
Abstract
Background
Soil ascomycete fungi produce plant-biomass-degrading enzymes to facilitate nutrient and energy uptake in response to exogenous stress. This is controlled by a complex signal network, but the regulatory mechanisms are poorly understood. An essential Zn2Cys6 transcription factor (TF) PoxCxrA was identified to be required for cellulase and xylanase production in Penicillium oxalicum. The genome-wide regulon and DNA binding sequences of PoxCxrA were further identified through RNA-Sequencing, DNase I footprinting experiments and in vitro electrophoretic mobility shift assays. Moreover, a minimal DNA-binding domain in PoxCxrA was recognised.
Results
A PoxCxrA regulon of 1970 members was identified in P. oxalicum, and it was displayed that PoxCxrA regulated the expression of genes encoding major plant cell wall-degrading enzymes, as well as important cellodextrin and/or glucose transporters. Interestingly, PoxCxrA positively regulated the expression of a known important TF PoxClrB. DNase I footprinting experiments and in vitro electrophoretic mobility shift assays further revealed that PoxCxrA directly bound the promoter regions of PoxClrB and a cellobiohydrolase gene cbh1 (POX05587/Cel7A-2) at different nucleic acid sequences. Remarkably, PoxCxrA autoregulated its own PoxCxrA gene expression. Additionally, a minimal 42-amino-acid PoxCxrA DNA-binding domain was identified.
Conclusion
PoxCxrA could directly regulate the expression of cellulase genes and the regulatory gene PoxClrB via binding their promoters at different nucleic acid sequences. This work expands the diversity of DNA-binding motifs known to be recognised by Zn2Cys6 TFs, and demonstrates novel regulatory mechanisms of fungal cellulase gene expression.
Similar content being viewed by others


Background
Soil microorganisms that drive biogeochemical processes, including nutrient cycling, disease suppression and water dynamics, are critical to ecological balance and climate change, and highly sensitive to precipitation seasonality. In soils of tropical and subtropical forests that are hot spots of global carbon cycling, fungal strains from Ascomycota (66.3%) and Basidiomycota (31.3%) are particularly abundant [1], among which Penicillium and Trichoderma are the dominant decomposers, producing carbohydrate-active enzymes (CAZymes) that degrade plant cell walls into soluble sugars for nutrient availability [2, 3].
Cells sense and respond to extracellular stimuli such as starvation and stress, and these processes are controlled by complex signalling networks, resulting in multi-faceted physiological and morphological changes, including cell metabolism and growth. During starvation, cells attempt to reduce energy and carbon requirements, recycle structural components and undergo specialized processes such as sporulation, autophagy, apoptosis or necrosis [4]. Fungal cells control CAZyme production by utilising complex nutrient-sensing pathways comprising numerous sensors and receptors such as kinases, transcription factors (TFs) and their targets [5]. However, the overall process remains poorly understood.
Penicillium oxalicum produces integrated cellulolytic enzymes that degrade insoluble cellulose, and displays a preference for particular carbon sources. When P. oxalicum grows in the presence of glucose, expression of cellulolytic enzyme-encoding genes is repressed via carbon catabolite repression (CCR). Among the TFs involved in CCR, the core zinc finger TF CreA/Cre-1 is the one best studied in filamentous fungi, including P. oxalicum. CreA directly or indirectly represses the expression of all major CAZyme genes and their regulatory genes that are involved in the degradation of plant cell walls in the presence of glucose [6, 7].
When cellulose is present as a sole carbon source, induction of cellulolytic genes in P. oxalicum is dependent on a few essential TFs including the Zn2Cys6 TFs PoxCxrA and PoxClrB. Individual deletion of PoxCxrA and PoxClrB results in almost no cellulase production by P. oxalicum [2, 8]. Clr2, a homolog of PoxClrB in Neurospora crassa, binds to a DNA sequence identical to that bound to by the Saccharomyces cerevisiae TF Gal4p (CGGN11CCG) [9]. Although PoxCxrA binds directly to the promotor regions of major cellulase and xylanase genes [8], the DNA element recognised by PoxCxrA remains unknown.
In the present study, we employed high-throughput sequencing of transcripts (RNA-seq) to analyse transcriptional levels of genes in P. oxalicum deletion mutant ∆PoxCxrA following exposure to Avicel in comparison with the parental strain ∆PoxKu70 to identify the regulon of PoxCxrA. Moreover, we identified two different DNA motifs recognised by PoxCxrA, as well as the minimal DNA-binding domain of PoxCxrA via in vitro DNase I footprinting and electrophoretic mobility shift assay (EMSA) experiments.
Results
PoxCxrA positively regulates the expression of most genes encoding plant-cell-wall-degrading enzymes in P. oxalicum
In previous work, an essential TF PoxCxrA was found to be required for cellulase production in P. oxalicum, when subjected to cellulose as a carbon source [2, 8]. However, its regulon is not yet fully understood. To comprehensively explore the regulatory roles of PoxCxrA in P. oxalicum, RNA-Seq was employed to analyse the transcriptomes of the P. oxalicum mutant strain ∆PoxCxrA and the parental strain ∆PoxKu70 cultured in medium containing Avicel as the sole carbon source for 24 h after a shift from glucose. A total of 21–23 million clean reads with a length of 100 bp were generated across all samples (Additional file 1: Table S1), > 90% of which were successfully mapped into the genome of P. oxalicum wild-type strain HP7-1 [2]. To evaluate the correlations among the three biological replicates for each strain, the Pearson’s correlation coefficient (r) was calculated. The resulting high r value (> 0.85; Additional file 2: Figure S1) suggests that the transcriptomic data were reliable and suitable for further analysis.
Comparative transcriptomic profiling identified 1970 DEGs in deletion mutant ∆PoxCxrA, compared with the parental strain ∆PoxKu70, according to the |log2 fold change| ≥ 1 and p value ≤ 0.05 thresholds (Additional file 3: Table S2), which were defined as the PoxCxrA regulon. The PoxCxrA regulon included 1010 genes down-regulated compared with ∆PoxKu70. Functional annotation based on Eukaryotic Orthologous Group (KOG) classification revealed that most of these DEGs were involved in primary and secondary metabolism (category E, amino acid transport and metabolism; category Q, secondary metabolite biosynthesis, transport and catabolism), and fell into the general function prediction only category (category R) (Fig. 1).

