindh (Fig. 1).

Map of Pakistan showing the districts (grey-coloured areas) included in this study. The names of districts include Jhelum (1), Okara (2), Layyah (3), Bahawalpur (4), Sukkur (5) and Thatta (6). Abbreviations: KPK, Khyber Pakhtunkhwa; FATA, Federally Administered Tribal Areas; AJ & K, Azad Jammu and Kashmir
Tick specimens from each animal were stored in separate tubes containing 70% ethanol. Subsequently, each tick was morphologically characterized under a dissecting microscope (Olympus SZ40, Japan) using dichotomous keys [42, 43]. Following morphological identification, ticks of the same species from the same animal were pooled in one tube. This resulted in a total of 234 tubes where 131 of those contained ticks from cattle whereas 103 were from buffaloes. DNA was extracted from one tick per tube as per the protocol described previously [Ghafar et al., 2019b, unpublished]. Morphological characterization of ticks was validated using PCR by amplifying cytochrome c oxidase subunit 1 (cox1) gene, 16S rRNA gene, and the second internal transcribed spacer and these results have been submitted for publication previously [Ghafar et al., 2019b, unpublished].
DNA pre-amplification
For DNA amplification, the Perfecta Preamp Supermix (Quanta Biosciences, Beverly, USA) was used according to the manufacturer’s guidelines. Primers (targeted all microorganisms) were pooled combining equal volumes (200 nM final each), and the reaction was performed in a final volume of 5 μl containing 1 μl Perfecta Preamp 5×, 1.25 μl pooled primers mix, 1.5 μl distilled water and 1.25 μl DNA. PCR cycling conditions were one cycle at 95 °C for 2 min followed by 14 cycles at 95 °C for 10 s and 60 °C for 3 min. At the end of the cycling program, the reactions were diluted as 1:10 and the amplicons were stored at – 20 °C until further use.
Microfluidic real-time PCR
High-throughput microfluidic amplification was performed for major TBPs and potential endosymbionts using 48.48 dynamics array in a Bio-MarkTM real-time PCR system (Fluidigm, California, USA). These chips dispensed 48 samples and 48 PCR mixes into individual wells, followed by on-chip real-time PCR reactions in individual chambers and thermal cycling, resulting in 2,304 individual reactions. For more details regarding the development of this high-throughput tool based on real-time microfluidic PCRs (test of sensitivity, specificity, and controls used), please see Michelet et al. 2014 [37].
Targeted microorganisms (and markers) were Borrelia spp. (23S), Bo. burgdorferi (rpoB), Bo. garinii (rpoB), Bo. afzelii (fla), Bo. valaisiana (ospA), Bo. lusitaniae (rpoB), Bo. spielmanii (fla), Bo. bissettii (rpoB), Bo. miyamotoi (glpQ), Bo. mayonii (fla), Bo. bavariensis (pyrG), Anaplasma spp. (16S), A. marginale (msp1), A. platys (groEL), A. phagocytophilum (msp2), A. ovis (msp4), A. centrale (groEL), A. bovis (groEL), Ehrlichia spp. (16S), E. canis (gltA), Neorickettsia mikurensis (groEL), Rickettsia spp. (gltA), R. conorii (ITS), R. slovaca (ITS), R. massiliae (ITS), R. helvetica (ITS), R. aeschlimannii (ITS), R. felis (orfB), Bartonella spp. (ssrA), Ba. henselae (pap31), Francisella spp. (tul4 and fopA), Coxiella spp. (IS1111 and icd), Babesia microti (CCTeta), B. canis (18S), B. ovis (18S), B. bovis (CCTeta), B. caballi (rap1), Babesia str. EU1 (18S), B. divergens (hsp70), B. vulpes (cox1), Theileria spp. (18S) and Hepatozoon spp. (18S). Briefly, amplifications were performed using 6-carboxyfluorescein (FAM)- and black hole quencher (BHQ1)-labelled TaqMan probes with TaqMan Gene expression master mix as per manufacturer’s recommendations (Applied Biosystems, Massachusetts, USA) [37]. PCR cycling conditions comprised of a denaturation step at 95 °C for 5 min followed by 45 cycles at 95 °C for 10 s, 60 °C for 15 s and 40 °C for 10 s. One negative control (water) was included per chip. Detection of ticks’ 16S gene served as a positive control for the confirmation of DNA extraction. To assess PCR inhibitory molecules present in tick DNA samples, DNA from Escherichia coli (EDL933 strain) was added to each sample as an internal inhibition control, and primers and probe specific for the eae gene of E. coli were used.
Validation of results by PCR and DNA sequencing
Microfluidic real-time PCR results were confirmed through conventional and nested PCR using different primers (see Additional file 1: Table S1) than those of the BioMarkTM system. Amplicons were sequenced by Eurofins MWG Operon (Ebersberg, Germany) and assembled using the Geneious Prime software (Biomatters Ltd, Auckland, New Zealand). An online BLAST (National Center for Biotechnology Information) was used to identify the sequenced organisms.
Phylogenetic analyses
The partial nucleotide sequences of microorganisms obtained for 18S rRNA, 16S rRNA and citrate synthase (gltA) genes were used to assess the genetic relationships with those of species of Babesia/Theileria, Ehrlichia and Rickettsia, respectively. Reference sequences were downloaded for each pathogen from GenBank and aligned separately (Babesia/Theileria over 582 bp; Ehrlichia over 618 bp; Rickettsia over 375 bp) with sequences obtained in this study, using ClustalW [44] in the software MEGA v7.00 [45]. The best-fit evolutionary models for each dataset were selected based on Corrected Akaikeʼs information criterion (cAIC) and Bayesian information criterion (BIC) using MEGA. Phylogenetic trees were constructed using the Neighbour-joining (NJ) and Maximum Likelihood (ML) methods in MEGA and Bayesian Inference (BI) method using Mr Bayes in Geneious Prime [46]. Each Bayesian analysis was run over 20,000,00 generations (ngen = 20,000,00) with two runs and every 400th tree was saved (samplefreq = 400). For the NJ tree estimations, evolutionary distances were computed using the p-distance method whereas for the ML method, initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log-likelihood value. All positions containing gaps and missing data were eliminated. Bootstrapping method (10,000 replicates) was used to assess the reliability of internal branches and all trees were visualized and edited using MEGA. Plasmodium falciparum (GenBank: M19172), A. marginale (GenBank: AF414872) and R. bellii (GenBank: AY362703) were used as outgroups for Babesia/Theileria, Ehrlichia and Rickettsia, respectively.
