
Abstract
Background
The accurate tracking of Cryptosporidium in faecal, water and/or soil samples in water catchment areas is central to developing strategies to manage the potential risk of cryptosporidiosis transmission to humans. Various PCR assays are used for this purpose. Although some assays achieve specific amplification from Cryptosporidium DNA in animal faecal samples, some do not. Indeed, we have observed non-specificity of some oligonucleotide primers in the small subunit of nuclear ribosomal RNA gene (SSU), which has presented an obstacle to the identification and classification of Cryptosporidium species and genotypes (taxa) from faecal samples.
Results
Using a novel bioinformatic approach, we explored all available Cryptosporidium genome sequences for new and diagnostically-informative, multi-copy regions to specifically design oligonucleotide primers in the large subunit of nuclear ribosomal RNA gene (LSU) as a basis for an effective nested PCR-based sequencing method for the identification and/or classification of Cryptosporidium taxa.
Conclusion
This newly established PCR, which has high analytical specificity and sensitivity, is now in routine use in our laboratory, together with other assays developed by various colleagues. Although the present bioinformatic workflow used here was for the specific design of primers in nuclear DNA of Cryptosporidium, this approach should be broadly applicable to many other microorganisms.
Similar content being viewed by others

Background
Animal faecal contamination in drinking water catchment areas is a global concern, and has the potential to lead to waterborne outbreaks of human gastrointestinal diseases including cryptosporidiosis, caused by members of the genus Cryptosporidium [1,2,3,4,5]. The identification and classification of Cryptosporidium species and genotypes (taxa) in faecal, water and/or soil samples in water catchment areas is central to assessing the risk of zoonotic transmission of cryptosporidiosis and to developing strategies to monitor and manage this risk [1,2,3,4,5].
As the oocysts and other developmental stages of different Cryptosporidium taxa cannot be unequivocally identified or differentiated microscopically, molecular tools can be used for their specific and genotypic identification and classification [6, 7]. A widely-used approach has been nested PCR-based sequencing or restriction fragment length polymorphism (RFLP) of particular gene markers within the actin, cowp, hsp70 and/or small subunit of nuclear ribosomal RNA (SSU) genes (e.g. [3, 7,8,9]). The sequencing of SSU has been particularly commonly used, because of its multi-copy nature [10], making PCR analytically sensitive, and because it contains diagnostically informative markers for the classification of Cryptosporidium taxa (cf. [3, 9, 11,12,13]). Indeed, to date, more than 34 species and 40 genotypes of Cryptosporidium have been classified using partial SSU sequence data [3, 5, 14,15,16,17].
Various PCR protocols are used for the amplification of regions within SSU (e.g. [8, 11, 12, 18]). When applied to faecal DNA samples from humans, most of these protocols appear to achieve relatively specific amplification from Cryptosporidium DNA for direct amplicon-sequencing. However, although not necessarily alluded to in the published literature, some PCR assays do not achieve high specificity when applied to faecal DNA samples originating from various animals, indicating that some primer sets designed are not entirely specific for Cryptosporidium. Indeed, we have observed ‘cross-amplification’ of SSU from DNA of some Cryptosporidium-related apicomplexans (e.g. [19]) as well as selected alveolates and/or dinoflagellates in animal faeces employing primers originally designed to Cryptosporidium spp. (Koehler et al., unpublished findings). Although we have designed and tested numerous new primer pairs, we did not achieve the desired level of specificity. Other primer sets to SSU [18] were designed to improve the sensitivity of primers over the leading primer set [8] by decreasing the regions targeted in the primary and secondary (nested) PCR amplification steps. Here, we used a novel bioinformatic approach to explore all available Cryptosporidium genome sequences (cf. [20]) for new and diagnostically-informative, multi-copy regions that would allow us to specifically design oligonucleotide primers to establish a nested PCR-based sequencing method with high analytical specificity and sensitivity for the identification and/or classification of Cryptosporidium species and genotypes.
Methods
Genomic DNAs from faecal deposits, and grouping of samples based on results from PCR-based sequencing of SSU
In the present study, we used a total of 332 genomic DNA samples from faecal samples from humans and 13 different animal species (mammals and birds) available from previous studies [2, 3, 21,22,23] or provided by colleagues (cf. Additional file 1: Table S1). The host origin of each sample was known or inferred using an authoritative field guide [24], and verified, as required, by PCR-based sequencing of a region of the mitochondrial cytochrome b gene from faecal DNA (cf. [25]). Genomic DNA was usually extracted directly from 0.25 g of each faecal sample using the PowerSoil kit (MoBio, Carlsbad, CA, USA) as described previously [3]; published evidence demonstrates that this method removes PCR-inhibitory components from faecal samples [26].