KOG annotation of DEGs in the PoxCxrA regulon. DEGs were selected as a |log2 fold change| ≥ 1 and a p value ≤ 0.05 as thresholds. DEGs differentially expressed genes, KOG Eukaryotic Orthologous Group
Expression of genes encoding three key module signal carriers (i.e. sugar transporters), TFs, and functional proteins (i.e. CAZymes) formed a complex signalling network, and this was investigated in detail. Among the 1970 DEGs, annotation revealed that 154 DEGs encoded CAZymes, including seven auxiliary activity families, six carbohydrate-binding module families, eight carbohydrate esterase families, 42 glycoside hydrolase families and 14 glycosyltransferase families, as well as two polysaccharide lyase families. Among them, 46 genes encoding putative plant cell wall-degrading enzymes (CWDEs) were detected. Importantly, most key cellulase and xylanase genes in P. oxalicum were included in this DEG set, such as three cellobiohydrolase (CBH) genes POX05587/Cel7A-2 (also called cbh1), POX02490/Cel7A-1 and POX04786/Cel6A, three endo-β-1,4-glucanase (EG) genes POX06147/Cel5A, POX02740 and POX06983, four β-glucosidase (BGL) genes POX00968, POX03062, POX07963 and POX08882, five xylanase (Xyn) genes POX00063/xyn10A, POX06783/xyn11A, POX06601, POX08484/Xyn11B and POX08990, two lytic polysaccharide monooxygenase POX02308/aa9A and POX08897 (Additional file 3: Table S2), which accounted for approximately 70% of cellulase and xylanase genes in the whole genome of P. oxalicum wild-type strain HP7-1 (Fig. 2).

Regulation of genes encoding carbohydrate-active enzymes and putative transcription factors by PoxCxrA. Histogram indicates relative transcription levels of DEGs in P. oxalicum mutant ∆PoxCxrA exposed to Avicel compared with the parental strain ∆PoxKu70. CWDEs plant cell walls-degrading enzymes, CAZymes carbohydrate-active enzymes
Of these 154 CAZyme-encoding genes, 95 were down-regulated (− 7.0 < log2 fold change < − 1.0) compared with the parental strain ∆PoxKu70. Interestingly, expression of all 17 cellulase and xylanase genes described above were down-regulated in the ∆PoxCxrA, except for two genes (POX00968 and POX02490/Cel7A-1) (Fig. 2).
PoxCxrA regulates the expression of genes encoding cellodextrin and glucose transporters
In complex signal transduction pathways, transporters/sensors play important roles, as demonstrated for cellodextrin transporters PoxCdtC and PoxCdtD in cellulase production in P. oxalicum [10]. Among the 1970 DEGs, 34 were annotated as sugar/inositol transporters (IPR003663) and/or major facilitators, or sugar transporter-like (IPR005828). Remarkably, two known genes (POX06051/PoxCdtC and POX05915/PoxCdtD) encoding cellodextrin transporters PoxCdtC and PoxCdtD, three genes (POX07576, POX07227 and POX08783) encoding N. crassa glucose transporter RCO3-like [11], and GLT1 and HGT-2 proteins [12], sharing 51–76% sequence identity, were included.
The regulon of PoxCxrA included 23 down-regulated genes encoding cellodextrin and glucose transporters (− 5.4 < log2 fold change < − 1.2). Among them, transcriptional levels of POX06051/PoxCdtC, POX05916/PoxCdtD and POX07576 were down-regulated in the ∆PoxCxrA compared with ∆PoxKu70, with a log2 fold change from − 4.4 to − 1.3 (Fig. 3 and Additional file 3: Table S2). In contrast, POX07227 and POX08783 transcript levels increased 4.5–3.1-fold in ∆PoxCxrA (Fig. 3).

Regulation of putative transporter genes by PoxCxrA. Values in heatmaps were calculated by log2 (Gene_FPKM in ∆PoxCxrA/Gene_FPKM in ∆PoxKu70). FPKM fragments per kilobase of exon per million fragments mapped
PoxCxrA regulates the expression of genes encoding TFs controlling cellulase gene expression
Genes encoding putative TFs among the 1970 DEGs were explored, and 88 candidates were identified, mostly encoding zinc finger proteins such as Zn2Cys6, C2H2 and CCHC families. Among the 88 TFs, 14 were known to regulate cellulase production in filamentous fungi, including eight activators (POX00331/FlbC, POX01184, POX01960/PoxClrB, POX02484, POX04420/PoxCxrB, POX05726, POX08415/NsdD and POX08910) and six repressors (POX00864, POX04860/PDE_07199, POX06534/BrlA, POX06759, POX07254/CreA and POX08375/Ace1) [2, 8, 13,14,15,16,17]. Comparative transcriptomics indicated that 45 down-regulated genes encoding putative TFs (− 9.6 < log2 fold change < − 1.0). Interestingly, deletion of PoxCxrA resulted in down-regulation of 11 known regulatory genes (POX00331/FlbC, POX01184, POX01960/PoxClrB, POX02484, POX04420/PoxCxrB, POX04860/PDE_07199, POX05726, POX06534/BrlA, POX06759, POX07254/CreA and POX08415/NsdD; − 6.8 < log2 fold change < − 1.1), and up-regulation of known regulatory genes POX08910, POX00864 and POX08375/Ace1 (1.1 < log2 fold change < 1.3), compared with the ∆PoxKu70 transcriptome (Fig. 2 and Additional file 3: Table S2).