Statistical analyses
Chi-square and Fisher’s exact tests were used to evaluate the overall prevalence of microorganisms and to compare their prevalence in different AEZs. Data were analyzed using GraphPad 5 Prism program (GraphPad Software Inc., La Jolla, CA, USA). The multiple correspondence analysis (MCA) was used to analyze the pattern of the association between infections (co-infections) and their distribution across provinces, districts and bovine host species. The inertia values were calculated by standard ‘Burt matrix’ method. The analyses were performed using the software, Statgraphics Centurion v. 16.1.03 (StatPoint technologies Inc, Warrenton, VA).
Results
Diversity of microorganisms detected in ticks
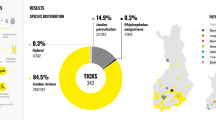
PCR detected DNA of at least one microorganism in each of 221 ticks tested (94.4%, 221/234) (Fig. 2; Table 1). Francisella-like endosymbionts (FLEs) were the most common (91.5%, 214/234) microorganisms detected in ticks (Table 1) followed by piroplasms (31.6%, 74/234), Ehrlichia spp. (20%, 47/234), A. marginale (7.7%, 18/234), Borrelia spp. (6.4%, 15/234), A. centrale (2.6%, 6/234), Rickettsia spp. (2.1%, 5/234), A. ovis (1.7%, 4/234), R. massiliae (1.7%, 4/234) and Coxiella-like endosymbionts (CLEs) (1.3%, 3/234). Furthermore, A. phagocytophilum, Bartonella spp., Babesia ovis and Hepatozoon spp. were also detected in 0.4% of ticks.

Information about microorganisms detected in ticks collected from bovines in different districts of two provinces, Punjab and Sindh, Pakistan
Overall, a high percentage of ticks was test-positive for DNA of microorganisms, with no significant variation (χ2 = 10.0, df = 5, P = 0.075) across districts (Sukkur and Layyah, 96%; Okara and Bahawalpur, 95%; Thatta, 93%; Jhelum, 87%) in different AEZs (Table 1). The prevalence of various pathogens (i.e. excluding endosymbionts) in ticks was high across districts (Bahawalpur, 60%; Thatta, 57%; Sukkur, 56%; Okara, 55%; Layyah, 50%; Jhelum, 33%) and it varied significantly (χ2 = 19.2, df = 5, P = 0.0018). The highest diversity of microorganisms was found in district Okara where DNA of eight of 14 (57.1%) microorganisms tested for was detected followed by Bahawalpur and Sukkur (7/14) and Layyah and Thatta (6/14) (Table 1). Ehrlichia spp., FLEs and piroplasms were found in all districts while Bartonella spp. and CLEs were only detected in Okara, B. ovis and Hepatozoon spp. in Layyah, and R. massiliae from Thatta. The prevalence of pathogens in ticks was significantly different between Jhelum and Bahawalpur (P = 0.0002), Jhelum and Thatta (P = 0.0010), Jhelum and Sukkur (P = 0.0017) Jhelum and Layyah (P = 0.0214) and Okara and Jhelum (P = 0.0027). At the provincial level, the diversity of microorganisms in ticks was higher in Punjab (11/14) than in Sindh (9/14) (Table 1). Based on the bovine host species, cattle ticks carried significantly higher piroplasms (41.2%, 54/131, P = 0.0011) than buffalo ticks (19.4%, 20/103); however, the overall prevalence of microorganisms in ticks did not vary significantly between both host species (Table 1).
Among four bovine tick species identified herein, the highest microorganism diversity (14/14) was found in Hy. anatolicum, with 96.2% (204/212) of them being test-positive for DNA of at least one microorganism, followed by Rh. microplus (4/14), Hy. hussaini (3/14) and Rh. annulatus (2/14) (Fig. 3, Table 2). However, no microorganism was detected in the single specimen of Hy. scupense analyzed (Table 2).

Information about microorganisms detected in different species of ticks collected from bovines in different districts of two provinces, Punjab and Sindh, Pakistan
Co-infections of microorganisms in ticks
Single infections with DNA of various microorganisms were present in 41% (96/234) of ticks, with FLEs (97.9%, 94/96) and piroplasm spp. (2.1%, 2/96) being the main microorganisms detected (Table 3). Among mixed infections of ticks, the highest percentage was found for double infections (36.8%, 86/234) followed by the triple (14.7%, 32/234), quadruple (2.1%, 5/234) and quintuple (0.9%, 2/234) infections. The most common double co-infection was with FLEs and piroplasm species (15.8%, 37/234) followed by FLEs and Ehrlichia spp. (9.4%, 22/234) whereas the most common triple co-infection was due to FLEs, piroplasm species and Ehrlichia spp. (6.8%, 16/234) (Table 3). Hyalomma anatolicum were positive for all types of single as well as mixed infections (i.e. single, double, triple, quadruple and quintuple) while the remaining tick species were positive for up to triple co-infections (Table 2). The distribution of tick infections with one or more microorganisms in various districts varied significantly, and ticks from Okara were positive for all five types of infections while those from Bahawalpur and Layyah had single, double, triple and quadruple infections. Ticks from Jhelum, Sukkur and Thatta were test-positive for DNA of one, two or three microorganisms only (Table 3).
