
Abstract
Background
Identification of the microflora of the sand fly gut and the environmental distribution of these bacteria are important components for paratransgenic control of Leishmania transmission by sand flies.
Methods
Biotic and abiotic bacterial communities of four compartments of a hyper-endemic focus of Zoonotic Cutaneous Leishmaniasis (ZCL) were investigated using 16S ribosomal DNA sequencing and phylogenetic tree construction. These compartments include Phlebotomus papatasi’s gut, skin and intestinal tract of great gerbil Rhombomys opimus, the gerbil nest supplies, and plant food sources of the vectors and reservoirs.
Results
Sequence homology analysis using nine available 16S rDNA data bases revealed 40, 24, 15 and 14 aerobic bacterial species from the vector guts, the gerbil bodies, the gerbil nests, and the plants, respectively. The isolated bacteria belong to wide ranges including aerobic to facultative anaerobic, pathogen to commensals, sand fly oviposition inducers, land to air and ocean habitats, animal and human probiotics, and plant growth-promoting rhizobacteria. Matching data analysis suggested that the adult P. papatasi gut bacteria could be acquired from three routes, adult sugar feeding on the plant saps, adult blood feeding on the animal host, and larval feeding from nest supplies. However, our laboratory experiment showed that none of the bacteria of the reservoir skin was transmitted to female sand fly guts via blood feeding. The microflora of sand fly guts were associated with the sand fly environment in which the predominant bacteria were Microbacterium, Pseudomonas, and Staphylococcus in human dwellings, cattle farms, and rodent colonies, respectively. Staphylococcus aureus was the most common bacterium in sand fly guts. Presence of some sand fly ovipoisition inducers such Bacillus spp. and Staphylococcus saprophyticus support association between gut flora and oviposition induction.
Conclusions
Results of this study showed that Bacillus subtilis and Enterobacter cloacae particularly subsp. dissolvens are circulated among the sand fly guts, the plants, and the sand fly larval breeding places and hence are possible candidates for a paratransgenic approach to reduce Leishmania transmission.
Similar content being viewed by others
Background
Leishmaniases are worldwide distributing sand fly-borne parasitic diseases with 1.4 million new cases and 20–30 thousand deaths annually. Due to complexity of life cycle of Leishmania spp. multifaceted intervention strategies are needed to prevent and control of the disease [1,2]. From the leishmaniasis spectrum, Zoonotic Cutaneous Leishmaniasis (ZCL), a neglected tropical disease, is a public health problem with a clear and disturbing increase in the number of cases in some areas of the world [3,4]. Leishmania major is widely distributed in various populations of rodents in arid and savannah regions [4,5] and transmitted by the Afro-Asian vector of ZCL, Phlebotomus papatasi Scopoli 1786, one of the most prevalent species among the Phlebotomus genus in indoor and outdoor places [6-10].
The disease is endemic in many rural districts of Iran, in 17 out of 31 provinces [11], so that it is still a great health problem and of research interest to many investigators. Rodents belonging to the subfamily Gerbillinae are the main reservoir hosts of ZCL in Iran and other countries where ZCL is endemic [12,13]. In general, gerbils are the most abundant mammals reported from natural ecosystems of Old World deserts [14].
The great gerbil, Rhombomys opimus (Cricetidae: Gerbillinae), is widely distributed in arid and semi arid habitats, mostly in sandy or clay desert areas throughout Central Asia, including Northwestern China, Mongolia, Russia, Kazakhstan, Iran, Afghanistan and western Pakistan [15-17]. In Iran it is widely distributed in central and northeast parts of the country [16,18,19]. Based on mitochondorial DNA cytochrome B (cytB) gene, at least two subspecies R. opimus sodalis and R. opimus sargadensis have been reported in Iran [20]. Because their burrows are a long-standing and important feature of the landscape, many other animal species such as Phlebtominae sand flies use them for shelter. Three coexisting Leishmania parasites, L. major, L. turanica, L. gerbilli and the bacterium Yersinia pestis have been isolated from this rodent and its corresponding insect vectors, which indicate that the great gerbil is a major reservoir that can maintain natural infection of leishmaniasis or plague [5,10,18,21,22]. The rate of infection of R. opimus by L. major is normally high and may vary from 55.8% to 92.5% in endemic areas [5,18]. The parasite can persist in the great gerbils for up to 25 months [23].
The primary diet of great gerbils is herbivorous (Folivore, Frugivore, and Granivore) and they cache these foods in complex tunnel systems. Living in desert habitats, this gerbil must rely on metabolic water found in succulent plants of family Chenopodiaceae (Climacoptera spp., Salsola spp., Suaeda spp.) [24,25]. Although their diet may vary according to the changes of plant species and coincides with the variations in the plants’ phenology [26], in central Iran, gerbils are commonly folivorous on Haloxylon spp. and Salsola spp. These plants constitute the main source of gerbil food because they have higher levels of water and mineral salts compared with other plants [27].
The insect alimentary canal is the main colonizing site of many microorganisms. Sand flies acquire bacteria at the larval stage from food and the breeding soil, and at the adult stage through contaminated sugar meals derived from plant leaves and fruits or aphid honeydew [28]. Sand fly females may also ingest bacteria while feeding on a blood meal; however, blood meals are usually sterile, while sugar meals from different sources may contain a variety of contaminating microorganisms [29]. These microbes are involved in many aspects of the host life including nutrition, reproduction, tolerance to environmental perturbations, maintenance and/or enhancement of host immune system homeostasis, defense, speciation, mucosal barrier fortification, xenobiotic metabolism, and pathogen transmission ability [29-35]. Among these, the role of midgut-associated bacteria in food digestion has been demonstrated in several insect species [34]. These indigenous bacteria are essential sources of carbohydrates improving digestion efficiency of plant-derived polymers such as lignin, hemicellulose and cellulose, xylan and pectin [36] and may also contribute to lipid and protein digestion [37].
Female sand flies need blood for egg production, but sugar is their main source of energy and the only food taken by males [38]. The sugar feeding behavior of sand flies, therefore, influences longevity and fecundity, dispersal, host seeking behavior and ultimately blood feeding and disease transmission [39-42]. According to the literature, sand flies obtain sugar meals mainly from honeydew excreted by aphids and coccids [43,44] and by feeding directly on tissues of plants in the field [45,46].
Generally, vector control is an effective and the simplest method to control vast majority of vector-borne diseases [47]. However in recent years, application of pesticides have been problematic because of their environmental toxicity, adverse effects on human health and the emergence of insecticide resistance in many countries [48].
Paratransgenesis is a Trojan-horse approach in which symbiotic bacteria, fungi, or viruses of the vector insect are genetically manipulated to deliver effector proteins that block development or transmission of the pathogen (vector competence). This approach attempts to decrease pathogen transmission without adverse effects on vectors themselves. Further, it is considered as a gene delivery mechanism to the host and indigenous bacterial flora of the host vector [34]. Bacterial symbionts of blood sucking bugs [49], tsetse flies [50], mosquitoes [51-55], American cockroach [56] and sand flies [57,58], as well as symbiotic viruses of An. gambiae [59] and Aedes aegypti [60], have been identified and in some cases successfully used to reduce or eliminate carriage of pathogens by host arthropods.
Multitrophic interactions are now recognized as being very important in understanding the complexity of the natural world. For example, during phytophagy or haematophagy, insects encounter microbiota on the surface of the host and their released metabolic products; likewise, the host is also exposed to microbial products released from both sides of the insect alimentary canal [34]. The role of microbiota as a fourth partner in behavioral aspects of vector-parasite-host interactions has been neglected for long time. Information gained from the study of these interactions can form the interface between biological control and restoration, which should be considered as part of biological control.