Genomic DNAs were subjected to PCR-based sequencing of SSU as described previously [21]. In brief, primary PCR (~850 bp) was conducted using primers XF2 (forward: 5′-GGA AGG GTT GTA TTT ATT AGA TAA AG-3′) and XR2 (reverse: 5′-AAG GAG TAA GGA ACA ACC TCC A-3′) (cf. [8]), followed by a secondary (nested) PCR (215 bp) using primers pSSUf (forward: 5′-AAA GCT CGT AGT TGG ATT TCT GTT-3′) and pSSUr (reverse: 5′-ACC TCT GAC TGT TAA ATA CRA ATG C-3′). All oligonucleotide primers (degenerate nucleotide positions indicated with an International Union of Pure and Applied Chemistry (IUPAC) code) were synthesized by GeneWorks, Thebarton, Australia.
For primary PCR, the cycling protocol was: 94 °C for 5 min (initial denaturation), followed by 30 cycles of 94 °C for 45 s (denaturation), 45 °C for 2 min (annealing) and 72 °C for 1.5 min (extension), with a final extension of 72 °C for 10 min. For secondary PCR, the protocol was: 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, with a final extension of 72 °C for 10 min. Amplicons were individually treated with ExoSAP-IT (Affymetrix, Santa Clara, CA, USA), according to the manufacturer’s instructions, and then subjected to bi-directional automated sequencing (BigDye® Terminator v.3.1 chemistry, Applied Biosystems, Foster City, CA, USA) using the same primers employed in the secondary (nested) PCR. Sequence quality was verified by comparison with corresponding electropherograms using the program Geneious v.9.1.4 [27]. Each SSU-derived sequence obtained was compared by BLASTn analysis with publicly available data (GenBank) (cf. [2, 3]). Based on the results, five distinct groups of genomic DNA samples were formed (Additional file 1: Table S1):
Group 1 (well-defined ‘positive’ samples containing DNA of particular Cryptosporidium taxa but without ‘cross-amplification’): Samples (n = 20; Additional file 1: Table S1; Fig. 1) containing one of the following species: C. avium, C. baileyi, C. bovis, C. canis, C. cuniculus, C. fayeri, C. felis, C. hominis, C. macropodum, C. meleagridis, C. muris, C. parvum, Cryptosporidium cf. ryanae, C. serpentis, Cryptosporidium cf. suis, C. ubiquitum, C. xiaoi, Cryptosporidium sp. “deer genotype”, Cryptosporidium sp. “duck-like genotype”, Cryptosporidium cf. fayeri EGK1, Cryptosporidium sp. “kangaroo genotype I”. Cryptosporidium sp. “mink genotype” or Cryptosporidium sp. “novel human genotype”.
Group 2 (samples with substantial cross-amplification and no evidence of the presence of Cryptosporidium): Samples (n = 13; Additional file 1: Table S1) from which one of the following microorganisms was amplified by PCR of SSU and sequenced: Adelina bambarooniae, Colpodella sp., Cryptosporidium struthionis (not recognized as a valid member of the genus Cryptosporidium [18]), Eimeria reichenowi, Gloeodinium montanum, Perkinsea sp., Placocista sp., “Uncultured eukaryote”, Chlorophytes; Chlorococcum vacuolatum, Colletotrichum aculatum, Monoraphidium minutum, dinoflagellate; Baldinia anauniensis; or fungus, “Uncultured fungus” (Fig. 1). These microorganisms have been commonly detected by SSU-PCR in a longitudinal study of waterborne pathogens in faecal samples from Australian wildlife (cf. [2, 3]).