PoxCxrA and PoxClrB dynamically regulates the expression of one another
Interestingly, PoxCxrA regulated the expression of a key regulatory gene PoxClrB through RNA-Seq. To further elucidate this regulation, RT-qPCR was employed. When ΔPoxCxrA was exposed to Avicel, transcription of PoxClrB was down-regulated to some extent (2.7- to 8.2-fold) after 4 h (p ≤ 0.05, Student’s t test) compared with that in ∆PoxKu70. In contrast, PoxCxrA expression increased by 1.5- to 2.1-fold in ∆PoxClrB during the latter stages of cultivation (24–48 h after induction; Fig. 4a).

Expression of PoxCxrA, PoxClrB and major cellulase genes in P. oxalicum, and their regulation. a Expression of PoxCxrA and PoxClrB in the mutant strain ∆PoxClrB and ∆PoxCxrA subjected to Avicel as the sole carbon source, respectively. b Transcriptional levels of genes PoxCxrA and PoxClrB in ∆PoxKu70 in the presence of different carbon sources. Gene expression levels were investigated in P. oxalicum strains cultivated on Avicel at four different time points (4, 12, 24 and 48 h) after a shift from glucose by real-time quantitative reverse-transcription PCR. Relative expression on the y-axis indicates differences in values for transcription between tested and reference genes, or between tested genes in deletion mutants and the parental strain ΔPoxKu70. *p ≤ 0.05 and **p ≤ 0.01 indicate differences among samples assessed by Student’s t-tests. All experiments were carried out with at least three independent replicates
Expression of both PoxCxrA and PoxClrB is induced by cellulose
When P. oxalicum strain ∆PoxKu70 was transferred into medium containing Avicel (induced state), PoxCxrA and PoxClrB exhibited similar transcriptional levels to those without a carbon source (de-repressed state) during the early induction stage (0–12 h), but only the transcriptional level of PoxClrB was higher than that without a carbon source during later stage (48 h). The expression of both PoxCxrA and PoxClrB on Avicel were higher than that on glucose (repressed state). Surprisingly, PoxCxrA expression on Avicel increased by ~ 70% compared with its expression without a carbon source, but only at 24 h. Expression level of PoxClrB in ∆PoxKu70 was higher than those of PoxCxrA during all states (induced, repressed and de-repressed). PoxCxrA expression under Avicel induction increased before 24 h, but reduced after 24 h (Fig. 4b).
PoxCxrA directly binds to the promoter regions of PoxClrB and PoxCxrA, and genes encoding cellodextrin and glucose transporters
To further confirm whether PoxCxrA directly or indirectly regulates PoxClrB expression, in vitro EMSA experiments were employed. The putative DNA-binding domain PoxCxrA17–150 was recombinantly expressed and purified by fusing with thioredoxin (Trx), His and S-tags. A 300-bp DNA fragment from the promoter region of PoxClrB tagged with 6-carboxyfluorescein (6-FAM) was amplified using specific primers (Additional file 4: Table S3) and used as the probe for EMSA experiments. A DNA fragment from the promoter region of the β-tubulin gene POX05989 was used as a control. The results revealed that a complex was formed between PoxCxrA17–150 [8] and the promoter region of PoxClrB, and its concentration increased with an increasing amount of fusion protein (1.0–2.0 µg). Bovine serum albumin (BSA) and Trx–His–S did not interact with the PoxClrB probe, and there was no interaction between PoxCxrA17–150 and the control POX05989 promoter region. Competitive EMSA experiments revealed that the concentration of the complex decreased gradually with an increasing amount of protein-binding DNA fragment without 6-FAM (Fig. 5), suggesting that PoxCxrA17–150 specifically bound the promoter region of PoxClrB.

Electrophoretic mobility shift assay (EMSA) analysis of the binding of PoxCxrA17–150 and PoxClrB1–120 to the promoter regions of PoxCxrA, PoxCxrB and POX05587/Cel7A-2
Each EMSA experiment comprised different DNA-binding domains (PoxCxrA17–150, PoxClrB1–120 or both; 0–2 µg) and 50 ng of the target probe labelled with 6-FAM. Probes without 6-FAM, and the β-tubulin gene POX05989, were used as competitive probes and negative controls, respectively. The fusion protein Trx–His–S purified from Escherichia coli cells harbouring the empty plasmid pET32a (+), and BSA alone, were used as protein controls.
The RT-qPCR data described above revealed that PoxClrB negatively regulated PoxCxrA expression during the latter stages of induction, but the regulatory mode was unclear. In vitro binding experiments showed that the putative DNA-binding domain of PoxClrB1–120 was unable to bind to the promoter region of the PoxCxrA gene (Fig. 5), indicating that PoxClrB indirectly regulates transcription of PoxCxrA in P. oxalicum. EMSA experiments also showed that PoxCxrA17–150 bound to the promoter region of its own gene, but PoxClrB1–120 did not, suggesting the autoregulation of PoxCxrA. When a mixture of PoxCxrA17–150 and PoxClrB1–120 was treated with probe POX05587/Cel7A-2, a band representing a larger protein–DNA complex than that formed by either PoxCxrA17–150 or PoxClrB1–120 individually was observed (Fig. 5), indicating that the binding motifs recognised by PoxCxrA17–150 and PoxClrB1–120 are distinct.
Intriguingly, genes encoding cellodextrin and glucose transporters were included in the PoxCxrA regulon. EMSA experiments revealed that PoxCxrA17–150 could bind to the promoter regions of genes encoding cellodextrin transporters PoxCdtC and PoxCdtD, and putative glucose transporters POX07576/RCO-3, POX08783/HGT-2, POX07209 and POX04369 (Fig. 6).