In this study, the presence and distribution of gut microbiota of male and female P. papatasi, the main vector of ZCL, were investigated from the following sources in the hyper-endemic focus of ZCL, Isfahan province, Iran: the exposed areas of skin surface, faeces and viscera of the great gerbil, R. opimus, the animal’s nest materials which include soil, vegetarian food residues, and two plants of Hyloxylon sp. and Salsola sp. as the food sources of both vector and reservoir. The results of this study may lead to identification of an appropriate bacterial candidate for genetic manipulation and delivery of effector molecules to diminish leishmaniasis transmission, using a paratransgenic strategy.
Methods
Study area
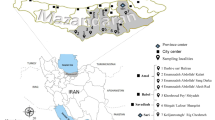
The study was conducted in five locations of four districts of the Isfahan province, a well-known hyper endemic ZCL focus in central Iran (Figure 1). Biological and non-biological samples were collected from different biotypes including excavated rodent colonies, semi-desolated cattle farm, uninhabited home, and deserts in the vicinity of villages and cities of the district.

Map of study area in the hyperendemic ZCL focus of Isfahan province, central of Iran. Locations are, A: Abbas Abad (Badrood), P: Parvaneh (Habibabad), S: Sejzi and V: Varzaneh. Solid dot: Isfahan city.
Sample collection
Sand fly collection
Initially funnel traps and sticky traps were used to evaluate the sand fly populations in the region. In order to study the sand fly gut mirobiota, live sand flies were collected using different methods including CDC light traps, car traps, and aspirator during the months of June and July 2012. The traps were set adjacent to rodent burrows and animal shelters between the hours of 20:00 PM to 4:00 AM next day. Car traps involved a just parked vehicle used to attract sand flies in the vicinity of rodent burrows at nighttime. By this method the sand flies landing on the car were collected using a mouth aspirator and battery operated torch. Mouth aspirator and battery operated torch also were used to collect sand flies from bathrooms and toilets within human dwellings. Sand flies were transferred alive to the National Institute of Health Research, Isfahan Health Research Station (NIHR-IHRS) laboratory. The specimens were divided into two groups: 1) sand flies were immediately microdissected and transferred into brain heart infusion (BHI) broth culture medium, and 2) sand flies were offered blood meals on their natural host, R. opimus, that were reared in the animal unit of the center. The guts of the second group were microdissected and cultured in the medium 24 hours after blood meal ingestion. The specimens were identified after microdissection and only P. papatasi specimens were tested for gut microbiota.
Rodent collection
Active colonies of gerbils were identified and animal collections were performed in summer season using a Sherman live-trap (30 cm × 15 cm × 15 cm wire mesh) baited with a sliced cucumber. Approximately 15–20 live traps per day were used in each location. The traps were set close to a burrow entrance 2–3 h before the initiation of diurnal activity in the morning and in the evening when the temperature was not very hot. The traps were checked at noon and in the late evening before sunset. Collected rodents were fed sliced carrots until microbiological studies.
The nest materials
To examine acquirement, association, and probable circulation of bacteria among the sand fly gut, rodent skin, and food sources within the rodent nest, the soil, food supplies, and wool fibres within the nest were collected from the same colony where sand flies and rodents were trapped. For this purpose the rodent burrows that were built on clay hills were selected for excavation. Sampling was performed from different parts of the nest for bacterial investigation.
Plants: hyloxylon sp. and salsola sp
Sampling of the plants was confined to the available plants of Haloxylon sp. and Salsola sp., the two prevalent plants in the central desert of Iran. The whole plant of Salsola sp. (bushes) and the leaves and green branches of Haloxylon sp. were cut and placed in a sterile clean plastic bag and labeled with the given rodent colony.
Isolation of bacteria
Sand fly guts
Isolation of sand fly guts was conducted in a sterile environment under a microbiological lab hood on a sterile glass slide. Before dissection, individual flies were surface sterilized for 2 min in 70% ethanol. The gut from each sand fly was micro-dissected and homogenized in test tubes with screw tops containing 5 cc brain heart infusion (BHI) broth medium. The rest of the dissected insect body was mounted on a slide for morphological identification.
The nest materials
The nest materials comprising the plant pieces, wool fibres, and soil samples were collected and transferred to the lab. Plant pieces and wool fibres were gathered in sterile bags and then were immersed in the BHI broth medium. For the soil samples, 0.7 ml of soil sample was collected using 0.7 ml micro tubes and then homogenized in the same medium.
The rodent body
The live field captured rhomboids and the laboratory reared R. opimus in the NIHR-IHRS animal unit, were anesthetized using intramuscular ketamine hydrochloride (60 mg/kg) and xylazine (5 mg/kg). Sterile cotton swabs (presented in a sterile sealed test tube) were used to swab the exposed area around the auricle, eyelid, muzzle or footpads of the animal, where sand flies choose to take blood meals. The swabs were then placed in BHI broth. Single rodent faeces were collected from the colonies in 0.7 ml micro tubes and then homogenized in the BHI medium. Also a rodent was dissected for swabbing throughout its alimentary canal. All experiments on the rodents were performed in accordance with the guidelines of the Ethical Board of Tehran University of Medical Sciences, Iran.
To test whether the rodent skin bacteria can enter/infect the sand fly gut via blood feeding, a group of unfed female sand flies were allowed to feed on laboratory R. opimus specimens mentioned above. The gut microflora of the fed sand flies were tested 24–48 hours post-blood meal as this coincides with the highest growth rate of bacteria as suggested in the literature [30,35].
Plants: Hyloxylon sp. and Salsoa sp.
To examine the surface bacterial flora of the diet plants (Epiphytes), portions of collected samples, were placed into the screw top tubes containing liquid medium. For isolation of the potential bacteria present in the tissue and sap (Endophytes) of the given plants, first about 10 grams of the these plants were surface sterilized with 70% alcohol for two minutes and then their juice was extracted by mortar. Finally the plant juice was poured directly into the BHI broth culture medium.
Bacteriological methods
Culture media
In this research we initially used BHI broth medium. It is a versatile liquid infusion medium, and was chosen as an enriched non-selective medium to promote growth of a diverse range of microbes including nutritionally fastidious and nonfastidious bacteria, aerobic and facultative anaerobic bacteria, from a variety of clinical and nonclinical materials. The transparent test tubes were incubated aerobically at 37°C overnight. After 24–48 hours, opaque test tubes considered as positive were sub cultured in BHI agar medium overnight at the same condition. A test tube containing BHI broth opened near the dissection area constituted our sterility control during the dissection process.
Purification of bacterial isolates
To obtain individual pure colonies, the grown bacteria were serially diluted or streaked on agar plates. After 18–24 hours incubation at 37°C area well-isolated discrete colonies were seen. Colonies with different phenotype and morphology were isolated and sub-cultured successively. Pure isolates were partially preserved and partially used for further identification procedures like Gram staining and molecular studies.
Molecular identification
16S rRNA gene amplification
The purified bacterial colonies isolated from different specimens were tested using sequence analysis of the 16S rRNA gene for molecular identification after initial classical phenotyping and biochemical identifications. Each purified colony was subjected to genomic DNA extraction using either a boiling method (STET buffer) and/or routine phenol/chloroform DNA extraction method for the isolates with hard cell walls that had not yielded proper DNA by the boiling method. The 16S rRNA universal primers 16suF: 5′-GAGTTTGATCCTGGCTCAG-3′ and 16suR: 5′-GTTACCTTGTTACGACTT-3′ [61] were used to amplify a 1.5 kilo base (kb) partial sequence of the 16S rRNA gene. The PCR amplification was performed using Maxime PCR PreMix Kit (i-Taq) Cat. No. 25026 in 20 μl reaction mixtures containing 1 μl of 10 μM both forward and reverse primers and 1–2 μl (~0.1 μg) of extracted genomic DNA. Double-distilled water and BHI agar medium were used as DNA extraction and PCR negative controls. The PCR conditions were set as an initial denaturation at 94°C for 10 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 57.5°C for 40 s, and extension at 72°C for 30 s, followed by a final extension at 72°C for 8 min. PCR products were visualized on a 1% agarose gel containing ethidium bromide and using an UV transilluminator.