Genetic relationship of DNA sequences of selected microorganisms
Three different phylogenetic methods (BI, ML and NJ) were used to analyze (separately) genetic relationships of 16S rRNA gene sequences of Ehrlichia spp., 18S rRNA gene sequences of Babesia and Theileria spp. and gltA sequences of Rickettsia spp. with those of respective previously published sequences. The topologies of all three trees for each target organism were similar; hence, only NJ trees are provided herein (Figs. 4, 5, 6). Two unique 16S rRNA sequences of Ehrlichia spp. (GenBank: MN726921 and MN726922) detected herein grouped with those previously published sequences from Thailand (GenBank:AF497581), Tibet, China (GenBank:AF414399) and Multan, Pakistan (GenBank:MH250197) (Fig. 4), and had a 99.8–100% similarity to these three reference sequences. Two unique 18S rRNA gene sequences of piroplasms (GenBank: MN726546 and MN726547) were identified and one of these grouped with T. annulata sequences from Pakistan (GenBank: JQ743630) and Turkey (GenBank: MK918607), whereas the second sequence clustered with that of B. occultans from South Africa (GenBank: U09834) (Fig. 5). For Rickettsia spp., only one unique gltA sequence (GenBank: MN728990) was found which grouped with R. aeschlimannii (GenBank: DQ235776) from Russia, R. rhipicephali (GenBank: U59721) from USA and Rickettsia sp. Bar (GenBank: U59720) from Spain (Fig. 6).

Genetic relationship of 16S rRNA gene sequences of Ehrlichia spp. identified in the present study (starred) with those of Ehrlichia spp. available on GenBank. The sequence data (618 bp) were analysed using Neighbour Joining (NJ), Maximum Likelihood (ML) and Bayesian Inference (BI) methods. There was a concordance among the topology of the BI, ML and NJ trees (not shown) and only NJ tree is presented here. Nodal support is given as a posterior probability of BI and bootstrap values for NJ and ML. The tree was rooted using A. marginale as outgroup. The scale-bar indicates the number of inferred substitutions per site

Genetic relationship of 18S rRNA gene sequences of Babesia/Theileria spp. identified in the present study (starred) with those of Babesia/Theileria spp. available on GenBank. The sequence data (582 bp) were analysed using Neighbour Joining (NJ), Maximum Likelihood (ML) and Bayesian Inference (BI) methods. There was a concordance among the topology of the BI, ML and NJ trees (not shown) and only NJ tree is presented here. Nodal support is given as a posterior probability of BI and bootstrap values for NJ and ML. The tree was rooted using Plasmodium falciparum as outgroup. The scale-bar indicates the number of inferred substitutions per site

Genetic relationship of gltA sequences of Rickettsia spp. identified in the present study (starred) with those of Rickettsia spp. available on GenBank. The sequence data (375 bp) were analysed using Neighbour Joining (NJ), Maximum Likelihood (ML) and Bayesian Inference (BI) methods. There was a concordance among the topology of the BI, ML and NJ trees (not shown) and only NJ tree is presented here. Nodal support is given as a posterior probability of BI and bootstrap values for NJ and ML. The tree was rooted using Rickettsia bellii as outgroup. The scale-bar indicates the number of inferred substitutions per site
Multiple correspondence analyses
Multiple correspondence analysis revealed no correlation between microorganisms and different districts, hosts or AEZs as microorganism clustered close to the centre of multidimensional matrices (Additional file 2: Figure S1, Additional file 3: Figure S2, Additional file 4: Figure S3).
Discussion
This is the first study to utilize a high-throughput microfluidic technique for the detection of TBPs in bovine ticks collected from five AEZs of Pakistan. This real-time PCR technique offers a unique ability to detect multiple pathogens in ticks [37,38,39]; here, we detected two protozoan genera (i.e. Babesia and Theileria) and 12 bacteria (species of Anaplasma, Bartonella, Borrelia, Coxiella, Ehrlichia, Francisella, Hepatozoon and Rickettsia) of veterinary and/or public health significance. Interestingly, this study revealed that bovine ticks from Pakistan have (i) a diverse range of endosymbionts, with a higher prevalence of FLEs (91.5%); (ii) a high prevalence of TBPs (127 positive of 234 ticks tested); and (iii) a high frequency of co-infections (56.6% of total infections were co-infections). Due to low cycle threshold (Cq) values, genetic characterization was not successful for some of the detected pathogens. As feeding ticks were collected from bovine hosts, we could not justify the bovine or tick origin of detected microorganisms. Likewise, this study does not argue the co-transmission of multiple microorganisms from a tick to the bovine host. However, such studies provide a snapshot of potential TBPs present in a region and help understanding disease(s) dynamics. Furthermore, all ticks tested in this study were collected during one season of the year, and therefore, this study does not inform about the seasonal variation of TBPs.
In this study, we detected the DNA of four Anaplasma spp. (A. centrale, A. marginale, A. ovis and A. phagocytophilum) in three species of ticks, Hy. anatolicum, Rh. microplus and Rh. annulatus. Bovine anaplasmosis is an endemic TBD in Pakistan, and is mainly caused by A. marginale [18, 21, 22]. We found the highest prevalence of A. marginale in ticks from Okara district which could be attributed to high prevalence of Rh. microplus in this region. Furthermore, this tick is the main vector for A. marginale [47]. This study reports the detection of A. phagocytophilum DNA in Hy. anatolicum for the first time from Pakistan. Anaplasma phagocytophilum is a zoonotic TBP infecting several mammalian species and can cause an acute febrile condition in humans, known as human granulocytic anaplasmosis [48]. In Pakistan, A. phagocytophilum is mainly responsible for equine granulocytic anaplasmosis and has been reported from equines [49]. Recently, Anaplasma species has been reported in bovines from Pakistan [50]. Main tick vectors for the transmission of A. phagocytophilum are Ixodes spp. but several other tick species have also been found positive for its DNA [51]. Additionally, A. ovis was detected in four specimens of Hy. anatolicum and it causes ovine anaplasmosis [52,53,54]. Previously, A. ovis has been detected in small ruminants from Khyber Pakhtunkhwa [55] and in three tick species (Hy. anatolicum, Hy. dromedarii and Rh. microplus) from Punjab, Pakistan [18]. The detection of A. ovis DNA in multiple tick species indicates the challenges associated with multi-species livestock farming by small-holder dairy farmers.