EMSA analysis of the interactions between the DNA-binding domain PoxCxrA17–150 and the promoter regions of genes encoding putative sugar transporters. Each EMSA experiment comprised PoxCxrA17–150 (0–2 µg) and 50 ng of target probes labelled with 6-carboxyfluorescein (6-FAM). Probes without 6-FAM, and the promoter of β-tubulin gene POX05989, were used as competitive probes and negative control, respectively. The fusion protein Trx–His–S purified from E. coli cells harbouring the empty plasmid pET32a (+), and BSA alone, were used as protein controls
Different PoxCxrA-binding motifs are present in the promoter regions of target genes POX05587/Cel7A-2 and PoxClrB
Based on the above results and those of previous work [8], PoxCxrA appears to regulate the expression of cbh gene POX05587/Cel7A-2 and TF gene PoxClrB by directly binding to their promoters. In vitro EMSA and DNase I footprinting experiments were, therefore, performed to identify protein-binding motifs (PBMs) in the promoters of the target genes. An initial DNase I footprinting experiment was performed using a 100-bp DNA fragment corresponding to the upstream flanking sequence (starting from the transcription initiation ATG codon) labelled with 6-FAM at the 3′-terminus to identify the PBM of PoxCxrA17–150 in the promoter region of POX05587/Cel7A-2. The results revealed that two putative PBMs (PBM1, 5′-ATCAGATCCTCAAAGA-3′; PBM2, 5′-TCATCTCCTCCACC-3′) were protected by PoxCxrA17–150 to various degrees (Fig. 7a). Further EMSA experiments confirmed that PoxCxrA17–150 bound to probes containing PBM1 or PBM1 plus PBM2, but not PBM2 (Fig. 7b), suggesting that PBM1 contains the binding motif of PoxCxrA in the POX05587/Cel7A-2 promoter region (PBM_POX05587).

The PoxCxrA17–150-binding sequence in the promoter region of cellobiohydrolase gene POX05587/Cel7A-2. a DNase I footprinting experiment results. b EMSA confirmation of PBM_POX05587. c Schematic diagram of PBM_POX05587 truncation. d Key nucleotides in PBM_POX05587 confirmed via EMSA experiments. Each EMSA experiment comprised 2 µg of PoxCxrA17–150 and 50 ng of truncated PBM_POX05587 as probe labelled with 6-carboxyfluorescein. The promoter of β-tubulin gene POX05989 was used as a probe control. The fusion protein Trx–His–S purified from E. coli cells harbouring the empty plasmid pET32a (+), and BSA alone, were used as protein controls. ‘−’ and ‘+’ represent EMSA experiments without and with DBD PoxCxrA17–150, respectively
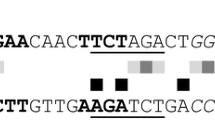
Meanwhile, a series of PoxClrB probes with different lengths (PoxClrB–375, PoxClrB–355, PoxClrB–330, PoxClrB–310, PoxClrB–295, PoxClrB–284 and PoxClrB–270 labelled with 6-FAM at the 5′-terminus) were amplified using specific primer pairs (Additional file 4: Table S3; Fig. 8a). EMSA experimental results revealed protein–DNA complexes between PoxCxrA17–150 and PoxClrB–375, PoxClrB–355, PoxClrB–330, PoxClrB–310 and PoxClrB–295, but not with the other probes or negative controls (Fig. 8b), suggesting that PoxCxrA binds the PBM_PoxCrlB in the promoter region of PoxClrB from positions 285–295 (5′-GCTGAGTCCTT-3′) (Fig. 8c). Compared with PBM_POX05587, three conserved sites were identified (i.e. GCTGAGTCCTT or ATCAGATCCTCAAAGA).

The PoxCxrA17–150-binding sequence in the promoter region of transcription factor gene PoxClrB. a Schematic diagram of putative PBM_PoxClrB truncation. b The PBM_PoxClrB sequence determined by in vitro EMSA. c EMSA analysis of key nucleotides in PBM_PoxClrB. Each EMSA experiments comprised 0–2 µg of PoxCxrA17–150 and 50 ng of truncated PBM_PoxClrB as probe labelled with 6-carboxyfluorescein. The promoter of β-tubulin gene POX05989 was used as a probe control. The fusion protein Trx–His–S purified from E. coli cells harbouring the empty plasmid pET32a (+), and BSA alone, were used as protein controls
Subsequently, to characterise the key nucleotides in PBM_PoxClrB and PBM_POX05587, we generated a series of truncated PBM_PoxClrB and PBM_POX05587 fragments as EMSA probes (Figs. 7c, 8c). The results revealed that protein–DNA complexes were formed between PoxCxrA17–150 and PBM_POX05587–3, PBM_POX05587–6, PBM_POX05587–3′, PBM_POX05587–6′, PBM_PoxClrB-3′, PBM_PoxClrB_6′ and PBM_PoxClrB_9′, and the concentrations of binding bands gradually decreased with the truncation of probe length (Figs. 7d, 8d). These results showed that the core nucleotides in PBM_POX05587 and PBM_PoxClrB were TCCT and GC, respectively, whereas their flanking sequences were also required for PoxCxrA binding.
Identification of a minimal DNA-binding domain of PoxCxrA
The putative DNA-binding domain (DBD) of PoxCxrA used above (residues 17–150) was serially truncated from the C-terminus to identify a minimal functional DBD. To facilitate the purification of the expressed recombinant protein in E. coli, we first fixed at 11th amino acid of N-terminus. Recombinant proteins PoxCxrA11–150, PoxCxrA11–114, PoxCxrA11–87, PoxCxrA11–58 and PoxCxrA11–31 were produced in E. coli cells and purified (Fig. 9a, b), and in vitro EMSA experiments were carried out to investigate their ability to bind a 6-FAM-labelled 300-bp DNA fragment of the POX05587/Cel7A-2 promoter region comprising the PoxCxrA binding site as EMSA probe. The results showed that each truncated protein bound the probe to form a protein–DNA complex that retarded electrophoretic mobility in gels, except for PoxCxrA11–31. The concentration of the protein–DNA complexes gradually increased with increasing protein loading (1.0–3.0 µg). Competitive EMSA experiments were simultaneously performed, and the results indicated that the amount of complex tended to reduce with increasing amounts of competitive probe without 6-FAM. BSA and Trx–His–S control proteins did not bind the POX05587/Cel7A-2 promoter region (Fig. 9b). PoxCxrA11–58 was confirmed to bind to PMB_POX05587 via in vitro ESMA experiments (Fig. 9c). Subsequently, the PoxCxrA17–58 was also expressed and purified. EMSA binding experiments indicated a clear band comprised of PoxCxrA17–58 and PMB_POX05587 on the gel (Fig. 9d). Thus, the minimal 42-amino-acid segment of PoxCxrA17–58 suffices for DNA binding.