16S rRNA gene sequencing and analysing
All successfully amplified 16S rRNA amplicons were bidirectionally sequenced via the same amplification primers by Bioneer Company (S. Korea). The probable chimeric sequences were checked with Mallard program [62] for all acquired sequences and the specimens with suspicious sequences removed from data. The consensus of confident sequences was analyzed using nine databases available for 16S rRNA genes of prokaryotes including Greengenes [63], EzTaxon-e [64], NCBI (16S rRNA sequences) [65], NCBI (Nucleotide collection) [66], EMBL [67], DDBJ [68], leBIBI [69], RDP [70] and Blast2Tree [71]. Sequence homology analysis was based on the number and quality of nucleotides in a given sequence and hence appropriate settings and defaults such as cultivable and or non-cultivable, type specimens and or non-type specimens were selected. In case of discrepancies among different databases, species identifications were based on either the most common nomenclature among the results of the nine databases or on the basis of the highest percentage similarity.
The MEGA5 software was used for phylogenetic analyses and tree construction. Position verifications were done using distance (neighbor joining) and parsimony (1000 bootstrap replicates) analyses. The sequences were deposited in GenBank database.
The DNA gyrB PCR method as described by Wang et al. [72], followed by RFLP using suitable restriction enzyme/s were used for identification of the isolates for which 16S rRNA sequences represented more than a single species such as Shigella flexneri/Escherichia coli, Stenotrophomonas maltophilia/Pseudomonas geniculata, closely related Bacillus species that share a similar genetic background but occupy different ecological niches (B. thurengiensis, B. anthracis and B. cereus), and subspecies of Bacillus subtilis. Those bacteria for which 16S rRNA sequences were identical were normally determined by the EzTaxon database.
Contamination controls
In order to verify the findings, bacterial contamination of other parts of the dissected sand flies (except for gut), rodent skin used for sand fly blood feeding, rodent viscera, and environmental bacterial contamination of culture media were examined.
Results
Collected samples
In total, 476 biotic and abiotic specimens of the ZCL compartments originated from five locations of Isfahan province were collected and their microflora were examined. They included 390 sand fly guts, 28 rodent skins, 11 rodent feces, 11 rodent nest soils, 12 plant pieces collected within the rodent nest, 2 wool fibre samples, 14 Haloxylon sp. samples and 8 Salsola sp. samples. Details of the collected samples are given in Table 1.
Identification of isolated bacteria
Initially, all isolates were identified according to their morphological characteristics. On the basis of the cell morphology (Gram staining) the isolates fell into two main categories of Gram-negative (n = 24) and Gram-positive (n = 45) bacteria. On the basis of the colony morphology (form, elevation, margin, surface, opacity, and chromogenesis) a large variation of bacterial isolates was described. Finally, sequence analysis of 16S rRNA gene revealed 12 isolates from male sand fly guts, 162 isolates from female sand fly guts, 47 isolates from the internal and external parts of rodent bodies, 31 isolates from rodent nest materials, 14 isolates from Haloxylon sp. and 7 isolates from Salsola sp. plants.
In total 273 16 rRNA PCR products were sequenced and the consensus data were deposited in GenBank. [GenBank: JX661713-JX661718 and GenBank: KF254489-KF254756] (Table 2). Molecular identification was performed according to the 16S rRNA gene sequence similarity rates between the amplified specimens and the available data in the nine data banks (Table 2). Molecular identification revealed presence of 40, 24, 15, and 14 bacterial species from vector midgut, reservoir host body, rodent nest supplies, and the vegetarian diet sources, respectively. Phylogenetic relationships of the bacteria species are shown in a diagrammatic representation in Figure 2. They belonged to 4 phyla, 16 families, and 28 genera of bacteria (Table 2). The relative abundance of the bacteria genera is shown in Figure 3. Herein we report 69 bacterial species from four phyla comprising 44% Firmicutes, 34% Proteobacteria, 20% Actinobacteria and 2% Bacteroidetes from the four main components of the hyper-endemic ZCL focus. From 476 biotic and abiotic specimens that were investigated, most specimens contained culturable bacteria; some had two or more species, but in sand fly vectors, 75% of females and 68% of males were gnotobiotic while four plant specimens and one wool fibre of nest material were sterile.

Phylogenetic relationship of the bacteria isolated from the partners of the ZCL Hyper-endemic focus of Isfahan province, Iran. Common and similar colors point out groups, complexes and close relatives. Solid and hollow signs represent the source of isolation and numbers around circles indicate bacterial phyla. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Numbers at the nodes indicate percent bootstrap values (1000 replicates).

Categorization and relative abundance of the identified isolated bacteria from the partners of the ZCL Hyper-endemic focus of Isfahan province, Iran. Pseudomonas geniculata isolates are grouped with Genus Stenotrophomonas.
Sand fly gut bacteria
We isolated 170 bacterial isolates from the guts of P. papatasi which included 40 distinct species (Table 3). The bacteria comprised pathogens, e.g. Acinetobacter calcoaceticus-baumannii complex, Escherichia coli, Enterobacter cloacae complex, Pseudomonas aeruginosa, Staphylococcus aureus, and Stenotrophomonas spp, whereas others were non-pathogenic or rare–pathogenic organisms. Members of families Microbacteriaceae, Staphylococcaceae and Xanthomonadaceae were the most prevalent bacteria in the sand flies guts. Also Microbacterium paraoxydans was generally found in the greatest abundance.
Results showed bacterial diversity among sand fly gut in the three regions studied where we found 24, 18, and 12 bacterial species from rodent colony, cattle farm and human dwellings, respectively. Staphylococcus aureus was the only common bacterium in guts of sand flies of three regions. Comparison of the bacteria isolated from guts of the three locations indicated that species of Microbacterium, Pseudomonas, and Staphylococcus genera were dominant in human dwellings, cattle farms, and rodent colonies, respectively.
Bacteria circulation in micro- and macroclimate levels
In this study we simultaneously identified microflora of all wild components of ZCL cycle presented in and around a single rodent colony (microclimate) located between the Abbas Abad village and the Agha-ali Abbas shrine. 83 aerobic bacterial strains were isolated from the biotic and abiotic parts of the colony where 59 isolates were from the sand fly guts and 24 isolates were from other compartments (Table 4, data shown in parentheses). Both Pseudomonas geniculata and Staphylococcus aureus were present in male and female guts. These two bacteria were also found on the surface of Haloxylon plants and internal tissues of Salsola plants. Staphylococcus epidermidis were found in the female midguts, the rodent faeces and the nest soils. Bacillus mojavensis was found in the female sand fly guts and internal tissues of Salsola plants. Different strains of Bacillus subtilis were found in the female sand fly guts as well as in the nest plant pieces, and as epiphytes on both Salsola and Haloxylon plants. Details of spatial distribution of the bacteria isolated from different partners of the rodent colony are shown in Table 4 (data shown in parentheses).
At the macroclimate level, analyses were performed on all isolates found in the whole area of study and the bacteria were assembled in Table 4 according to their isolation origins. Comparative analysis revealed that in 16 cases the bacterial isolates were present in both micro- and macroclimates (Table 4).
Analyses of bacteria at the macroclimate level simulated bacterial circulation pattern among four ZCL operators. In this way selection of suitable candidates and their possible application routes were disclosed. This model demonestrates how bacteria are circulated among other ZCL partners by sand flies (Figure 4).