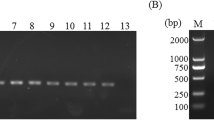
Agarose gel showing results for each of the three groups of (individual) genomic DNA samples produced by nested PCR of regions of the small (SSU) and large (LSU) subunits of the nuclear ribosomal RNA genes. Group 1: Well-defined ‘positive’ samples containing DNA of particular Cryptosporidium taxa, with no ‘cross-amplification’. Group 2: Representative samples with substantial cross-amplification but no evidence of the presence of Cryptosporidium. Group 3: Representative ‘negative’ samples with no cross-amplification or evidence of the presence of Cryptosporidium DNA (cf. Methods section). The marker is a 100 bp ladder, plus a 1500 bp band (top). No-template controls were included in primary and secondary PCR runs (negative controls 1 and 2, respectively). For full description of samples and methods, see Methods section
Group 3 (‘negative’ samples without cross-amplification with no evidence of the presence of Cryptosporidium DNA): Samples (n = 15; Additional file 1: Table S1; Fig. 1) from which no SSU amplicons were produced, and for which there was no evidence of PCR inhibition or background amplification in SSU-PCR.
Group 4 (samples with substantial cross-amplification and containing Cryptosporidium DNA): Samples (n = 9; Additional file 1: Table S1) from human faecal samples known to contain Cryptosporidium taxa. Attempts at sequencing the SSU locus from these “challenging” samples yielded indeterminate sequence data (not shown). To obtain unambiguous sequence data to classify the Cryptosporidium taxa within these samples, we amplified part of the 60 kDa-glycoprotein gene (gp60) using a standard method and sequenced the products (cf. [21]).
Group 5 (samples without cross-amplification and containing DNA of closely-related genotypes or subtypes of Cryptosporidium): Samples (n = 24; Additional file 1: Table S1) each containing a particular genotype of C. fayeri, C. macropodum, C. ryanae and C. ubiquitum (n = 15) or a particular subtype of C. parvum or C. hominis (n = 9).
Bioinformatic workflow for the specific design of new primers
Focused on designing highly specific oligonucleotide primers for use in PCR, we interrogated all publicly available genomic data available for Cryptosporidium species for multi-copy DNA (copy number = 4; [10]), and identified LSU as the key candidate with the greatest potential to achieve high analytical sensitivity. As only one full-length LSU sequence (accession number AF040725; [28]) was available for C. parvum in GenBank, we used a bioinformatic approach to identify homologous sequences in other Cryptosporidium data sets as well as similar LSU sequences in other microorganisms, and created an alignment from which oligonucleotide primers could be designed (described below).
In brief, genome sequence files (FASTA) and the general feature format (GFF) files for available Cryptosporidium species (February 2016) as well as relatively ‘closely related’ alveolates and dinoflagellates (for which LSU data were available; cf. Additional file 1: Table S1) were downloaded from GenBank, the Cryptosporidium Genomic Resources Database release 27 (cryptoDB.org) and the Silva rRNA Database Project (www.arb-silva.de), respectively: i.e. Cryptosporidium baileyi (JIBL01000085); C. hominis (JRXJ01000009); C. meleagridis (JIBK01000002); C. muris (DS989727); C. parvum (AF040725); Cryptosporidium sp. “chipmunk genotype” (JXRN01000036); Eimeria tenella (CBUW010002483); Colpodella angusta (KU159286); Perkinsus chesapeaki (AY305326); P. olseni (AF509333); “Uncultured marine dinoflagellate” (FJ032679); Vitrella brassicaformis (HM245049); and Voromonas pontica (EF681910).
For species for which only genome sequence files were available, modelled repeats were combined with a collection of known repeats in Repbase v.17.02 [29] and soft-masked using RepeatMasker v.open-3.3.0 [30]. Repeats identified as representing LSU were selected and reverse-complemented (as required) and then converted into the FASTA format. An alignment of LSU sequences (over ~3800 bp) (Additional file 2: Figure S1) was made using MAFFT in Mesquite [31] and transferred to the program Geneious v9.1.4 [27] where outer and inner (nested) pairs of primers were designed to the flanking regions of the variable D8 domain of LSU (cf. [32]) with the aid of the program Primer3 (http://primer3.ut.ee/) [33] (Additional file 2: Figure S1). The primers designed were: LSU2040F (forward: 5′-CGA ATA GCG TTA TCT TTG CTA TTT-3′) and LSU3020R (reverse: 5′-GTC TTC CGC GAA GAT CAG-3′), followed by secondary (nested) PCR (~500 bp) using primers LSU2065F (forward: 5′-TTA CCA TGG AAT (C/T)AG TTC AGC-3′) and LSU2557R (reverse: 5′-AAC ACC ATT TTC TGG CCA TC-3′).