The DNA-binding domain (DBD) of PoxCxrA. a Schematic diagram of the PoxCxrA protein containing a Zn2Cys6 domain, and DBD truncation. b The minimal DBD11–58 determined via in vitro EMSA. c Interaction between PoxCxrA11–58 and PBM_POX05587. d Interaction between PoxCxrA17–58 and PBM_POX05587. Each EMSA experiment comprised 2 µg of the truncated PoxCxrA-binding domain and 50 ng of PBM_POX05587 labelled with 6-carboxyfluorescein. The promoter of β-tubulin gene POX05989 and PBM2 were used as control probes. The fusion protein Trx–His–S purified from E. coli cells harbouring the empty plasmid pET32a (+), and BSA alone, were used as protein controls. ‘−’ and ‘+’ represent EMSA without and with DBD of PoxCxrA11–58. ‘*’indicated the mutated amino acids into alanine in the mutated DBD of PoxCxrA11–58. e Alignment of DBD17–58 with known Zn2Cys6-containing transcription factors; PoxCxrA and PoxClrB are from P. oxalicum strain HP7-1 (accession numbers KY368172 and KU597415); Pho7, XlnR, Clr1 and Gal4 are from Schizosaccharomyces pombe, P. oxalicum strain 114-2 (EPS32714), N. crassa strain OR74A (XP_011394265) and Saccharomyces cerevisiae S288C (NP_015076), respectively. f Interaction between the mutated PoxCxrA17–58 and PBM_POX05587
Subsequently, an alignment analysis of PoxCxrA17–58 with the DBDs in known Zn2Cys6-containing TFs including PoxClrB, Pho7, XlnR, Clr1 and Gal4 that came from P. oxalicum HP7-1 [2], Schizosaccharomyces pombe [18], P. oxalicum strain 114-2 [7], N. crassa strain OR74A [19] and Saccharomyces cerevisiae S288C [9], was respectively performed. The results found three pairs of highly conserved zinc-coordinating cysteines that are essential for protein binding [18] and relatively conserved flanking amino acids such as arginine (R), lysine (K), aspartic acid (D) and proline (P; Fig. 9e). The retained amino acids showed high diversity. Several relatively conserved amino acids (18th R, 19th R, 27th Q, 30th K, 32th K and 38th P) were respectively replaced by alanine to generate a mutated PoxCxrA17–58. In vitro EMSA further displayed that all the mutated PoxCxrA17–58 lost the ability to bind the PM_POX05587 (Fig. 9f).
Discussion
PoxCxrA is known to be a critical transcriptional activator in P. oxalicum exposed to cellulose as a carbon source, but its regulatory mechanism is unclear. Herein, we found that PoxCxrA regulates cellulase production in P. oxalicum by controlling the expression of PoxClrB, and further elucidated their regulatory network. PoxCxrA autoregulated the transcription of its own PoxCxrA gene, but PoxClrB did not. Moreover, the DNA-binding domain of PoxCxrA17–150 bound to the promoter regions of PoxClrB and POX05587/Cel7A-2 at different binding sites (5′-ATCAGATCCTCAAAGA-3′ and 5′-GCTGAGTCCTT-3′, respectively) according to in vitro DNase I footprinting and EMSA experiments. A minimal functional DBD (residues 17–58) of PoxCxrA was identified. These findings provide novel insights into the regulatory mechanism governing cellulase gene expression in P. oxalicum.
The PoxCxrA regulon was identified, which included a number of members involved in primary and secondary metabolism. To withstand starvation caused by insoluble cellulose as the sole carbon source, expression of major cellulase genes in P. oxalicum, including genes encoding CBH1 (GH7) and EGs (GH5 and GH12), was rapidly up-regulated, resulting in abundant intra- and extracellular cellodextrin production in fungal cells [20,21,22]. Accumulated intracellular cellodextrin triggered signalling cascades that activated the expression of PoxCxrA and PoxClrB, subsequently resulting in activation of the expression of cellulase genes such as POX05587/Cel7A-2.
Intriguingly, PoxCxrA also directly activated PoxClrB expression, whereas PoxClrB indirectly repressed the transcription of PoxCxrA in P. oxalicum. Regrettably, exactly how PoxCxrA expression is repressed by PoxClrB remains unknown (Fig. 10). In the early phase of P. oxalicum exposed to Avicel, both PoxCxrA and PoxClrB were transcribed at a background level. Expression of PoxCxrA was first up-regulated at the middle of the culture period, and then reduced in the latter stages due to autoinhibition or repression by PoxClrB (Fig. 10). In contrast, PoxClrB expression gradually increased in the middle and later stages.

Postulated modules of the PoxCxrA and PoxClrB networks regulating cellulase gene expression in P. oxalicum. Lines with arrows indicate positive regulation, lines with flat ends indicate negative regulation, and dashed lines with flat ends represent unknown repressive expression. I–IV indicates predicted regulatory process over time
Moreover, PoxCxrA up-regulated the expression of genes involved in cellodextrin transportation, such as POX06051/PoxCdtC and POX05915/PoxCdtD [10] and retarded the expression of genes involved in glucose transportation such as POX07576/GLT-1 and POX08783/HGT-2 [12] that caused CCR at high concentration. PoxCxrA stimulated cellulase gene expression via two pathways; direct binding, and through key TF mediators such as PoxClrB (Fig. 10), PoxCxrB and/or PoxNsdD [8].
Differences in regulatory networks occurred in different host cells. In N. crassa, cellobiose-activated CLR1 was necessary for the increased expression of clr2, a homolog of PoxClrB. CLR1 also induced most cellulase genes, thereby positively affecting enzyme production [19]. In the present study, we found that PoxCxrA was required for PoxClrB expression, but not POX03837, which encodes a homolog of CLR1 in P. oxalicum. Knockout of POX03837 had no effect on cellulase production in P. oxalicum HP7-1 cultured on Avicel (data not shown).