Natural circulations of bacteria among the partners of ZCL focus in Isfahan, Iran. Operators P. papatasi, R. opimus and plant food sources (Hyloxylon and Salsola) indicated by graphics. One or two way arrows show how the bacteria are acquired and circulated. Continuous and non-continuous lines represent proved and doubtful transmission routes respectively. Bacteria phyla engrave close to the isolated source.
Sand fly oviposition inducing bacteria
In this study we found 12 isolates of soil bacteria comprising eight bacilli and four coagulase-negative staphylococci that are known to be involved in the inter-kingdom communication of eukaryotic sand flies/plants and prokaryotic bacteria. Details of the bacteria that may be considered to enhance the oviposition response of gravid P. papatasi females are shown in Table 5.
Potential routes of bacterial acquisition by sand flies
Adult P. papatasi midgut bacteria might be acquired from three general routes: adult sugar feeding on the plant saps, adult blood feeding on the animal host and larval feeding in their breeding places that would be transmitted transstadially from larvae to adult. There were five common bacteria present in both male and female guts: Pseudomonas geniculata (male = 3, female = 17), P. aeruginosa (1, 12), Bacillus vallismortis (1, 1), B. flexus (1, 1), and Staphylococcus aureus (3, 19). The habits of feeding on plant saps by both adult female and male sand flies and of feeding on organic materials by larvae can explain the presence of those common bacteria in their digestive tracts. Comparison of the bacterial content of the nest soil, rodent feces, the imported plant diets, and the wool fibres specimens with the bacteria found in the gut of adult sand flies verified the possibility of a transstadial transmission mode of 7 isolates during transition from breeding places materials (immature midgut) to adult midgut (Table 6).
Concerning the sand fly midgut bacteria and the routes that allow them to enter during insect feeding, it was revealed that 5 species of Acinetobacter calcoaceticus, Enterobacter ludwigii, Bacillus amyloliquefaciens, Enterococcus gallinarum and Escherichia coli might be acquired when blood feeding on the reservoir host and 7 species of B. pumilus, B. mojavensis, B. subtilis, S. aureus, Stenotrophomonas maltophilia, Pseudomonas geniculata and Planomicrobium spp. might be obtained when feeding on the plant saps (Table 6).
The bacterium Microbacterium paraoxydans was isolated from different physiological states (unfed, fed, semi-gravid and gravid) of female sand flies caught from an uninhabited home in the Parvaneh village of Habib-abad district, indicating that the isolate bacterium could tolerate blood digestion and gonotrophic processes (Table 7). Bacterial flora comparisons before and after blood feeding showed that some bacterial strains remain after blood digestion but in general there were variations in bacterial compositions (Table 8).
Possible bacterial acquirement of sand fly gut via blood feeding
A group of sand flies were allowed to feed on rodents and 24 hours after blood ingestion their gut contents were examined for the presence of the bacteria originally isolated on the rodent skin. None of the rodent skin bacteria was found in the female gut. Details of the bacteria that were isolated from the control specimens are shown in Table 9. Comparison of skin surface bacteria of field and lab rodents showed no resemblance except for the presence of Enterococcus faecalis.
Discussion
In this study we investigated the microbiology of the biotic and abiotic compartments of a natural ZCL cycle, including the gut of the sand fly vector P. papatasi, skin and internal organs of the animal reservoir R. opimus, natural plants normally used as food for both vector and reservoir, soils and other materials present in rodent nests and sand fly larval breeding places. Data analyses showed that bacteria flora encompass a wide range of aerobic to facultative anaerobic, harmless commensals to important pathogens, inter-cellular to intra-cellular, environmental to nosocomial pathogens, skin surface to gut lumen bacteria, endophytes to epiphytes, extremophiles to mesophiles or neutrophiles, land to air and ocean habitat, animal and human probiotics to plant growth-promoting rhizobacteria (PGPR) (Table 2).
This study shows an association between the microbiota of the sand fly gut and the places they live in; a number of the isolates identified in the sand fly guts were also present in the R. opimus nest materials/sand fly larval breeding places and the plants which were used by vectors as sugar sources or by rodents for food and water. The association between the microbiota of the sand fly gut and larval breeding sites supports transstadial transmission of some bacteria; however, some authors argue against the transstadial route and believe that full gut turnover occurs during pupation [73]. Environmental acquisition of sand fly gut bacteria has been reported by other investigators [57,74]. These studies suggest that the sand fly gut microbiota is a reflection of both the environment in which the sand fly larvae reside and the food sources of larvae and adults.
Comparison of bacterial diversity in the sand fly guts from three regions revealed that the microflora were largely environmental; the predominant bacteria were species of Microbacterium, Pseudomonas, and Staphylococcus in the human dwellings, cattle farm, and rodent colony, respectively. This diversity may be partly due to the kind and accessibility of sand fly hosts. The available hosts for sand flies of the human dwellings (Parvaneh village) were pigeons, sheep, and or humans, whereas available hosts for sand flies trapped from rodent’s colony and the cattle farm were rodent and cow, respectively. The effect of blood meal components on the growth and persistence of some microbes has been proven [75,76]. Other factors such as intestine-specific structure, pH, redox, digestive enzymes, and food sources are determinants of microbial colonization in insect guts [34,77,78]. It was noted that in insects with various diets, microbial growth kinetics are dissimilar and different types of bacteria are present in their guts. Dillon et al. [77] showed that the number of bacteria present in the gut of P. papatasi changes during the lifetime of a female. Volf et al. [29] showed that the highest bacterial counts occurred two days after blood ingestion. The protein rich bolus of the blood presumably caused rapid growth of midgut bacteria and when digestion is completed (on day 4–5) most bacteria were defecated with blood remains. Seven days after blood feeding the bacterial count returned to the pre-feeding level. It is suggested that blood digestion, the development of Leishmania parasites in the competent vector and bacterial population fluctuations are closely related to each other. Sant’Anna et al. [79] implied colonisation resistance in the Lutzomyia longipalpis and investigated the balance of microbiota and Leishmania in the insect gut. They found a reduction in the number of flies harboring a Leishmania population that had been pre-fed with Pseudozyma sp. and Asaia sp. or Ochrobactrum intermedium. Also they discovered that L. mexicana protects Lu. longipalpis from Serratia marcescens infection. They concluded that Leishmania-vector association might benefit for both the sand fly and parasite [79].
In the current study we found that Microbacteriaceae were the most frequently isolated bacteria (27%) in the P. papatasi digestive tract, which is in agreement with the previous study of sand fly gut microbiology of P. papatasi and P. duboscqi [28]. Also we observed presence of Microbacterium sp in different physiologic states of P. papatasi indicating resistance to trypsins and chymotrypsin enzymes during blood digestion (Table 8). These bacteria have already been found in the adult guts of P. duboscqi and P. papatasi [28]. Also presence of Microbacterium in the 2nd and 4th instar larvae, pupae, male and female adults of P. duboscqi suggested transstadial transmission. Microbacterium species was also reported in the guts of field trapped P. argentipes from India and of Ixodes ricinus [57,80]. Interestingly, various strains of Microbacterium isolated from Musca domestica guts have been shown to support its larval development [81]. It is demonstrated that gut microbiota also influence the sand fly immune systems. For example, the concentration of regulating gut-microbe homeostasis such as reactive oxygen species (ROS) will change in Lu. longipalpis midguts in response to Leishmania parasite or to insect pathogen Serratia marcescens [82]. Also it is shown that the rate of defensin expression in Lu. longipalpis upon bacterial and Leishmania infection will vary based on the bacterial species and the routes of Leishmania infection [83]. Further studies warrant showing the effect of gut microbiota on the immune system of P. papatasi the most important vector of ZCL in the Old World.