Protocol for the newly established nested PCR
The final method is described in the following: The nested PCR was carried out in 50 μl using a standard reaction buffer, 3.0 mM of MgCl2, 200 μM of each dNTP, 50 pmol of each primer and 1 U of Taq polymerase (MangoTaq, Bioline, London, UK). Primary PCR of the D8 region (~1000 bp) of the LSU gene was conducted using the primer pair LSU2040F/LSU3020R, followed by secondary (nested) PCR (~500 bp) using the primer pair LSU2065F/LSU2557R. The conditions of the primary PCR were: 94 °C for 5 min (initial denaturation), followed by 35 cycles of 94 °C for 30 s (denaturation), 58 °C for 30 s (annealing) and 72 °C for 50 s (extension), with a final extension of 72 °C for 5 min. The conditions of secondary PCR were the same, except that the extension step was 30 s instead of 50 s. Except for the no-template controls, 2 μl of genomic DNA were added to the primary PCR, from which 1 μl was carried over to the secondary PCR. No-template (negative) controls were included at all steps, and no-template controls were carried over from the primary to the secondary (nested) PCR. A well-known positive control sample (C. parvum DNA) was included in each PCR run. The sequencing of LSU amplicons was performed (as described for SSU amplicons) using primers LSU2065F and LSU2557R.
Phylogenetic analysis of LSU and SSU sequence data
Sequences were aligned using the program MAFFT [34], and alignments were manually adjusted using the program Mesquite v.3.10 [31] (cf. Additional file 2: Figure S2). Sequences were then compared with those available in GenBank using BLASTn (NCBI) (see Additional file 1: Tables S2 and S3 for additional sequence data used for comparative purposes). Colpodella angusta was chosen as a suitable outgroup due to its proposed close relatedness to Cryptosporidium (cf. [19]). Specifically, phylogenetic analysis of sequence data was conducted by Bayesian inference (BI) using Monte Carlo Markov Chain (MCMC) analysis in MrBayes v.3.2.6 [35]. The likelihood parameters set for BI analysis of LSU data were based on the Akaike Information Criteria (AIC) test in jModeltest v.2.1.10 [36]. For trees constructed using (partial) LSU or SSU sequence data, the number of substitutions (Nst) was set at 6, with a gamma-distribution. For SSU, the proportion of invariable sites was also considered. Posterior probability (pp) values were calculated by running 2,000,000 generations with four simultaneous tree-building chains. Trees were saved every 100th generation. At the end of each run, the standard deviation of split frequencies was <0.01, and the potential scale reduction factor approached one. A 50% majority rule consensus tree for each analysis was constructed based on the final 75% of trees generated by BI. Analyses were run three times to ensure convergence and insensitivity to priors.
Results and discussion
Establishing the new assay using primers designed employing a bioinformatic approach
Using the present bioinformatic workflow (see Methods section), we identified specific regions in LSU (flanking the D8 domain) and designed primers LSU2040F (forward: 5′-CGA ATA GCG TTA TCT TTG CTA TTT-3′) and LSU3020R (reverse: 5′-GTC TTC CGC GAA GAT CAG-3′) to amplify a region of ~800 bp in primary PCR, and primers LSU2065F (forward: 5′-TTA CCA TGG AAT YAG TTC AGC-3′) and LSU2557R (reverse: 5′-AAC ACC ATT TTC TGG CCA TC-3′) to amplify a region of ~500 bp in secondary (nested) PCR. Having designed these primer pairs, we proceeded to establish the nested PCR assay, following authoritative guidelines and recommendations for optimization [37]. The final protocol/method is described in the Methods section.