The PoxCxrA DBD resembles those of Gal4-like TFs (e.g. Gal4 in S. cerevisiae, CLR1 and XlnR in N. crassa, and PoxClrB). PoxCxrA17–58 was found to be the minimal functional DBD, and it comprises three pairs of zinc-coordinating cysteines and several conserved amino acids that are essential for protein binding [18]. The amino acids flanking these cysteines display high diversity, which might determine the binding motifs. In the literature, many binding motifs of Gal4-like proteins have been characterised, including CGGN5CGGNCCG (CLR1), CGGN11CCG (Gal4 or CLR2), GGNTAAA (XlnR) [9], TCG(G/C)(A/T)NNTTNAA (Pho7) [18] and 5′-ATCAGATCCTCAAAGA-3′ or 5′-GCTGAGTCCTT-3′ determined in this study. This suggests that the amino acids flanking the cysteines are also required for binding.
Moreover, screening PoxCxrA-binding sequences in other target genes confirmed by in vitro EMSA experiments using PBM_POX05587 and PBM_PoxClrB revealed that the binding sequences were highly diverse, which supports the enormous regulon of PoxCxrA that includes 1970 members, accounting for almost a quarter of the entire P. oxalicum strain HP7-1 genome. However, identification and analysis of all PoxCxrA-binding sequences in the genome requires further study.
In summary, the present study determined regulon for the essential TF PoxCxrA that is required for cellulase production of P. oxalicum. PoxCxrA regulates cellulase gene expression via two mechanisms: regulating key TF mediator PoxClrB, and directly binding cellulase genes with diverse binding motifs. This work provides novel insights into the regulatory mechanisms of fungal cellulase gene expression.
Methods
Strains and culture conditions
Penicillium oxalicum mutant strains ΔPoxCxrA (no. 12965) and ΔPoxClrB (no. 3.15649), and the parental strain ΔPoxKu70 (no. 3.15650) were obtained from the China General Microbiological Culture Collection (CGMCC) [8]. Spores were collected after maintaining strains on potato dextrose agar (PDA) plates at 28 °C for 6 days with buffer containing 0.1% Tween-80. In general, an aliquot of conidial suspension (1 × 108 conidia per milliliter) was inoculated into 100 mL of modified culture medium (MMM; pH 5.5) containing (g/L) (NH4)2SO4 (4.0), KH2PO4 (4.0), CaCl2 (0.6), MgSO4·7H2O (0.60), FeSO4·7H2O (0.005), MnSO4 (0.0016), ZnCl2 (0.0017), CoCl2 (0.002), and 1 mL of Tween-80, containing either 1.0% glucose or 2.0% Avicel (Sigma-Aldrich, St. Louis, MO, USA) as the sole carbon source. Mixtures were incubated on an orbital shaker at 28 °C at 180 rpm for 6 days.
For transfer cultivation, P. oxalicum strains were first pre-cultured for 24 h on MMM supplemented with 1.0% glucose as the carbon source at 28 °C with shaking at 180 rpm. Mycelia of pre-cultured P. oxalicum strains were transferred into MMM containing 2.0% Avicel and cultured for 4–24 h at 28 °C with shaking at 180 rpm. Mycelia were harvested and total RNA was extracted for RNA-Seq and real-time quantitative reverse transcription-PCR (RT-qPCR) assays.
RNA extraction
For total RNA extraction, mycelia harvested from three replicate independent cultures at each time point were frozen, ground under liquid nitrogen using a pestle and mortar, and RNA was purified using a TRIzol RNA Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA concentration and quality were determined from the ratio of the absorbance at 260 nm/280 nm measured using a Nanodrop ND-2000 spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA).
RNA-Seq
Total RNA was sequenced as described previously by Zhao et al. [2], and a cDNA library for each total RNA sample was constructed and assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). After confirming eligibility, cDNA libraries were sequenced using an Illumina HiSeq 4000 system. After quality control, clean reads were mapped onto the genome of the wild-type P. oxalicum HP7-1 strain to assess sequence homology and functional annotation using BWA v0.7.10-r789 [23] and Bowtie2 v2.1.0 [24]. RSEM v1.2.12 software [25] was used to analyse gene expression levels using the fragments per kilobase of exon per million mapped reads (FPKM) method. Differentially expressed genes (DEGs) were screened using the DESeq tool [26] with |log2 fold change| ≥ 1 and p value ≤ 0.05 as thresholds. The reliability of RNA-Seq data was assessed by Pearson’s correlation coefficients for three biological replicates for each sample. BLAST v2.2.26 (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for sequence homology and functional annotation analyses. DEGs detected by comparative assays were functionally analysed based on Eukaryotic Orthologous Group (KOG) annotation to the P. oxalicum HP7-1 genome [2].
Expression and purification of truncated PoxCxrA constructs
Recombinant expression in Escherichia coli and protein purification were carried out as described previously by Yan et al. [8]. DNA sequences encoding a series of truncated PoxCxrA constructs (PoxCxrA11–150, PoxCxrA11–114, PoxCxrA11–87, PoxCxrA11–58, PoxCxrA17–58, PoxCxrA11–50 and PoxCxrA11–31) were amplified by PCR with specific primer pairs (Additional file 4: Table S3) and digested using appropriate restriction endonucleases. Digested DNA fragments were inserted into the expression vector pET32a (+) digested with the corresponding restriction endonucleases to generate recombinant plasmids that were subsequently introduced into competent E. coli cells by chemical recombination. After confirmation, cells harbouring constructs were pre-cultured for 12 h at 37 °C, then induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside, with culturing being continued for 20 h at 28 °C to produce fusion proteins possessing thioredoxin (TRX), His and S tags. Fusion proteins were purified by affinity chromatography on TALON Metal Affinity Resin (Clontech, Palo Alto, CA, USA). Cells harbouring empty pET32a (+) vector were cultured as described above and the resulting Trx–His–S protein was used as a negative control.