We found the DNA of Ehrlichia species in 47 specimens of Hy. anatolicum from all six districts, with the highest percentage in Layyah (11/26) and lowest in Jhelum (1/24). Both districts are located within the same AEZ but hold a diverse topography because Jhelum district has plain and mountainous areas while Layyah district is comprised of desert and plain lands. Furthermore, in Layyah, summer temperatures are higher, and the livestock population there is three times larger than in Jhelum, thereby possibly providing more suitable host and environmental factors for the growth of ticks and the transmission of TBPs. Sequence and phylogenetic analyses of 16S rRNA gene fragments of Ehrlichia spp. detected in this study showed a close similarity to those previously published from China (Ehrlichia sp. Tibet; [56]), Pakistan (Ehrlichia sp. Multan; [18]) and Thailand (Ehrlichia sp. EBm52; [57]) (Fig. 4). Although studies from Pakistan have reported ehrlichiosis in dogs using blood smear examination [58] and molecular tools [38, 59], no information is available on the epidemiology of ehrlichiosis in bovines from Pakistan. Recently, one study investigated TBPs in bovine ticks from Punjab, Pakistan, and reported a high prevalence of Ehrlichia spp. (21%) [18]. The high prevalence of Ehrlichia spp. in ticks reported in our study as well as in the previous study [18] suggests that Hyalomma ticks might be transmitting ehrlichiosis in bovines in Pakistan; however, this hypothesis warrants further investigation.
A high number of piroplasms (Theileria/Babesia spp.; 31.6%) were detected in three tick species (i.e. Hy. anatolicum, Rh. microplus and Rh. annulatus). Microfluidic results were validated using piroplasm-specific primers targeting 18S rRNA gene and revealed that out of seven Theileria/Babesia-positive samples sequenced, five identical sequences belonged to T. annulata while two remaining identical sequences were identified as B. occultans. Since sequencing was not undertaken for all samples, we were not able to determine individual prevalence rates for Babesia and Theileria spp. in ticks. Babesia ovis was also found in one tick sample using species-specific primers. All of these pathogens have been previously reported from Pakistan [18, 30, 31, 60]. Theileriosis is an economically important disease affecting domestic and wild ruminants in tropical and subtropical regions of the world [61]. In Pakistan, this disease is mainly caused by the host cell-transforming sporozoan T. annulata and is responsible for high economic losses due to reduced production and mortalities, particularly in exotic and cross-bred animals [7]. Additionally, T. orientalis has been reported in bovines and ticks from Pakistan [19, 31] but clinical cases associated with the Theileria species complex still remain to be seen. Bovine babesiosis is another important TBD in Pakistan and is mainly caused by B. bovis and B. bigemina [7]. However, we did not detect B. bovis DNA in any tick sample using species-specific primers and no tick was tested for B. bigemina, both of which were previously reported to occur in Pakistan [17, 18, 30]. However, during validation using Babesia/Theileria genera-specific primers, B. occultans was found in two Hy. anatolicum samples. Since its first detection in South Africa in 1981, B. occultans had been considered as apathogenic with its distribution limited to sub-Saharan countries [62, 63]. However, recently it was associated with a clinical babesiosis outbreak in Italy [64] and was also detected in canine blood from India [65] and ticks from Pakistan [18] and China [66]. As climate change can impact the distribution and occurrence of vector-borne diseases [67], B. occultans might become an important TBP for bovines in future. Furthermore, we found that ticks from cattle carried significantly higher piroplasms compared to those collected buffaloes which might be due to the natural variation in the susceptibility of the hosts to different pathogens as buffaloes are known to be asymptomatic reservoirs of Babesia spp., which are pathogenic to cattle [68].
This study detected, for the first time, DNA of Borrelia in 15 tick specimens of Hy. anatolicum (n = 14) and Rh. microplus (n = 1) in Pakistan. However, none of the seven species-specific primer pairs employed in this study could verify species identity, and therefore, the pathogenic and/or zoonotic potential of the detected Borrelia species could not be established. Borrelia species include spirochetes belonging to Lyme borreliosis and relapsing fever spirochete groups as well as intermittent clades and are transmitted by the body louse and hard and soft ticks [69, 70]. There is no surveillance and/or diagnostic system in place for borreliosis in animals or humans in Pakistan; thereby it remained unknown until now. Further testing is required to characterise Borrelia spp. and to assess their pathogenic and zoonotic potential.
We found that one Hy. hussaini and three Hy. anatolicum ticks contained DNA of R. massiliae, whereas, five ticks (one Hy. hussaini and four Hy. anatolicum) were positive for the DNA of unidentified Rickettsia spp. DNA sequencing followed by phylogenetic analyses revealed that gltA sequences of Rickettsia spp. determined herein clustered with those of R. aeschlimannii (Fig. 6). This study provides the first report of Rickettsia spp. in bovine ticks from Sindh Province of Pakistan as they have previously been detected in ticks from Punjab [18, 71], and Islamabad and Azad Jammu and Kashmir, Pakistan [17]. Rickettsia massiliae and R. aeschlimannii belong to spotted fever group and both species are of public health significance [72, 73] as reported in the USA, Africa, Asia and Europe [74, 75]. Overall, rickettsial infections rank second after dengue in Southeast Asia as the cause of non-malarial febrile illnesses [76]. However, due to the lack of clinical testing facilities in Pakistan and other developing countries, these infections in animals and humans either remain unreported or underreported. We also found Bartonella spp. and Hepatozoon spp. in Hy. anatolicum ticks from Okara and Layyah districts, respectively. However, species identification of these organisms could not be established, and therefore, the pathogenic or zoonotic potential could not be assessed.