Performance of the newly established nested PCR
To evaluate the specificity of the PCR assay, we produced LSU amplicons from all genomic DNA samples in Groups 1–4 and sequenced them. The PCR and sequencing results for individual samples are given in Additional file 1: Table S1. The sequencing of the amplicons derived from Group 1-samples (Fig. 1) yielded sequences that exclusively/specifically represented Cryptosporidium. Using the nested LSU-PCR, no products of ~500 bp were amplified from any of the samples in Groups 2 or 3, showing the specificity of the newly designed primers (Fig. 1). The sequencing of LSU amplicons derived from all individual Group 4-samples allowed the unequivocal identification of Cryptosporidium in each sample, without any complication of background- or cross-amplification, in contrast to results obtained using PCR-based sequencing of SSU (Additional file 1: Table S1). In conclusion, LSU amplicons produced from individual genomic DNAs were consistently abundant and of the expected size (~500 bp) on agarose gels, and sequences derived from these amplicons consistently and unambiguously matched those of Cryptosporidium, which represented a substantial improvement over results achieved previously using SSU-PCR (see Methods section). Thus, using the LSU-PCR, there was no instance of background- or cross-amplification of a ~500 bp product from any microbial taxon other than Cryptosporidium (based on direct sequencing of amplicons). Using serial titration experiments, the analytical sensitivity of the LSU-PCR was estimated at < 1 pg of Cryptosporidium DNA (data not shown). All results obtained were reproducible (at least three times) on different days.
Ability of PCR-based sequencing of LSU to assign species and/or genotypes of Cryptosporidium
Here, we used 24 samples from Group 5 to assess the ability of PCR-based sequencing of LSU to differentiate closely related genotypes or subtypes of select Cryptosporidium (cf. Additional file 1: Table S1). We compared the magnitude of sequence variation in LSU within each of four species with that in SSU for the same species. Specifically, maximum sequence variation in LSU within C. fayeri (2.6%), C. macropodum (0%), C. ryanae (5.9%) and C. ubiquitum (0.8%), using 2–5 samples per species (cf. Additional file 1: Tables S2 and S3), was for the most part, less than that recorded in SSU for the similar samples (5.5%, 1.9%, 3.4% and 3.2%, respectively) (cf. Additional file 2: Figure S2). Subsequently, we used the remaining 9 genomic DNAs from human faecal samples from Group 5 to assess sequence variability in LSU among selected subtypes of both C. parvum and C. hominis, and then compared the magnitude of variation in LSU with that derived from a comparative set of SSU sequences representing the same taxa (i.e. species and subtypes) from GenBank. Sequence variation in LSU within C. parvum (0%) and C. hominis (1.0%) was similar to that recorded for the comparative set of SSU sequences (0% and 1.8%, respectively) (cf. Additional file 2: Figure S2).
Then, a phylogenetic tree built using the LSU sequence data (Additional file 1: Table S4) showed that it could differentiate Cryptosporidium variants (genotypes) with similar resolution to the tree constructed using SSU data (Additional file 1: Table S5; Figure 2) - the latter locus being that most extensively used for phylogenetic studies of Cryptosporidium to date [7, 9]. Subsequently, we assessed whether phylogenetic resolution would differ significantly when LSU are used instead of SSU data. For the important human-affiliated species, C. hominis and C. parvum, an analysis of the LSU data did not provide more resolution than the SSU data (also for samples previously characterized using the gp60 locus; cf. [21]). Although statistically unsupported, genotypes of C. parvum and C. hominis clustered separately, showing that they can be readily distinguished, both in an alignment of LSU sequence data (Additional file 2: Figure S2) and in the LSU tree (Fig. 2). Nonetheless, the sequence of the D8 domain of LSU does not discern some of the genotypes or subtypes that could be identified/differentiated using the SSU locus (cf. Fig. 2). For example, C. ryanae was not divided in the LSU tree, whereas for SSU, multiple C. ryanae-like genotypes (e.g. MW1, MW4 and MW7; [2]) could be distinguished. Additionally, differences between genotypes of some Cryptosporidium species, such as C. ubiquitum, which have several nucleotide alterations in SSU in Cryptosporidium from wombats compared with more common subtypes [38], were retained when the LSU locus was used (see C. ubiquitum in Fig. 2). In our opinion, less variation (resolution) might actually be advantageous in some instances, particularly when genetically discriminating among Cryptosporidium species, as the D8 domain of LSU tends to ‘combine’ closely related genotypes, rather than divide them up. As noted by Xiao et al. [6], it is challenging to predict what degree of genetic variability is needed for the differentiation of species, and the question arises at what point should genotypes and/or subtypes be coined (cf. [38]).