It is shown that oviposition by gravid P. papatasi is influenced by microbial flora of the environment. Radjame et al. [84] and Mukhopadhyay et al. [74] introduced bacteria to breeding sites, thereby attracting sand flies [74,84]. It was implied that the gravid sand flies found oviposition sites through attraction cues of four bacillus species B. pumilus, B. cereus, B. firmus, B. licheniformis and one Coagulase-negative staphylococcus, Staphylococcus saprophyticus. The current study revealed presence of these five oviposition-inducing bacteria in the study area (Table 5), however only two species of B. pumilus and S. saprophyticus were found in the great gerbil nest materials where sand flies lay eggs.
We found some bacteria in ZCL partners that can cause super infection in human lesions, which may hinder or prevent the healing process of ZCL. In a rural area of north Isfahan, the bacteria were isolated from 66.8% of ZCL and 64.7% of non-ZCL lesions. The most common species were Staphylococcus aureus and S. epidermidis followed by Bacillus sp., Streptococcus pyogenes, Escherichia coli, Klebsiella sp., Proteus sp., Enterobacter sp. and Pseudomonas aeroginosa [85].
Results of this study established the presence of Enterobacter cloacae subsp. dissolvens and Bacillus subtilis in the digestive tract of sand flies as well as at the larval breeding sites in the great gerbil nest materials and plants that were part of rodent and insect diets. The association of these organisms with the sand flies makes them good candidates for use in a model of paratransgenesis. The two bacteria are commensal sand fly bacteria and could be transformed to deliver antileishmanial peptides within sand fly guts to prevent or to reduce Leishmania transmission. The transformed bacteria could be delivered easily on the plants and or sand fly larval breeding sites such as great gerbil nests, pigeon nests, and sheep and cattle sheds. The sand fly would encounter and be infected with the bacteria either at the larval stage in their breeding sites while feeding on organic materials or at the adult stage while taking sugar meals on plants. The phlebotomine sand flies, require sugar for survival and several different sources of sugar meals of insect origin (honeydew), and of plant origin, have been identified [42,86,87]. These sugar meals are often taken by feeding directly on tissues of plant organs including stems, leaves, and flowers [42,45,46]. Hurwitz et al. [88] showed transstadial passage of some bacteria in P. argentipes sand fly by introducing an engineered Bacillus subtilis expressing Green Fluorescent Protein (GFP) in sterilized larval chow and retrieved the glowing bacteria in the adult.
Bacillus subtilis harbour metabolites that exhibit activity against both the larval and pupal stages of mosquitoes [89] as well as plant pathogens [90]. It is one of the main bacteria used in industrial production of enzymes, antibiotics, fermented foods and vitamins [91,92]. Enterobacter cloacae is a member of the normal gut flora of many insects such as symbiotic or entomopathogenic and in the surface of vegetables. Several reports have been made with E. cloacae strains in the biological control of plant pathogens, such as Phytium spp., Sclerotinia sp., Rhizopus sp., Fusarium spp. and many others [93]. Also it was shown that the bacterium significantly is able to block the Plasmodium vivax sporogonic development in Anopheles albimanus [93]. Currently Eappen et al. [94] showed that E. cloacae strongly induce expression of components of the mosquito immune response in the An. stephensi midgut.
Enterobacter cloacae have already been tested to deliver, express, and spread foreign genes in termite colonies [95]. Watanabe et al. [96] transformed E. cloacae with an ice nucleation gene to reduce the mulberry pyralid moth, Glyphodes pyloalis. Also Kuzina et al. [97] transformed E. gergoviae with the Bacillus thuringiensis toxin gene to control pink bollworm, (Pectinophora gossypiella) (Lepidoptera: Gelechiidae).
The present study and literature review revealed that E. cloacae subsp. dissolvens belong to the natural and stable flora of P. papatasi, and are amenable to isolation, culture and transformation with foreign genes. Although some species of Enterobacter, including E. cloacae, are potential human pathogens, these species have also been reported from rhizosphere of various crops exhibiting plant growth promoting abilities; just as E. cloacae subsp. dissolvens was found associated with rhizosphere of soybean under natural field conditions [98]. It was isolated from orchard soil samples in China, and is a potential industrial candidate for 2,3-Butanediol productions, which could produce more than 100 g/liter 2,3-BD from glucose [99]. E. cloacae subsp. dissolvens was also found in the endosphere of rice plants without causing apparent harm to the host plant [100,101]. Study of Rodrigues Neto et al. [102] showed low level of pathogenicity of the type strain of E. cloacae dissolvens on the onion.
Crucial requirements for the application of paratransgenesis in control of Leishmania is the ability to transform the isolate bacteria and to then test their potentially colonization rates in the sand flies and finally to assess their antileishmanial effect in laboratory and field conditions. There are some available antileishmanial molecules such as histatin 5, racemoside A, monoclonal antibodies, defensin A, and temporins [103-107]. We are currently started to transform the E. cloacae subsp. dissolvens isolate with defensin gene to test its efficacy against L. major in in-vitro condition. Our study is ongoing to enhance the expression and to assess the efficacy of the antileishmanial molecule in this genetically modified bacterium (unpublished data; data not shown). We have tested the transstadial situation of the candidate bacteria, and found that E. cloacae subsp. dissolvens was transstadial and transfer from larvae to adult stage that would permit delivery of transformed bacteria to the breeding sites of sand fly larvae.
Conclusions
On the basis of our knowledge this study is the first culture-dependent molecular analysis of four important partners of ZCL cycle and could be used as an effective platform for future efforts to prevent leishmaniasis. This study revealed possible routes of acquisition of sand fly bacteria, which can provide proper application of transformed bacteria in the field. Also here we introduced two bacterial species of Entrobacter cloacae subsp. dissolvens and Bacillus subtilis, which are good candidates for paratransgenic control of the ZCL in the hyperendemic focus in central Iran.
Abbreviations
- ZCL:
-
Zoonotic Cutaneous Leishmaniasis
- NIHR-IHRS:
-
National institute of health research, Isfahan health research station
- BHI:
-
Brain heart infusion
- PGPR:
-
Plant growth-promoting rhizobacteria
References
Dostálová A, Volf P: Leishmania development in sand flies: parasite-vector interactions overview. Parasit Vectors 2012, 5:276.
WHO: Fact sheet n° 375. 2014. updated January 2014 [http://www.who.int/mediacentre/factsheets/fs375/en/]
Desjeux P: Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis 2004, 27:305–318.
Gramiccia M, Gradoni L: The current status of zoonotic leishmaniases and approaches to disease control. Int J Parasitol 2005, 35:1169–1180.
Hajjaran H, Mohebali M, Mamishi S, Vasigheh F, Oshaghi MA, Naddaf SR, Teimouri A, Edrissian GH, Zarei Z: Molecular identification and polymorphism determination of cutaneous and visceral leishmaniasis agents isolated from human and animal hosts in Iran. Biomed Res Int 2013, 2013:789326.
Akhavan AA, Yaghoobi-Ershadi MR, Hasibi F, Jafari R, Abdoli H, Arandian MH, Soleimani H, Zahraei-Ramazani AR, Mohebali M, Hajjaran H: Emergence of cutaneous leishmaniasis due to Leishmania major in a new focus of southern Iran. Iran J Arthropod-Borne Dis 2007, 1:1–8.
Boussaa S, Pesson B, Boumezzough A: Phlebotomine sand flies (Diptera: Psychodidae) of Marrakech city, Morocco. Ann Trop Med Parasitol 2007, 101(8):715–724.
Rassi Y, Oshaghi MA, Azani SM, Abaie MR, Rafizadeh S, Mohebai M, Mohtarami F, Zeinali MK: Molecular detection of Leishmania infection due to Leishmania major and Leishmania turanica in the vectors and reservoir host in Iran. Vector Borne Zoonotic Dis 2011, 11(2):145–150.
Latrofa MS, Annoscia G, Dantas-Torres F, Traversa D, Otranto D: Towards a rapid molecular identification of the common phlebotomine sand flies in the Mediterranean region. Vet Parasitol 2012, 184(2–4):267–270.