In vitro EMSA assays
In vitro EMSA experiments were performed as described previously by He et al. [17]. Briefly, DNA fragments of different lengths harbouring the putative promoter regions of PoxClrB and POX05578/Cel7A-2, labelled with 6-carboxyfluorescein (6-FAM) at the 3′ terminus, were amplified by PCR using specific primer pairs (Additional file 4: Table S3), and used as probes for in vitro EMSA experiments. Meanwhile, the same DNA fragments without 6-FAM and a 300-bp DNA fragment from the promoter region of the β-tubulin gene POX05989 served as competitive and negative probes, respectively.
For EMSA experiments, mixtures containing ~ 50 ng EMSA probes and various amounts (0–2.0 µg) of fusion proteins in binding buffer consisting of 20 mM TRIS–HCl (pH 8.0), 0.1 mg/mL bovine serum albumin (BSA), 5% (v/v) glycerol, 50 mM KCl, 1 mM dithiothreitol and 1.0 µg sheared salmon sperm DNA) were cultured for 30 min at 30 °C, then separated by 4% polyacrylamide-TRIS–acetic acid-ethylene diamine tetraacetic acid (EDTA) gel electrophoresis. Gels were observed using a Bio-Rad ChemiDoc MP imaging system (BIO-RAD, Hercules, CA, USA) at an excitation wavelength range of 489–506 nm. Competitive binding experiments were performed as described above, except that probes were replaced with competitive probes.
DNase I footprinting
DNase I footprinting experiments were carried out as reported by Wang et al. [27] with minor modifications. For each assay, 350 ng of each probe (100-bp DNA fragments corresponding to the region upstream from the start codon ATG of POX05587/Cel7A-2) were separately incubated with 6.6 µg of recombinant PoxCxrA11–150 protein for 20 min at 30 °C. Subsequently, 0.015 U DNase I (Promega, Beijing, China) and 100 nM CaCl2 were added and the reaction was incubated for 1 min at 30 °C. DNase I stop solution, containing 200 mM unbuffered sodium acetate, 30 mM EDTA and 0.15% sodium dodecyl sulphate (SDS), was used to stop the enzymatic reaction, and DNA was extracted and sequenced.
RT-qPCR assays
RT-qPCR assays were carried out according to a previously reported method [8]. Briefly, total RNA was extracted from P. oxalicum deletion mutant ∆PoxCxrA grown on Avicel, and from the parental strain ∆PoxKu70 that served as a control. First-strand cDNA was synthesized from the extracted RNA as template using a PrimeScript RT Reagent Kit (TaKaRa Bio Inc., Dalian, China) according to the manufacturer’s instructions. Each target gene was subjected to PCR amplification using the first-strand cDNA as template in a 20 µL reaction mixture containing 10 µM of each primer (0.8 µL; Additional file 4: Table S3), first-strand cDNA (0.2 µL) and SYBR Premix Ex Taq II (10 µL; TaKaRa Bio Inc.). Thermal cycling included 35 cycles at 95 °C for 3 s and 60 °C for 30 s. Fluorescent signals were measured at the end of each extension step at 80 °C. Transcriptional levels of each gene in deletion mutants ∆PoxCxrA and ∆PoxClrB were calculated relative to that of the control gene POX09428 encoding actin, and relative expression levels were normalised against levels in the ΔPoxKu70 strain. All RT-qPCR assays were performed independently in at least three replicates.
Network construction
Network was constructed using Cytoscape v.3.6.1 software [28].
Statistical analysis
Experimental data were statistically analysed by Student’s t tests using Microsoft Excel within Office 2016 (Microsoft, Redmond, WA, USA).
Accession number
Transcriptomic data have been deposited in the Sequence Read Archive database under Accession Numbers SRR8377263–SRR8377265 for the ∆PoxCxrA and SRR8377258, SRR8377259 and SRR8377266.
Abbreviations
- 6-FAM:
-
6-carboxyfluorescein
- BSA:
-
bovine serum albumin
- BGL:
-
β-glucosidase
- CAZymes:
-
carbohydrate-active enzymes
- CBH:
-
cellobiohydrolase
- CCR:
-
carbon catabolite repression
- CMCase:
-
carboxymethylcellulase
- CWDEs:
-
plant cell wall-degrading enzymes
- DBD:
-
DNA-binding domain
- DEGs:
-
differentially expressed genes
- EG:
-
endo-β-1,4-glucanase
- EMSA:
-
electrophoretic mobility shift assay
- FPase:
-
filter paper cellulase
- FPKM:
-
fragments per kilobase of exon per million fragment mapped
- MMM:
-
modified culture medium
- PBMs:
-
protein-binding motifs
- PDA:
-
potato dextrose agar
- pNPCase:
-
p-nitrophenyl-β-cellobiosidase
- pNPGase:
-
p-nitrophenyl-β-glucopyranosidase
- RT-qPCR:
-
real-time quantitative reverse-transcription PCR
- TFs:
-
transcriptional factors
- Xyn:
-
xylanase
References
He D, Shen WJ, Eberwein J, Zhao Q, Ren LJ, Wu QLL. Diversity and co-occurrence network of soil fungi are more responsive than those of bacteria to shifts in precipitation seasonality in a subtropical forest. Soil Biol Biochem. 2017;115:499–510.
Zhao S, Yan YS, He QP, Yang L, Yin X, Li CX, et al. Comparative genomic, transcriptomic and secretomic profiling of Penicillium oxalicum HP7-1 and its cellulase and xylanase hyper-producing mutant EU2106, and identification of two novel regulatory genes of cellulase and xylanase gene expression. Biotechnol Biofuels. 2016;9:203.
Zhang Z, Liu JL, Lan JY, Duan CJ, Ma QS, Feng JX. Predominance of Trichoderma and Penicillium in cellulolytic aerobic filamentous fungi from subtropical and tropical forests in China, and their use in finding highly efficient β-glucosidase. Biotechnol Biofuels. 2014;7:107.
Caro-Maldonado A, Muñoz-Pinedo C. Dying for something to eat: how cells respond to starvation. Open Cell Signal J. 2011;3:42–51.