a Relationships among Cryptosporidium taxa inferred from the phylogenetic analysis of sequence data for the variable D8 domain of the large subunit of nuclear ribosomal RNA gene (LSU) by Bayesian inference (BI). b Relationships among Cryptosporidium taxa inferred from the phylogenetic analysis of sequences from the small subunit of the nuclear ribosomal RNA gene (SSU) by Bayesian inference (BI). Posterior probabilities are indicated at all major nodes. Bold-type indicates Cryptosporidium species or genotypes characterized from faecal DNA samples in this study. Individual identification codes and accession numbers follow the species or genotype designation. Scale-bar represents the number of substitutions per site. The alveolate Colpodella angusta was used as an outgroup
Conclusions
Here, we have shown the benefit of using a bioinformatics-guided approach for the specific design of primers for PCR. In this case, our focus was on finding regions in multi-copy ribosomal DNA to ensure that any PCR developed would achieve high analytical sensitivity. The laboratory-based evaluations conducted using well-defined groups of genomic (faecal) DNAs showed unequivocally that both analytical specificity and sensitivity of this nested PCR are very high, allowing the genetic characterization and classification of Cryptosporidium species and genotypes by phylogenetic means. This newly established PCR is now in routine use in our laboratory, together with other assays originally developed by various colleagues [8, 11]; having tested > 900 animal faecal DNA samples to date, the performance of our nested LSU-PCR is entirely consistent with that achieved here and results are reproducible (Wang and Koehler, unpublished findings). The specificity and sensitivity of this new assay need to be continuously monitored over time. It will also be relevant to reassess these parameters if the present assay were deployed to other geographical regions or applied to faecal samples from animal species distinct from those tested herein. Although we have not yet had the opportunity to test genomic DNA from the recently described piscine species of Cryptosporidium (e.g. C. huwi; [14]), we anticipate that the present LSU-PCR assay will specifically amplify DNA from this taxon. We hope that this assay could eventually be added to the currently recommended tool-kit (using markers in the cowp and SSU genes) for the genetic characterization of Cryptosporidium species and genotypes (cf. [7, 39]). In conclusion, although the present bioinformatic workflow was used for the specific design of reagents (primer pairs) nuclear DNA of Cryptosporidium, we believe that this approach will be applicable to a wide range of genes in the genomes of many other microorganisms.
Abbreviations
- AIC:
-
Akaike information criteria
- BI:
-
Bayesian inference
- GFF:
-
general feature format
- LSU :
-
large subunit ribosomal RNA
- MCMC:
-
Monte Carlo Markov chain
- RFLP:
-
restriction fragment length polymorphism
- SSU :
-
small subunit ribosomal RNA
References
Smith HV, Nichols RA. Cryptosporidium: detection in water and food. Exp Parasitol. 2010;124(1):61–79.
Nolan MJ, Jex AR, Koehler AV, Haydon SR, Stevens MA, Gasser RB. Molecular-based investigation of Cryptosporidium and Giardia from animals in water catchments in southeastern Australia. Water Res. 2013;47(5):1726–40.
Koehler AV, Haydon SR, Jex AR, Gasser RB. Cryptosporidium and Giardia taxa in faecal samples from animals in catchments supplying the city of Melbourne with drinking water (2011 to 2015). Parasit Vectors. 2016;9(1):315.
Zahedi A, Monis P, Aucote S, King B, Paparini A, Jian F, et al. Zoonotic Cryptosporidium species in animals inhabiting Sydney water catchments. PLoS One. 2016;11(12):e0168169.
Zahedi A, Paparini A, Jian F, Robertson I, Ryan U. Public health significance of zoonotic Cryptosporidium species in wildlife: critical insights into better drinking water management. Int J Parasitol Parasites Wildl. 2016;5(1):88–109.
Xiao L, Fayer R, Ryan UM, Upton SJ. Cryptosporidium taxonomy: recent advances and implications for public health. Clin Microbiol Rev. 2004;17(1):72–97.
Ryan U, Fayer R, Xiao L. Cryptosporidium species in humans and animals: current understanding and research needs. Parasitology. 2014;141(13):1667–85.
Xiao L, Morgan UM, Limor J, Escalante A, Arrowood M, Shulaw W, et al. Genetic diversity within Cryptosporidium parvum and related Cryptosporidium species. Appl Environ Microbiol. 1999;65(8):3386–91.
Jex AR, Smith HV, Monis PT, Campbell BE, Gasser RB. Cryptosporidium - biotechnological advances in the detection, diagnosis and analysis of genetic variation. Biotechnol Adv. 2008;26(4):304–17.
Le Blancq SM, Khramtsov NV, Zamani F, Upton SJ, Ribosomal WTW. RNA gene organization in Cryptosporidium parvum. Mol Biochem Parasitol. 1997;90(2):463–78.