Bakhshi H, Oshaghi MA, Abai MR, Rassi Y, Akhavan AA, Sheikh Z, Mohtarami F, Saidi Z, Mirzajani H, Anjomruz M: Molecular detection of Leishmania infection in sand flies in border line of Iran-Turkmenistan: restricted and permissive vectors. Exp Parasitol 2013, 135(2):382–387.
Yaghoobi-Ershadi MR: Phlebotomine sand flies (Diptera: Psychodidae) in Iran and their role on Leishmania transmission. J Arthropod-Borne Dis 2012, 6:1–17.
Yaghoobi-Ershadi MR, Javadian E: Epidemiological study of reservoir hosts in an endemic area of Zoonotic Cutaneous Leishmaniasis in Iran. Bull World Health Org 1996, 74:587–590.
Yaghoobi-Ershadi MR, Akhavan AA, Mohebali M: Meriones libycus and Rhombomys opimus (Rodentia:Gerbillidae) are the main reservoir hosts in a new focus of zoonotic cutaneous leishmaniasis in Iran. Trans R Soc Trop Med Hyg 1996, 90:503–504.
Dubrovsky YA: Biology of great gerbil–the principal carrier of zoonotic cutaneous leishmaniasis. WHO Traveling Seminar on leishmaniasis control. ex-USSR Ministry of Health; 1979.
Dennis DT, Gage KL, Gratz N, Poland JD, Tikhomirov E: Plague Manual: Epidemiology, Distribution, Surveillance and Control. Geneva, Switzerland: World Health Organization; 1999.
Abai MR, Oshaghi MA, Tajedin L, Rassi Y, Akhavan AA: Geographical distribution and ecological features of the great gerbil subspecies in the main zoonotic cutaneous leishmaniasis foci in Iran. Asian Pac J Trop Med 2010, 3:800–803.
Kausrud KL, Begon M, Ben Ari T, Viljugrein H, Esper J, Büntgen U, Leirs H, Junge C, Yang B, Yang M, Xu L, Stenseth NC: Modeling the epidemiological history of plague in Central Asia: palaeoclimatic forcing on A disease system over the past millennium. BMC Biol 2010, 8:112.
Akhavan AA, Mirhendi H, Khamesipour A, Alimohammadian MH, Rassi Y, Bates P, Kamhawi S, Valenzuela JG, Arandian MH, Abdoli H, Jalali-zand N, Jafari R, Shareghi N, Ghanei M, Yaghoobi-Ershadi MR: Leishmania species: detection and identification by nested PCR assay from skin samples of rodent reservoirs. Exp Parasitol 2010, 126:552–556.
Bakhshi H, Oshaghi MA, Abai MR, Rassi Y, Akhavan AA, Mohebali M, Hajaran H, Mohtarami F, Mirzajani H, Maleki-Ravasan N: MtDNA cytb structure of Rhombomys opimus (Rodentia: Gerbellidae), the main reservoir of cutaneous leishmaniasis in the borderline of Iran-Turkmenistan. J Arthropod-Borne Dis 2013, 7:173–184.
Oshaghi MA, Rassi Y, Tajedin L, Abai MR, Akhavan AA, Enayati A, Mohtarami F: Mitochondrial DNA diversity in the populations of great gerbils, Rhombomys opimus , the main reservoir of cutaneous leishmaniasis. Acta Trop 2011, 119(2–3):165–171.
Davis S, Begon M, De Bruyn L, Ageyev VS, Klassovskiy NL, Pole SB, Viljugrein H, Stenseth NCHR, Leirs H: Predictive thresholds for plague in Kazakhstan. Science 2004, 304:736–738.
Parvizi P, Ready PD: Nested PCRs and sequencing of nuclear ITS-rDNA fragments detects three Leishmania species of gerbils in sand flies from Iranian foci of zoonotic cutaneous leishmaniasis. Trop Med Int Health 2008, 13:1159–1171.
Strelkova MV, Eliseev LN, Ponirovsky EN, Dergacheva TI, Annacharyeva DK, Erokhin PI, Evans DA: Mixed leishmanial infections in Rhombomys opimus : a key to the persistence of Leishmania major from one transmission season to the next. Ann Trop Med Parasit 2001, 95:811–819.
Dubrovsky YA: Ecological causes of predominance of some mammals as reservoirs of Leishmania tropica major in Turanian deserts. Folia Parasitol (Praha) 1975, 22:163–169.
Rogovin K, Randall JA, Kolosova I, Moshkin M: Social correlates of stress in adult males of the great gerbil. Rhombomys opimus , in years of high and low population densities. Horm Behav 2003, 43:132–139.
Wei L, Wenxuan X, Weikang Y, Cong G, David B, Canjun X, Jie L, Feng X, Honghai Q: Food habits of the great gerbil ( rhombomys opimus ) in the southern Gurbantunggut desert, Xinjiang, China. Pakistan J Zool 2012, 44:931–936.
Towhidi A, Saberifar T, Dirandeh E: Nutritive value of some herbage for dromedary camels in the central arid zone of Iran. Trop Anim Health Prod 2011, 43(3):617–622.
Guernaoui S, Garcia D, Gazanion E, Ouhdouch Y, Boumezzough A, Pesson B, Fontenille D, Sereno D: Bacterial flora as indicated by PCR-temperature gradient gel electrophoresis (TGGE) of 16S rDNA gene fragments from isolated guts of phlebotomine sand flies (diptera: psychodidae). J Vector Ecol 2011, 36(Suppl 1):144–147.
Volf P, Kiewegova A, Nemec A: Bacterial colonization in the gut of phlebotomus duboscqi (diptera psychodidae): transtadial passage and the role of female diet. Folia Parasitol 2002, 49:73–77.
O’Neill SL, Hoffmann AA, Werren JH: Influential Passengers. Inherited Microorganisms and Arthropod Reproduction. New York: Oxford Univer In sity Press; 1997.
Baumann P, Moran NA, Baumann L: Bacteriocyte-Associated Endosymbionts of Insects. In The Prokaryotes. Edited by Dworkin M. New York: Springer; 2000.
Bourtzis K, Miller TA: Insect Symbiosis. Boca Raton, FL: CRC Press; 2003.
Rio RVM, Hu Y, Aksoy S: Strategies of the home team: symbioses exploited for vector-borne disease control. Trends Microbiol 2004, 12:325–336.
Dillon RJ, Dillon VM: The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol 2004, 49:71–92.
Weiss B, Aksoy S: Microbiome influences on insect host vector competence. Trends Parasitol 2011, 27:514–522.
Douglas AE: The microbial dimension in insect nutritional ecology. Funct Ecol 2009, 23:38–47.
Visôtto LE, Oliveira MGA, Guedes RNC, Ribon AOB, Good-God PIV: Contribution of gut bacteria to digestion and development of the velvet bean caterpillar, anticarsia gemmatalis . J Insect Physiol 2009, 55:185–191.
Killick-Kendrick R: The biology and control of phlebotomine sand flies. Clin Dermatol 1999, 17:279–289.
Dye CM, Guy W, Elkins DB, Wilkes TJ, Killick-Kendrick R: The life expectancy of phlebotomine sand flies: first field estimates from southern France. Med Vet Entomol 1987, 1:417–425.
Gibb PA, Anderson JC, Dye C: Are nulliparous flies light shy? Trans R Soc Trop Med Hyg 1988, 82:342–343.
Yuval B, Warburg A, Schlein Y: Leishmaniasis in the Jordan Valley. V. Dispersal characteristics of the sand fly Phlebotomus papatasi . Med Vet Entomol 1988, 2:391–395.
Müller GC, Schlein Y: Nectar and honeydew feeding of Phlebotomus papatasi in a focus of Leishmania major in Neot Hakikar oasis. J Vector Ecol 2004, 29:154–158.