Huberman LB, Coradetti ST, Glass NL. Network of nutrient-sensing pathways and a conserved kinase cascade integrate osmolarity and carbon sensing in Neurospora crassa. Proc Natl Acad Sci USA. 2017;114:E8665–74.
Huberman LB, Liu J, Qin LN, Glass NL. Regulation of the lignocellulolytic response in filamentous fungi. Fungal Biol Rev. 2016;30:101–11.
Li ZH, Yao GS, Wu RM, Gao LW, Kan QB, Liu M, et al. Synergistic and dose-controlled regulation of cellulase gene expression in Penicillium oxalicum. PLoS Genet. 2015;11:e1005509.
Yan YS, Zhao S, Liao LS, He QP, Xiong YR, Wang L, et al. Transcriptomic profiling and genetic analyses reveal novel key regulators of cellulase and xylanase gene expression in Penicillium oxalicum. Biotechnol Biofuels. 2017;10:279.
Craig JP, Coradetti ST, Starr TL, Glass NL. Direct target network of the Neurospora crassa plant cell wall deconstruction regulators CLR-1, CLR-2, and XLR-1. mBio. 2015;6:e01452.
Li J, Liu G, Chen M, Li Z, Qin Y, Qu Y. Cellodextrin transporters play important roles in cellulase induction in the cellulolytic fungus Penicillium oxalicum. Appl Microbiol Biotechnol. 2013;97:10479–88.
Madi L, McBride SA, Bailey LA, Ebbole DJ. rco-3, a gene involved in glucose transport and conidiation in Neurospora crassa. Genetics. 1997;146:499–508.
Wang B, Li JG, Gao JF, Cai PL, Han XY, Tian CG. Identification and characterization of the glucose dual-affinity transport system in Neurospora crassa: pleiotropic roles in nutrient transport, signalling, and carbon catabolite repression. Biotechnol Biofuels. 2017;10:17.
Qin YQ, Bao LF, Gao MR, Chen M, Lei YF, Liu GD, et al. Penicillium decumbens BrlA extensively regulates secondary metabolism and functionally associates with the expression of cellulase genes. Appl Microbiol Biotechnol. 2013;97:10453–67.
Aro N, Ilmen M, Saloheimo A, Penttilä M. ACEI of Trichoderma reesei is a repressor of cellulase and xylanase expression. Appl Environ Microbiol. 2003;69:56–65.
Xiong Y, Sun J, Glass NL. VIB1, a link between glucose signaling and carbon catabolite repression, is essential for plant cell wall degradation by Neurospora crassa. PLoS Genet. 2014;10:e1004500.
Yao GS, Li ZH, Wu RM, Qin YQ, Liu GD, Qu YB. Penicillium oxalicum PoFlbC regulates fungal asexual development and is important for cellulase gene expression. Fungal Genet Biol. 2016;86:91–102.
He QP, Zhao S, Wang JX, Li CX, Yan YS, Wang L, et al. Transcription factor NsdD regulates the expression of genes involved in plant biomass-degrading enzymes, conidiation, and pigment Biosynthesis in Penicillium oxalicum. Appl Environ Microbiol. 2018;84:e01039–010418.
Schwer B, Sanchez AM, Garg A, Chatterjee D, Shuman S. Defining the DNA binding site recognized by the fission yeast Zn2Cys6 transcription factor Pho7 and its role in phosphate homeostasis. mBio. 2017;8:e01218.
Coradetti ST, Craig JP, Xiong Y, Shock T, Tian CG, Glass NL. Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. Proc Natl Acad Sci USA. 2012;109:7397–402.
Znameroski EA, Coradetti ST, Christine M, Tsai JC, Iavarone AT, Cate JHD, et al. Induction of lignocellulose-degrading enzymes in Neurospora crassa by cellodextrins. Proc Natl Acad Sci USA. 2012;109:6012–7.
Tian C, Beeson WT, Lavarone AT, Sun JP, Marletta MA, Cate JHD, et al. Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc Natl Acad Sci USA. 2009;106:22157–62.
Delmas S, Pullan ST, Gaddipati S, Kokolki M, Malla S, Blythe MJ, et al. Uncovering the genome-wide transcriptional responses of the filamentous fungus Aspergillus niger to lignocellulose using RNA sequencing. PLoS Genet. 2012;8:e1002875.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA seq data with DESeq2. Genome Biol. 2014;15:550.
Wang Y, Cen XF, Zhao GP, Wang J. Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J Bacteriol. 2012;194:5237–44.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Authors’ contributions
JXF conceived, supervised this study, wrote and revised the manuscript. SZ codesigned and co-supervised this study, and was involved in the data analysis and manuscript revision. LSL conducted in vitro binding experiments, identification of the binding sequences of PoxCxrA in POX05587 and its DNA-binding domain. CXL carried out bioinformatic analyses. FFZ performed identification of the binding sequences of PoxCxrA in PoxClrB. YSY conducted RT-qPCR experiments. XML were involved in preparation of experimental materials and the analysis of experimental data. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors consent for publication.
Ethics approval and consent to participate
Not applicable.
Funding
This work was financially supported by the Grants from the National Natural Science Foundation of China (Grant Nos. 31760023 and 31660305), and the ‘One Hundred Person’ Project of Guangxi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Summary of transcriptomic data generated from Penicillium oxalicum.
Additional file 2: Figure S1.
Pearson’s correlation analysis of transcriptomes from Penicillium oxalicum deletion mutant ∆PoxCxrA and the parental strain ∆PoxKu70. Total RNA was extracted from P. oxalicum strains cultivated in medium containing Avicel as the sole carbon source for 24 h after a shift from glucose, then sequenced.
Additional file 3: Table S2.
PoxCxrA regulon in Penicillium oxalicum when subjected to Avicel as the sole carbon source.
Additional file 4: Table S3.
Primers used in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liao, LS., Li, CX., Zhang, FF. et al. How an essential Zn2Cys6 transcription factor PoxCxrA regulates cellulase gene expression in ascomycete fungi?. Biotechnol Biofuels 12, 105 (2019). https://doi.org/10.1186/s13068-019-1444-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-019-1444-5