Ryan UM, Xiao L, Read C, Zhou L, Lal AA, Pavlasek I. Identification of novel Cryptosporidium genotypes from the Czech Republic. Appl Environ Microbiol. 2003;69(7):4302–7.
Nolan MJ, Jex AR, Haydon SR, Stevens MA, Gasser RB. Molecular detection of Cryptosporidium cuniculus in rabbits in Australia. Infect Genet Evol. 2010;10(8):1179–87.
Xiao L. Molecular epidemiology of cryptosporidiosis: an update. Exp Parasitol. 2010;124(1):80–9.
Ryan U, Paparini A, Tong K, Yang R, Gibson-Kueh S, O'Hara A, Lymbery A, Xiao L. Cryptosporidium huwi n. sp. (Apicomplexa: Eimeriidae) from the guppy (Poecilia reticulata). Exp Parasitol. 2015;150:31–5.
Holubova N, Sak B, Horcickova M, Hlásková L, Kvetonova D, Menchaca S, et al. Cryptosporidium avium n. sp. (Apicomplexa: Cryptosporidiidae) in birds. Parasitol Res. 2016;115(6):2243–51.
Ježková J, Horcickova M, Hlásková L, Sak B, Kvetonova D, Novak J, et al. Cryptosporidium testudinis sp. n., Cryptosporidium ducismarci Traversa, 2010 and Cryptosporidium tortoise genotype III (Apicomplexa: Cryptosporidiidae) in tortoises. Folia Parasitol (Praha). 2016;63:1–10.
Kváč M, Havrdová N, Hlásková L, Daňková T, Kanděra J, Ježková J, et al. Cryptosporidium proliferans n. sp.(Apicomplexa: Cryptosporidiidae): molecular and biological evidence of cryptic species within gastric Cryptosporidium of mammals. PLoS One. 2016;11(1):e0147090.
Silva SO, Richtzenhain LJ, Barros IN, Gomes AM, Silva AV, Kozerski ND, et al. A new set of primers directed to 18S rRNA gene for molecular identification of Cryptosporidium spp. and their performance in the detection and differentiation of oocysts shed by synanthropic rodents. Exp Parasitol. 2013;135(3):551–7.
Mikhailov KV, Tikhonenkov DV, Janouskovec J, Diakin AY, Ofitserov MV, Mylnikov AP, Aleshin VV. Primary structure of 28S rRNA gene confirms monophyly of free-living heterotrophic and phototrophic apicomplexans (Alveolata). Biochem Mosc. 2015;80(11):1492–9.
Heiges M, Wang H, Robinson E, Aurrecoechea C, Gao X, Kaluskar N, et al. CryptoDB: a Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 2006;34(Database issue):D419–22.
Koehler AV, Bradbury RS, Stevens MA, Haydon SR, Jex AR, Gasser RB. Genetic characterization of selected parasites from people with histories of gastrointestinal disorders using a mutation scanning-coupled approach. Electrophoresis. 2013;34:1720–8.
Koehler AV, Whipp M, Hogg G, Haydon SR, Stevens MA, Jex AR, Gasser RB. First genetic analysis of Cryptosporidium from humans from Tasmania, and identification of a new genotype from a traveller to Bali. Electrophoresis. 2014;35(18):2600–7.
Ebner J, Koehler AV, Robertson G, Bradbury RS, Jex AR, Haydon SR, et al. Genetic analysis of Giardia and Cryptosporidium from people in northern Australia using PCR-based tools. Infect Genet Evol. 2015;36:389–95.
Triggs B. Tracks, scats and other traces: a field guide to Australian mammals. South Melbourne: Oxford University Press; 2004.
Dalén L, Götherström A, Angerbjörn A. Identifying species from pieces of faeces. Conserv Genet. 2004;5(1):109–11.
Roeber F, Jex AR, Gasser RB. Comparative evaluation of two DNA isolation techniques for PCR-based diagnosis of gastrointestinal nematode infections in sheep. Mol Cell Probes. 2013;27(3–4):153–7.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9.
Zhu G, Keithly JS, Philippe H. What is the phylogenetic position of Cryptosporiidum? Int J Syst Evol Microbiol. 2000;50(4):1673–81.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–7.