MacVicker JAK, Moore JS, Molyneux DH, Maroli M: Honeydew sugars in wild caught Italian phlebotomine sand flies (diptera: psychodidae) as detected by high performance liquid chromatography. Bull Entomol Res 1990, 80:339–344.
Wallbanks KR, Moore JS, Bennet LR, Soren R, Molyneux DH, Carlin JM, Perez JE: Aphid derived sugars in the neotropical sand fly lutzomyia peruensis . Trop Med Parasitol 1991, 42:60–62.
Schlein Y, Jacobson RL: Mortality of Leishmania major in Phlebotomus papatasi caused by plant feeding of the sand flies. Am J Trop Med Hyg 1994, 50:20–27.
Schlein Y, Muller G: Assessment of plant tissue feeding by sand flies (diptera: psychodidae) and mosquitoes (diptera: culicidae). J Med Entomol 1995, 32:882–887.
WHO: Global Insecticide use for Vector-Borne Disease Control. 5th edition. Geneva, Switzerland: WHO Pesticide Evaluation Scheme (WHOPES), World Health Organization; 2011.
Ranson H, N’Guessan R, Lines J, Moiroux N, Nkuni Z, Corbel V: Pyrethroid resistance in African anopheline mosquitoes: what are the implications for malaria control? Trends Parasitol 2011, 27:91–98.
Durvasula RV, Gumbs A, Panackal A, Kruglov O, Aksoy S, Merrifield RB, Richards FF, Beard CB: Prevention of insect-borne disease: an approach using transgenic symbiotic bacteria. Proc Natl Acad Sci U S A 1997, 94:3274–3278.
Medlock J, Atkins KE, Thomas DN, Aksoy S, Galvani AP: Evaluating paratransgenesis as a potential control strategy for African trypanosomiasis. PLoS Negl Trop Dis 2013, 7:e2374.
Favia G, Ricci I, Damiani C, Raddadi N, Crotti E, Marzorati M, Rizzi A, Urso R, Brusetti L, Borin S, Mora D, Scuppa P, Pasqualini L, Clementi E, Genchi M, Corona S, Negri I, Grandi G, Alma A, Kramer L, Esposito F, Bandi C, Sacchi L, Daffonchio D: Bacteria of the genus asaia stably associate with anopheles stephensi , an asian malarial mosquito vector. Proc Natl Acad Sci U S A 2007, 104:9047–9051.
Jin C, Ren X, Rasgon JL: The virulent wolbachia strain wMelPop efficiently establishes somatic infections in the malaria vector anopheles gambiae . Appl Environ Microbiol 2009, 75:3373–3376.
Chavshin AR, Oshaghi MA, Vatandoost H, Pourmand MR, Raeisi A, Enayati AA, Mardani N, Ghoorchian S: Identification of bacterial microflora in the midgut of the larvae and adult of wild caught anopheles stephensi : a step toward finding suitable paratransgenesis candidates. Acta Trop 2012, 121:129–134.
Chavshin AR, Oshaghi MA, Vatandoost H, Yakhchali B, Raeisi A, Zarenejad F: Escherichia coli expressing a green fluorescent protein (GFP) in anopheles stephensi : a preliminary model for paratransgenesis. Symbiosis 2013, 60:17–24.
Chavshin AR, Oshaghi MA, Vatandoost H, Pourmand MR, Raeisi A, Terenius O: Isolation and identification of culturable bacteria from wild anopheles culicifacies , a first step in a paratransgenesis approach. Parasit Vectors 2014, 7:419.
Akbari S, Oshaghi MA, Hashemi-Aghdam SS, Hajikhani S, Oshaghi G, Shirazi MH: Aerobic bacterial community of american cockroach periplaneta americana , a step toward finding suitable paratransgenesis candidates. J Arthropod-Borne Dis 2015, 9(1):35–48.
Hillesland H, Read A, Subhadra B, Hurwitz I, McKelvey R, Ghosh K, Das P, Durvasula R: Identification of aerobic gut bacteria from the kala azar vector, phlebotomus argentipes : a platform for potential paratransgenic manipulation of sand flies. Am J Trop Med Hyg 2008, 79:881–886.
Maleki-Ravasan N, Oshaghi MA, Hajikhani S, Saeidi Z, Akhavan AA, Gerami-Shoar M, Shirazi MH, Yakhchali B, Rassi Y, Afshar D: Aerobic microbial community of insectary population of phlebotomus papatasi . J Arthropod-Borne Dis 2014, 8:69–81.
Ren X, Hoiczyk E, Rasgon JL: Viral paratransgenesis in the malaria vector anopheles gambiae . PLoS Pathog 2008, 4:e1000135.
Carlson J, Suchman E, Buchatsky L: Densoviruses for control and genetic manipulation of mosquitoes. Adv Virus Res 2006, 68:361–392.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ: 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 1991, 173:697–703.
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ: New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 2006, 72:5734–5741.
Greengenes: 16S rDNA data and tools. http://greengenes.lbl.gov/cgi-bin/nph-index.cgi.
EzTaxon-e: identification of prokaryotes based on 16S ribosomal RNA gene sequences. http://eztaxon-e.ezbiocloud.net.
NCBI BLAST, search set: 16S rRNA sequences. http://blast.ncbi.nlm.nih.gov/Blast.cgi.
NCBI BLAST, search set: nucleotide collection. http://blast.ncbi.nlm.nih.gov/Blast.cgi.
EMBL, ENA: The European nucleotide archive. http://www.ebi.ac.uk/ena.
DDBJ: DNA data bank of Japan. http://blast.ddbj.nig.ac.jp/?lang=en.
leBIBI: Bio informatics bacteria identification. http://umr5558-sud-str1.univ-lyon1.fr/lebibi/lebibi.cgi.
RDP: Ribosomal database project. http://rdp.cme.msu.edu/seqmatch/seqmatch_intro.jsp.
Blast2Tree: Blast server for the identification of prokaryotes. http://bioinfo.unice.fr/blast/.
Wang L, Lee F, Tai C, Kasai H: Comparison of gyrB gene sequences, 16S rRNA gene sequences and DNA-DNA hybridization in the bacillus subtilis group. Intl J Syst Evol Microbiol 2007, 57:1846–1850.
Moll RM, Romoser WS, Modrakowski MC, Moncayo AC, Lerdthusnee K: Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (diptera: culicidae ) metamorphosis. J Med Entomol 2001, 38:29–32.
Mukhopadhyay J, Braig HR, Rowton ED, Ghosh K: Naturally occurring culturable aerobic gut flora of adult Phlebotomus papatasi , vector of Leishmania major in the old world. PLoS One 2012, 7:e35748.
Anand C, Rhonda G, Helene S, Fonseca K, Olsen M: Pig and goat blood as substitutes for sheep blood in blood-supplemented agar media. J Clin Microbiol 2000, 38:591–594.
Tresierra-Ayala A, Navas M, Flores J, Perea R, Huanaquiri J, Bendayán M, Fernández H: Growth capacity of thermo tolerant campylobacters in culture media supplemented with pig and cow blood. Braz Arch Biol Technol 2010, 53:1087–1091.
Dillon RJ, Kordy EE, Lanee RP: The prevalence of a microbiota in the digestive tract of phlebotomus papatasi . Ann Trop Med Parasitol 1996, 90:669–673.
Lehane M: The Biology of Blood-Sucking in Insects. 2nd edition. Cambridge: Cambridge University Press; 2005.
Sant’Anna MRV, Diaz-Albiter H, Aguiar-Martins K, Al Salem WS, Cavalcante RR, Dillon VM, Bates P, Genta FA, Dillon RJ: Colonisation resistance in the sand fly gut: Leishmania protects Lutzomyia longipalpis from bacterial infection. Parasit Vectors 2014, 7:329.