In this study, a high prevalence of endosymbionts such as FLEs (91.5%) was inferred, for the first time, in Hy. anatolicum, Rh. microplus and Hy. hussaini ticks from Pakistan. Previously, a study by Karim et al. [17] investigated tick microbiomes of specimens collected from different livestock species from various parts of Pakistan and reported endosymbionts, Francisella (0.2% in Hy. anatolicum ticks from buffaloes) and Coxiella (7.9% in ticks belonging to the genera Rhipicephalus, Haemphysalis, Hyalomma and Ornithodoros) for the first time from Pakistan. Endosymbionts are non-pathogenic mutualistic and/or commensal microbes, and they are also abundant in ticks. Main tick endosymbionts belong to the genera of Rickettsia, Francisella and Coxiella [36, 77]. Endosymbionts can (i) have multiple effects (detrimental or beneficial) on their carriers [77,78,79]; (ii) cause diseases to humans [80, 81]; and (iii) interact with other TBPs and affect their colonization and transmission [82,83,84]. The composition of endosymbionts can vary significantly among ticks in different parts of the world and it can be affected by several factors such as environment [85], season [86], geographical location [87], tick species [86], tick life stage [88], feeding status [85] and co-existing pathogens [89]. Given that endosymbionts could play role in the prevalence and transmission of various pathogens, further investigations are required to explore the endosymbiotic communities in ticks infesting animals in Pakistan. Mixed infections with FLEs and piroplasm spp. and/or Ehrlichia spp. were most commonly detected in ticks across all AEZs. Ticks co-infected with multiple pathogens might pose a greater risk to animals as well as humans because of increased risk of co-infections which would ultimately increase the clinical complexity of diseases. High co-infection rates of microorganisms in ticks highlights the need of such studies to be conducted on larger populations to further assess pathogen communities and their potential interactions as well as their pathogenic or zoonotic potential.
Conclusions
This study reports that multiple TBPs of animal and public health significance are carried by bovine tick population in Pakistan. Co-infections with multiple pathogens are common, and endosymbionts are ubiquitously present. Overall, the prevalence of TBPs does not vary significantly across different AEZs of the country, but some pathogens may be restricted to particular regions or AEZs. Future studies are required to characterize endosymbionts further to explore their possible interaction(s) with pathogens transmitted by ticks collected from a larger animal population across different seasons in various AEZs.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
Keirans J, Durden L. Tick systematics and identification. In: Goodman JL, Dennis DT, Sonenshine DE, editors. Tick-borne diseases of humans. Washington: ASM Press; 2005.
Sonenshine DE, Roe RM. Biology of ticks, vol. 1. 2nd ed. Oxford: Oxford University Press; 2014.
Anderson JF, Magnarelli LA. Biology of ticks. Infect Dis Clin N Am. 2008;22:195–215.
Uilenberg G. Veterinary significance of ticks and tick-borne diseases. In: Fivaz B, Petney T, Horak I, editors. Tick vector biology: medical and veterinary aspects. Berlin: Springer; 1992. p. 23–33.
de Castro JJ. Sustainable tick and tickborne disease control in livestock improvement in developing countries. Vet Parasitol. 1997;71:77–97.
Dantas-Torres F. Climate change, biodiversity, ticks and tick-borne diseases: the butterfly effect. Int J Parasitol Parasites Wildl. 2015;4:452–61.
Jabbar A, Abbas T, Sandhu ZD, Saddiqi HA, Qamar MF, Gasser RB. Tick-borne diseases of bovines in Pakistan: major scope for future research and improved control. Parasites Vectors. 2015;8:283.
Zia UE, Mahmood T, Ali MR. Dairy development in Pakistan. Rome: Food and Agriculture Organization of the United Nations; 2011.
Anonymous. Pakistan Economic Survey. Government of Pakistan: Ministery of Finance, Islamabad; 2018–19. p. 11–33. http://www.finance.gov.pk/survey_1819.html. Accessed 16 Aug 2019.
Farooqi SH, Ijaz M, Saleem MH, Rashid MI, Oneeb M, Khan A, et al. Distribution of ixodid tick species and associated risk factors in temporal zones of Khyber Pakhtunkhwa Province, Pakistan. Pak J Zool. 2017;49:2011–7.
Iqbal A, Sajid MS, Khan MN, Khan MK. Frequency distribution of hard ticks (Acari: Ixodidae) infesting bubaline population of district Toba Tek Singh, Punjab, Pakistan. Parasitol Res. 2013;112:535–41.
Rehman A, Nijhof AM, Sauter-Louis C, Schauer B, Staubach C, Conraths FJ. Distribution of ticks infesting ruminants and risk factors associated with high tick prevalence in livestock farms in the semi-arid and arid agro-ecological zones of Pakistan. Parasites Vectors. 2017;10:190.
Sajid MS, Iqbal Z, Khan MN, Muhammad G. Point prevalence of hard ticks (ixodids) infesting domestic ruminants of lower Punjab, Pakistan. Int J Agric Biol. 2008;10:349–51.
Sajid MS, Iqbal Z, Khan MN, Muhammad G, Khan MK. Prevalence and associated risk factors for bovine tick infestation in two districts of lower Punjab, Pakistan. Prev Vet Med. 2009;92:386–91.
Ali A, Khan MA, Zahid H, Yaseen PM, Qayash Khan M, Nawaz F, et al. Seasonal dynamics, record of ticks infesting humans, wild and domestic animals and molecular phylogeny of Rhipicephalus microplus in Khyber Pakhtunkhwa Pakistan. Front Physiol. 2019;10:793.
Atif M, Saqib A, Ikram R, Sarwar MR, Scahill S. The reasons why Pakistan might be at high risk of Crimean Congo haemorrhagic fever epidemic; a scoping review of the literature. Virol J. 2017;14:63.
Karim S, Budachetri K, Mukherjee N, Williams J, Kausar A, Hassan MJ, et al. A study of ticks and tick-borne livestock pathogens in Pakistan. PLoS Negl Trop Dis. 2017;11:e0005681.
Rehman A, Conraths FJ, Sauter-Louis C, Krücken J, Nijhof AM. Epidemiology of tick-borne pathogens in the semi-arid and the arid agro-ecological zones of Punjab Province, Pakistan. Transbound Emerg Dis. 2019;66:526–36.