Smit AFA, Hubley R, Green P. RepeatMasker. Version open-3.3.0. 1996–2010. http://www.repeatmasker.org. Accessed 20 Feb 2016.
Maddison WP, Maddison DR. Mesquite. A modular system for evolutionary analysis. Version 3.04. 2015. http://mesquiteproject.org. Accessed 20 Feb 2016.
Ki JS, Han MS. Informative characteristics of 12 divergent domains in complete large subunit rDNA sequences from the harmful dinoflagellate genus, Alexandrium (Dinophyceae). J Eukaryot Microbiol. 2007;54(2):210–9.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3 new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
Katoh K, Misawa K, Kuma K, Miyata TMAFFT. A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66.
Huelsenbeck JP, Ronquist FMRBAYES. Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–5.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Hillis DM, Moritz C, Mable BK. Molecular systematics. Sunderland, Massachusetts: Sinauer Associates; 1996.
Koehler AV, Haydon SR, Jex AR, Gasser RBI. Cryptosporidium from the common wombat (Vombatus ursinus) a new species and distinct from Cryptosporidium ubiquitum? Infect Genet Evol. 2016;44:28–33.
Šlapeta J. Cryptosporidium: identification and genetic typing. Curr Protoc Microbiol. 2017;44:20B.1.1–20B.1.17.
Acknowledgements
We thank Professor Una Ryan and Dr Alireza Zahedi for providing some samples.
Funding
This study supported by the Australian Research Council (ARC; LP160101299) and Melbourne Water Corporation.
Availability of data and materials
All data generated or analysed in this study are included in this published article and its additional files. The sequence data from this study are accessible in the GenBank database under accession nos. KY882313–KY882353 and MF621049–MF621052.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study and supervised the project: AVK, NDY, PKK and RBG. Undertook the study and data analysis: AVK, NDY, PKK and TW. Contributed through materials, tools, analyses and/or interpretations: AVK, NDY, PKK, TW SRH and RBG. Wrote the paper: AVK and RBG, with input from co-authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Relevant information pertaining to small subunit ribosomal RNA gene (SSU) sequences obtained from amplicons produced from selected faecal DNA samples using a nested PCR method ([2]; see Methods section). These DNA samples were employed to assess the analytical specificity of large subunit ribosomal RNA gene (LSU) nested PCR assay established in this study (see also Methods section) (cf. Fig. 1). Sequence similarities (90–100%) were calculated with reference to the closest matched sequence in the GenBank database using BLASTn. Table S2. Pairwise comparison of sequence difference (%) in the variable D8 domain of the large subunit of the nuclear ribosomal RNA gene (LSU) used for the construction of the phylogenetic tree. Table S3. Pairwise comparison of sequence difference (%) in the region of the small subunit of the nuclear ribosomal RNA gene (SSU) used for the construction of the phylogenetic tree. Table S4. Salient information pertaining to the sequences of the variable D8 domain of the nuclear large subunit ribosomal RNA gene (LSU) used for the construction of the phylogenetic tree (cf. Fig. 2a). Sequences produced in this study are shown in bold-type. Table S5. Salient information pertaining to the sequences from the small subunit of the nuclear ribosomal RNA gene (SSU) used for the construction of the phylogenetic tree (cf. Fig. 2b). Sequences produced in this study are shown in bold-type. (XLSX 78 kb)
Additional file 2: Figure S1.
Alignment of sequences of the variable D8 domain of the large subunit of nuclear ribosomal RNA gene (LSU) representing Cryptosporidium and closely related apicomplexans, alveolates and dinoflagellates. Oligonucleotide primers (LSU2040F, LSU3020R; LSU2065F and LSU2557R) designed specifically to regions flanking the variable D8 domain are indicated in green. Nucleotide differences from the majority consensus of the alignment are highlighted. Figure S2. Alignment of sequences of the variable D8 domain of the large subunit of nuclear ribosomal RNA gene (LSU) representing Cryptosporidium derived from 45 faecal DNA samples. Nucleotide differences from the majority consensus of the alignment are highlighted. Colpodella angusta was included as an outgroup. (PDF 222 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Koehler, A.V., Korhonen, P.K., Hall, R.S. et al. Use of a bioinformatic-assisted primer design strategy to establish a new nested PCR-based method for Cryptosporidium . Parasites Vectors 10, 509 (2017). https://doi.org/10.1186/s13071-017-2462-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2462-4