Rudolf I, Mendel J, Sikutova S, Svec P, Masarikova J, Novakova D, Bunkova L, Sedlacek I, Hubalek Z: 16S rRNA gene-based identification of cultured bacterial flora from host-seeking ixodes ricinus , dermacentor reticulatus and haemaphysalis concinna ticks, vectors of vertebrate pathogens. Folia Microbiol (Praha) 2009, 54:419–428.
Zurek L, Schal C, Watson DW: Diversity and contribution of the intestinal bacterial community to the development of musca domestica (diptera: muscidae) larvae. J Med Entomol 2000, 37:924–928.
Diaz-Albiter H, Sant’Anna MR, Genta FA, Dillon RJ: Reactive oxygen species-mediated immunity against Leishmania mexicana and Serratia marcescens in the phlebotomine sand fly Lutzomyia longipalpis . Biol Chem 2012, 287(28):23995–24003.
Telleria EL, Sant’Anna MRV, Alkurbi MO, Pitaluga AN, Dillon TJ, Traub-Csekö YM: Bacterial feeding, Leishmania infection and distinct infection routes induce differential defensin expression in Lutzomyia longipalpis . Parasit Vectors 2013, 6:12.
Radjame K, Srinivasan R, Dhanda V: Oviposition response of phlebotomid sand fly phlebotomus papatasi to soil bacteria isolated from natural breeding habitats. Indian J Exp Biol 1997, 35:59–61.
Doudi M, Setorki M, Narimani M: Bacterial superinfection in Zoonotic Cutaneous Leishmaniasis. Med Sci Monit 2012, 18:356–361.
Schlein Y, Warburg A: Phytophagy and the feeding cycle of phlebotomus papatasi (diptera: psychodidae) under experimental conditions. J Med Entomol 1986, 23:11–15.
Moore JS, Kelly TB, Killick-Kendrick R, Killick-Kendrick M, Wallbanks KR, Molyneux DH: Honeydew sugars in wild-caught phlebotomus ariasi detected by high performance liquid chromatography (HPLC) and gas chromatography (GC). Med Vet Ent 1987, 1:427–434.
Hurwitz I, Hillesland H, Fieck A, Das P, Durvasula R: The paratransgenic sand fly: a platform for control of Leishmania transmission. Parasit Vectors 2011, 2011(4):1–9.
Geetha I, Manonmani AM, Prabakaran G: Bacillus amyloliquefaciens : a mosquitocidal bacterium from mangrove forests of andaman & nicobar islands, india. Acta Trop 2011, 120:155–159.
Chen XH, Scholz R, Borriss M, Junge H, Mögel G, Kunz S, Borriss R: Difficidin and bacilysin produced by plant associated bacillus amyloliquefaciens are efficient in controlling fire blight disease. J Biotechnol 2009, 140:38–44.
Schallmey M, Singh A, Ward OP: Developments in the use of bacillus species for industrial production. Can J Microbiol 2004, 50:1–17.
Cutting SM: Bacillus probiotics. Food Microbiol 2011, 28:214–220.
Gonzalez-Ceron L, Santillan F, Rodriguez MH, Mendez D, Hernandez-Avila JE: Bacteria in midguts of field-collected anopheles albimanus block plasmodium vivax sporogonic development. J Med Entomol 2003, 40:371–374.
Eappen AG, Smith RC, Jacobs-Lorena M: Enterobacter -activated mosquito immune responses to plasmodium involve activation of SRPN6 in anopheles stephensi . PLoS One 2013, 8:e62937.
Husseneder C, Grace JK: Genetically engineered termite gut bacteria ( enterobacter cloacae ) deliver and spread foreign genes in termite colonies. Appl Microbiol Biotechnol 2005, 68:360–367.
Watanabe K, Abe K, Sato M: Biological control of an insect pest by gut-colonizing enterobacter cloacae transformed with ice nucleation gene. J Appl Microbiol 2000, 88:90–97.
Kuzina LV, Miller ED, Ge B, Miller TA: Transformation of enterobacter gergoviae isolated from pink bollworm (lepidoptera: gelechiidae) gut with bacillus thuringiensis toxin. Curr Microbiol 2002, 44:1–4.
Ramesh A, Sharma SK, Sharma MP, Yadav N, Joshi OP: Plant growth-promoting traits in enterobacter cloacae subsp. Dissolvens MDSR9 isolated from soybean rhizosphere and its impact on growth and nutrition of soybean and wheat upon inoculation. Agric Res 2014, 3:53–66.
Xu Y, Wang A, Tao F, Su F, Tang H, Ma C, Xu P: Genome sequence of enterobacter cloacae subsp. Dissolvens SDM, an efficient biomass-utilizing producer of platform chemical 2,3-butanediol. J Bacteriol 2012, 194:897–898.
Prakamhang J, Minamisawa K, Teamtaisong K, Boonkerd N, Teaumroong N: The communities of endophytic diazotrophic bacteria in cultivated rice ( oryza sativa L.). Appl Soil Ecol 2009, 42:141–149.
Hardoim PR, Nazir R, Sessitsch A, Elhottová D, Korenblum E, van Overbeek LS, van Elsas JD: The new species enterobacter oryziphilus sp. nov. and enterobacter oryzendophyticus sp. nov. are key inhabitants of the endosphere of rice. BMC Microbiol 2013, 13:164.
Rodrigues Neto J, Yano T, Beriam LOS, Destéfano SAL, Oliveira VM, Rosato YB: Comparative RFLP-ITS analysis between enterobacter cloacae strains isolated from plants and clinical origin. Arq Inst Biol 2003, 70:367–372.
Mangoni ML, Saugar JM, Dellisanti M, Barra D, Simmaco M, Rivas L: Temporins, small antimicrobial peptides with leishmanicidal activity. J Biol Chem 2005, 280:984–990.
Anjili C, Langat B, Ngumbi P, Mbati PA, Githure J, Tonui WK: Effects of anti- Leishmania monoclonal antibodies on the development of Leishmania major in Phlebotomus duboscqi (Diptera: Psychodidae). East Afr Med J 2006, 83:72–78.
Dutta A, Ghoshal A, Mandal D, Mondal NB, Banerjee S, Sahu NP, Mandal C: Racemoside a, an anti-leishmanial, water-soluble, natural steroidal saponin, induces programmed cell death in Leishmania donovani . J Med Microbiol 2007, 56:1196–1204.
Luque-Ortega JR, van’t Hof W, Veerman EC, Saugar JM, Rivas L: Human antimicrobial peptide histatin 5 is a cell penetrating peptide targeting mitochondrial ATP synthesis in Leishmania . FASEB J 2008, 22:1817–1828.
Pretzel J, Mohring F, Rahlfs S, Becker K: Antiparasitic peptides. Adv Biochem Eng Biotechnol 2013, 135:157–192.
Acknowledgements
We wish to thank all staffs of NIHR-IHRS for providing requirements in the field of Isfahan province. This research was supported financially by the Tehran University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NMR, MAO, AAA and YR designed the experiments in collaboration with the other authors. NMR, MHA, KA and RAFV collected sand flies. NMR, AAA, MHA and RJ dissected the midgut of the sand flies. DA, SH, BY, MHS helped in the insects microbiology part. NMR drafted the first version of this manuscript. MAO and RD completed final version of this manuscript. NMR completed the processing of the data, analyzed and interpreted the results, and revised and finalized the manuscript in consultation with the other authors. MAO and BY supervised the thesis and implementation of this study. All authors revised and approved the final version of this manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Maleki-Ravasan, N., Oshaghi, M.A., Afshar, D. et al. Aerobic bacterial flora of biotic and abiotic compartments of a hyperendemic Zoonotic Cutaneous Leishmaniasis (ZCL) focus. Parasites Vectors 8, 63 (2015). https://doi.org/10.1186/s13071-014-0517-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-014-0517-3