Ahmad I, Khawja A, Shams S, Ayaz S, Khan S, Akbar N. Detection of babesiosis and identification of associated ticks in cattle. Int J Bioassays. 2014;3:3195–9.
Ali Z, Maqbool A, Muhammad K, Khan M, Younis M. Prevalence of Theileria annulata infected hard ticks of cattle and buffalo in Punjab, Pakistan. J Anim Plant Sci. 2013;23:20–6.
Ashraf QU, Khan AU, Khattak RM, Ali M, Shaikh RS, Ali M, et al. A report on the high prevalence of Anaplasma sp. in buffaloes from two provinces in Pakistan. Ticks Tick Borne Dis. 2013;4:395–8.
Atif F, Khan M, Muhammad F, Ahmad B. Sero-epidemiological study of Anaplasma marginale among cattle. J Anim Plant Sci. 2013;23:740–4.
Atif FA, Khan MS, Iqbal HJ, Arshad GM, Ashraf E, Ullah S. Prevalence of Anaplasma marginale, Babesia bigemina and Theileria annulata infections among cattle in Sargodha District, Pakistan. Afr J Agric Res. 2012;7:3302–7.
Bhutto B, Gadahi JA, Khuhro A, Rajput HM, Bhutto F, Rajput MA, et al. A Survey on haemo-protozoan parasites in buffaloes of Landhi Dairy Colony, Karachi-Pakistan. Int J Agro Vet Med Sci. 2012;6:73–6.
Iqbal F, Khattak R, Ozubek S, Khattak M, Rasul A, Aktas M. Application of the reverse line blot assay for the molecular detection of Theileria and Babesia sp. in sheep and goat blood samples from Pakistan. Iran J Parasitol. 2013;8:289.
Irshad N, Qayyum M, Hussain M, Khan M. Prevalence of tick infestation and theileriosis in sheep and goats. Pak Vet J. 2010;30:178–80.
Naz S, Maqbool A, Ahmed S, Ashraf K, Ahmed N, Saeed K, et al. Prevalence of theileriosis in small ruminants in Lahore-Pakistan. J Vet Anim Sci. 2012;2:16–20.
Qayyum M, Farooq U, Samad H, Chauhdry H. Prevalence, clinicotherapeutic and prophylactic studies on theileriosis in district Sahiwal (Pakistan). J Anim Plant Sci. 2010;20:266–70.
Sajid M, Siddique R, Khan S, Iqbal Z, Khan M. Prevalence and risk factors of anaplasmosis in cattle and buffalo populations of district Khanewal, Punjab. Pak Glob Vet. 2014;12:146–53.
Hassan MA, Liu J, Rashid M, Iqbal N, Guan G, Yin H, et al. Molecular survey of piroplasm species from selected areas of China and Pakistan. Parasites Vectors. 2018;11:457.
Hassan MA, Liu J, Sajid MS, Mahmood A, Zhao S, Abbas Q, et al. Molecular detection of Theileria annulata in cattle from different regions of Punjab, Pakistan, by using recombinase polymerase amplification and polymerase chain reaction. J Parasitol. 2018;104:196–201.
Farooqi SH, Ijaz M, Rashid MI, Aqib AI, Ahmad Z, Saleem MH, et al. Molecular epidemiology of Babesia bovis in bovine of Khyber Pakhtunkhwa, Pakistan. Pak Vet J. 2017;37:275–80.
Khan MK, He L, Hussain A, Azam S, Zhang WJ, Wang LX, et al. Molecular epidemiology of Theileria annulata and identification of 18S rRNA gene and ITS regions sequences variants in apparently healthy buffaloes and cattle in Pakistan. Infect Genet Evol. 2013;13:124–32.
Salih DA, El Hussein AM, Singla LD. Diagnostic approaches for tick-borne haemoparasitic diseases in livestock. J Vet Med Anim Health. 2015;7:45–56.
Duron O, Noël V, Mccoy KD, Bonazzi M, Sidi-Boumedine K, Morel O, et al. The recent evolution of a maternally-inherited endosymbiont of ticks led to the emergence of the Q fever pathogen, Coxiella burnetii. PLoS Pathog. 2015;11:e1004892.
Moutailler S, Moro CV, Vaumourin E, Michelet L, Tran FH, Devillers E, et al. Co-infection of ticks: the rule rather than the exception. PLoS Negl Trop Dis. 2016;10:e0004539.
Michelet L, Delannoy S, Devillers E, Umhang G, Aspan A, Juremalm M, et al. High-throughput screening of tick-borne pathogens in Europe. Front Cell Infect Microbiol. 2014;4:103.
Cabezas-Cruz A, Allain E, Ahmad AS, Saeed MA, Rashid I, Ashraf K, et al. Low genetic diversity of Ehrlichia canis associated with high co-infection rates in Rhipicephalus sanguineus (s.l.). Parasites Vectors. 2019;12:12.
Sprong H, Fonville M, van Leeuwen AD, Devillers E, Ibañez-Justicia A, Stroo A, et al. Detection of pathogens in Dermacentor reticulatus in northwestern Europe: evaluation of a high-throughput array. Heliyon. 2019;5:e01270.
Liu J, Hansen C, Quake SR. Solving the “world-to-chip” interface problem with a microfluidic matrix. Anal Chem. 2003;75:4718–23.
Khan AG. The characterization of the agro ecological context in which FAnGR (farm animal genetic resource) are found. ILRI. 2004. https://cgspace.cgiar.org/handle/10568/17258. Accessed 15 Oct 2019.
Barker SC, Walker AR. Ticks of Australia. The species that infest domestic animals and humans. Zootaxa. 2014;3816:1–144.
Walker A, Bouattour A, Camicas J, Estrada-Pena A, Horak I, Latif A, et al. Ticks of domestic animals in Africa. A guide to identification of species. Edinburgh: Bioscience Reports; 2003.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Kocan KM, de la Fuente J, Blouin EF, Coetzee JF, Ewing S. The natural history of Anaplasma marginale. Vet Parasitol. 2010;167:95–107.
Dumler JS, Barbet AF, Bekker C, Dasch GA, Palmer GH, Ray SC, et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‛HGE agentʼ as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51:2145–65.
Saleem S, Ijaz M, Farooqi SH, Rashid MI, Khan A, Masud A, et al. First molecular evidence of equine granulocytic anaplasmosis in Pakistan. Acta Trop. 2018;180:18–25.
Iqbal N, Mukhtar MU, Yang J, Sajid MS, Niu Q, Guan G, et al. First molecular evidence of Anaplasma bovis and Anaplasma phagocytophilum in bovine from central Punjab, Pakistan. Pathogens. 2019;8:155.
Battilani M, De Arcangeli S, Balboni A, Dondi F. Genetic diversity and molecular epidemiology of Anaplasma. Infect Genet Evol. 2017;49:195–211.
Shabana I, Alhadlag N, Zaraket H. Diagnostic tools of caprine and ovine anaplasmosis: a direct comparative study. BMC Vet Res. 2018;14:165.
Renneker S, Abdo J, Salih D, Karagenç T, Bilgiç H, Torina A, et al. Can Anaplasma ovis in small ruminants be neglected any longer? Transbound Emerg Dis. 2013;60:105–12.
Cabezas-Cruz A, Gallois M, Fontugne M, Allain E, Denoual M, Moutailler S, et al. Epidemiology and genetic diversity of Anaplasma ovis in goats in Corsica, France. Parasites Vectors. 2019;12:3.
Talat R, Khanum T, Hayat A. Studies on mammalian haematozoan parasites of NWFP Pakistan. Pak J Biol Sci. 2005;8:726–9.
Wen B, Jian R, Zhang Y, Chen R. Simultaneous detection of Anaplasma marginale and a new Ehrlichia species closely related to Ehrlichia chaffeensis by sequence analyses of 16S ribosomal DNA in Boophilus microplus ticks from Tibet. J Clin Microbiol. 2002;40:3286–90.
Parola P, Cornet JP, Sanogo YO, Miller RS, Van Thien H, Gonzalez JP, et al. Detection of Ehrlichia spp., Anaplasma spp., Rickettsia spp., and other eubacteria in ticks from the Thai-Myanmar border and Vietnam. J Clin Microbiol. 2003;41:1600–8.
Abbas G, Mughal M, Munir A, Azeem W. Lahore canine fever in a racing greyhound. Adv Anim Vet Sci. 2015;3:332–3.
Malik MI, Qamar M, Ain Q, Hussain MF, Dahmani M, Ayaz M, et al. Molecular detection of Ehrlichia canis in dogs from three districts in Punjab (Pakistan). Vet Med Sci. 2018;4:126–32.
Shahzad W, Haider N, Mansur-ud-Din A, Munir R, Saghar MS, Mushtaq MH, et al. Prevalence and molecular diagnosis of Babesia ovis and Theileria ovis in Lohi sheep at livestock experiment station (LES), Bahadurnagar, Okara, Pakistan. Iran J Parasitol. 2013;8:570.
Bishop R, Musoke A, Morzaria S, Gardner M, Nene V. Theileria: intracellular protozoan parasites of wild and domestic ruminants transmitted by ixodid ticks. Parasitology. 2004;129(Suppl. 1):271–83.
Gray J, De Vos A. Studies on a bovine Babesia transmitted by Hyalomma marginatum rufipes Koch, 1844. Onderstepoort J Vet Res. 1981;48:215–23.
Ros-García A, García-Pérez A, Verdera J, Juste R, Hurtado A. Monitoring piroplasms infection in three cattle farms in Minorca (Balearic Islands, Spain) with previous history of clinical piroplamosis. Vet Parasitol. 2012;190:318–25.
Decaro N, Larocca V, Parisi A, Losurdo M, Lia RP, Greco MF, et al. Clinical bovine piroplasmosis caused by Babesia occultans in Italy. J Clin Microbiol. 2013;51:2432–4.
Mandal M, Banerjee P, Garg R, Ram H, Kundu K, Kumar S, et al. Genetic characterization and phylogenetic relationships based on 18S rRNA and ITS1 region of small form of canine Babesia spp. from India. Infect Genet Evol. 2014;27:325–31.
Sun M, Wang J, Liu Z, Guan G, Li Y, Liu J, et al. First molecular evidence of Babesia occultans and Theileria separata infection in ticks and sheep in China. Exp Appl Acarol. 2019;78:223–9.
Caminade C, McIntyre KM, Jones AE. Impact of recent and future climate change on vector-borne diseases. Ann N Y Acad Sci. 2019;1436:157.
Benitez D, Mesplet M, Echaide I, de Echaide ST, Schnittger L, Florin-Christensen M. Mitigated clinical disease in water buffaloes experimentally infected with Babesia bovis. Ticks Tick Borne Dis. 2018;9:1358–63.
Barbour AG, Adeolu M, Gupta RS. Division of the genus Borrelia into two genera (corresponding to Lyme disease and relapsing fever groups) reflects their genetic and phenotypic distinctiveness and will lead to a better understanding of these two groups of microbes (Margos et al. (2016) There is inadequate evidence to support the division of the genus Borrelia Int. J. Syst. Evol. Microbiol. doi: 10.1099/ijsem.0.001717). Int J Syst Evol Microbiol. 2017;67:2058–67.
Margos G, Gofton A, Wibberg D, Dangel A, Marosevic D, Loh SM, et al. The genus Borrelia reloaded. PLoS ONE. 2018;13:e0208432.
Robertson RG, Wisseman CL Jr. Tick-borne Rickettsiae of the spotted fever group in west Pakistan: ii. serological classification of isolates from west Pakistan and Thailand: evidence for two new species. Am J Epidemiol. 1973;97:55–64.
Vitale G, Mansueto S, Rolain J-M, Raoult D. Rickettsia massiliae human isolation. Emerg Infect Dis. 2006;12:174.
Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, et al. Update on tick-borne rickettsioses around the world: a geographic approach. Clin Microbiol Rev. 2013;26:657–702.
Yu P, Liu Z, Niu Q, Yang J, Abdallah MO, Chen Z, et al. Molecular evidence of tick-borne pathogens in Hyalomma anatolicum ticks infesting cattle in Xinjiang Uygur autonomous region, Northwestern China. Exp Appl Acarol. 2017;73:269–81.
de Mera IGF, Zivkovic Z, Bolaños M, Carranza C, Pérez-Arellano JL, Gutierrez C, et al. Rickettsia massiliae in the Canary Islands. Emerg Infect Dis. 2009;15:1869.
Acestor N, Cooksey R, Newton PN, Menard D, Guerin PJ, Nakagawa J, et al. Mapping the aetiology of non-malarial febrile illness in Southeast Asia through a systematic review—terra incognita impairing treatment policies. PLoS ONE. 2012;7:e44269.
Ahantarig A, Trinachartvanit W, Baimai V, Grubhoffer L. Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol. 2013;58:419–28.
Gray JS, Dautel H, Estrada-Pena A, Kahl O, Lindgren E. Effects of climate change on ticks and tick-borne diseases in europe. Interdiscip Perspect Infect Dis. 2009;2009:593232.
Guizzo MG, Parizi LF, Nunes RD, Schama R, Albano RM, Tirloni L, et al. A Coxiella mutualist symbiont is essential to the development of Rhipicephalus microplus. Sci Rep. 2017;7:17554.
Apperson CS, Engber B, Nicholson WL, Mead DG, Engel J, Yabsley MJ, et al. Tick-borne diseases in North Carolina: is “Rickettsia amblyommii” a possible cause of rickettsiosis reported as Rocky Mountain spotted fever? Vector Borne Zoonotic Dis. 2008;8:597–606.
Sanchez JL, Candler WH, Fishbein DB, Greene CR, Coté TR, Kelly DJ, et al. A cluster of tick-borne infections: association with military training and asymptomatic infections due to Rickettsia rickettsii. Trans R Soc Trop Med Hyg. 1992;86:321–5.
Lively CM, Clay K, Wade MJ, Fuqua C. Competitive co-existence of vertically and horizontally transmitted parasites. Evol Ecol Res. 2005;7:1183–90.
de la Fuente J, Blouin EF, Kocan KM. Infection exclusion of the rickettsial pathogen Anaplasma marginale in the tick vector Dermacentor variabilis. Clin Diagn Lab Immunol. 2003;10:182–4.
Macaluso KR, Sonenshine DE, Ceraul SM, Azad AF. Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second Rickettsia. J Med Etomol. 2002;39:809–13.
Heise SR, Elshahed M, Little S. Bacterial diversity in Amblyomma americanum (Acari: Ixodidae) with a focus on members of the genus Rickettsia. J Med Entomol. 2010;47:258–68.
Lalzar I, Harrus S, Mumcuoglu KY, Gottlieb Y. Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities. Appl Environ Microbiol. 2012;78(12):4110–6.
Van Overbeek L, Gassner F, Van Der Plas CL, Kastelein P, Nunes–da Rocha U, Takken W. Diversity of Ixodes ricinus tick-associated bacterial communities from different forests. FEMS Microbiol Ecol. 2008;66:72–84.
Moreno CX, Moy F, Daniels TJ, Godfrey HP, Cabello FC. Molecular analysis of microbial communities identified in different developmental stages of Ixodes scapularis ticks from Westchester and Dutchess Counties, New York. Environ Microbiol. 2006;8:761–72.
Abraham NM, Liu L, Jutras BL, Yadav AK, Narasimhan S, Gopalakrishnan V, et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc Natl Acad Sci USA. 2017;114:E781–90.
Acknowledgements
The authors are thankful to the AVCCR dairy beef project (LPS/2016/011) in Pakistan for providing logistic support during the sample collection. AG is grateful to the Australian Society for Parasitology for providing ASP Network Researcher Exchange, Training and Travel Award. Authors are also grateful to farmers and local animal-health professionals for allowing and assisting in collection of ticks.
Funding
The financial assistance for this project was provided by the University of Melbourne as part of AG’s PhD project and AVCCR Dairy beef project (LPS/2016/011) in Pakistan. AG is a grateful recipient of the Australian Government Research Training Scholarship through the University of Melbourne.
Author information
Authors and Affiliations
Contributions
AJ, ACC, and AG conceived the idea and designed the study. AG conducted field work and sample processing. CG and AG performed lab experiments. ACC and SM provided technical help during experiments. AG, ACC, RBG and AJ drafted the manuscript. DO contributed to the statistical analyses. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The collection of tick specimens from cattle and buffaloes was approved by the Animal Ethics Committee, Faculty of Veterinary and Agricultural Science, The University of Melbourne (Ethics ID: 1714216).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Primers used for validation of microfluidic real-time PCR results.
Additional file 2: Figure S1.
Multiple correspondence analyses maps from the projections of the first two dimensions, showing the associations between infections (co-occurrence), and their distributions among six districts in Punjab and Sindh provinces of Pakistan. Percentage in each dimension (axis) indicates the fraction of the inertia that each principal component explains. Analyses are shown for correlation between infections and their prevalence in different provinces.
Additional file 3: Figure S2.
Multiple correspondence analyses maps from the projections of the first two dimensions, showing the associations between infections (co-occurrence), and their distributions among six districts in Punjab and Sindh provinces of Pakistan. Percentage in each dimension (axis) indicates the fraction of the inertia that each principal component explains. Analyses are shown for correlation between infections and their prevalence in different districts.
Additional file 4: Figure S3.
Multiple correspondence analyses maps from the projections of the first two dimensions, showing the associations between infections (co-occurrence), and their distributions among six districts in Punjab and Sindh provinces of Pakistan. Percentage in each dimension (axis) indicates the fraction of the inertia that each principal component explains. Analyses are shown for correlation between infections and their prevalence in bovine host species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ghafar, A., Cabezas-Cruz, A., Galon, C. et al. Bovine ticks harbour a diverse array of microorganisms in Pakistan. Parasites Vectors 13, 1 (2020). https://doi.org/10.1186/s13071-019-3862-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3862-4