
Abstract
Background
Clostridium acetobutylicum and Clostridium saccharobutylicum are Gram-positive, spore-forming, anaerobic bacterium capable of converting various sugars and polysaccharides into solvents (acetone, butanol, and ethanol). The sequencing of their genomes has prompted new approaches to genetic analysis, functional genomics, and metabolic engineering to develop industrial strains for the production of biofuels and bulk chemicals.
Results
The method used in this paper to knock-out, knock-in, or edit genes in C. acetobutylicum and C. saccharobutylicum combines an improved electroporation method with the use of (i) restrictionless Δupp (which encodes uracil phosphoribosyl-transferase) strains and (ii) very small suicide vectors containing a markerless deletion/insertion cassette, an antibiotic resistance gene (for the selection of the first crossing-over) and upp (from C. acetobutylicum) for subsequent use as a counterselectable marker with the aid of 5-fluorouracil (5-FU) to promote the second crossing-over. This method was successfully used to both delete genes and edit genes in both C. acetobutylicum and C. saccharobutylicum. Among the edited genes, a mutation in the spo0A gene that abolished solvent formation in C. acetobutylicum was introduced in C. saccharobutylicum and shown to produce the same effect.
Conclusions
The method described in this study will be useful for functional genomic studies and for the development of industrial strains for the production of biofuels and bulk chemicals.
Similar content being viewed by others

Background
In recent years, solventogenic Clostridia have been of interest in the postgenomic era due to the complete sequencing and annotation of their genome [1, 2], supplying a wealth of information regarding the metabolism of these industrially important strains. This global knowledge has prompted new approaches to genetic analysis, functional genomics, and metabolic engineering to develop industrial strains for the production of biofuels and bulk chemicals.
To this end, several reverse genetic tools have been developed for solventogenic Clostridia, including gene inactivation systems based on nonreplicative [3,4,5] and replicative plasmids [6,7,8,9,10] and the group II intron gene inactivation system [11, 12]. All methods based on electroporation for in frame deletions use a replicative plasmid (typically containing a pIMP13 origin of replication from Bacillus subtilis that is functional in Clostridia) due to the low frequency of transformation of solventogenic Clostridia [13, 14]. Two families of methods have been developed to allow deletion and/or the introduction of genes at their normal chromosomal context without maintaining an antibiotic marker.
The first family [7, 10] uses a replicative vector containing (i) a replacement cassette consisting of an antibiotic resistance gene (ThR) flanked by two FRT sequences, (ii) two sequences homologous to the selected regions around the target DNA sequence, and (iii) a counterselectable marker made either of the codon-optimized mazF toxin gene from Escherichia coli (under the control of a lactose-inducible promoter) or the upp gene [which encodes an uracil phosphoribosyl-transferase and leads to 5-fluorouracil (5-FU) toxicity] to allow the direct positive selection of double-crossover allelic exchange mutants. After this first step, a second plasmid system expressing the FLP recombinase must be introduced, enabling efficient deployment of the FLP–FRT system to generate markerless deletion or integration mutants. A scar consisting of an FRT site remains at the target site, which can potentially act as a transcriptional terminator [15] or create a large chromosomal DNA deletion or inversion when several FRT sites are present on the chromosome [16, 17].
The second family [9, 18] also uses a replicative vector containing (i) a replacement cassette consisting of two sequences homologous to the selected regions around the target DNA sequence and (ii) a counterselectable marker made either of the codA [18] gene or the pyrE [9] gene. However, as the replacement cassette does not include an antibiotic resistance gene, and as this method uses a replicative plasmid, its stable single integration in the chromosome will be a rare event that cannot be selected for. When the counterselection is then applied, most of the clones will lose the plasmid and have a wild-type phenotype.
Creating a method for the rapid deletion, insertion, or modification of genes would require the use of a small suicide vector (to improve the transformation efficiency by electroporation), a replacement cassette consisting of two sequences homologous to the selected regions around the target DNA sequence and a counter selection marker such as upp, codA, or pyrE. One way to increase the transformation efficiency of solventogenic Clostridia is to remove the restriction modification system naturally present in the bacterium [5, 10, 14, 19]. Restrictionless, markerless mutants of solventogenic Clostridia have already been constructed for two species, C. acetobutylicum [10] and C. saccharobutylicum [5]. Although a transformation efficiency of 104/μg DNA has previously been reported when using electroporation for a restrictionless mutant of C. acetobutylicum [10], the transformation efficiency of a restrictionless mutant of C. saccharobutylicum has not been measured [5].
In the present study, we further improved the transformation efficiency of the two restrictionless mutants by weakening the cell wall using a lysozyme treatment before electroporation. We then constructed small suicide vectors containing the catP or the mlsR genes for the selection of chromosomal integration and the upp gene to select, in combination with the 5-fluorouracil (5-FU) system, for the second crossing-over. These plasmids, the restrictionless strains with a upp deletion and the improved transformation protocol were successfully used to develop a method for gene knock-in, knock-out, and editing in C. acetobutylicum and C. saccharobutylicum.
Results and discussion
Transformation efficiency of different industrially relevant solventogenic Clostridia
In a previous study [10], we demonstrated that a restrictionless mutant of C. acetobutylicum could be transformed by electroporation with unmethylated pCons2.1 at very high efficiency (6 × 104 transformants/μg of unmethylated DNA). However, when we evaluated the transformation efficiency of most of the non-sporulating, metabolically engineered strains, we noticed that the transformation efficiency of unmethylated pCons2.1 drastically decreased to values as low as 85 transformants/μg of unmethylated DNA. To improve the transformation efficiency of these industrially important, non-sporulating strains, we used as a prototype a C. acetobutylicum ∆cac1502 ∆cac3535∆upp∆pSOL mutant that no longer sporulated or produced solvent. This mutant was obtained by spreading the C. acetobutylicum ∆cac1502 ∆cac3535∆upp strain on an RCA plate and selecting clones that no longer produced a halo of starch hydrolysis after iodine staining [20]. The loss of pSOL1 was demonstrated by PCR analysis. The initial transformation efficiency of this strain with unmethylated pCons2.1 was low at approximately 142 ± 47 transformants/μg of unmethylated DNA (Table 1). Changing the voltage or the time constant did not significantly improve the transformation efficiency (data not shown). It was then decided to evaluate the use of cell wall weakening agents to facilitate DNA entry during the electroporation step. Such treatments, such as the use of lysozyme, have been shown previously [21,22,23] to improve the transformation efficiency of other Gram-positive bacteria. Lysozyme treatment, at concentrations ranging from 15 to 1500 μg/ml, was initially applied in the electroporation buffer for 30 min at 4 °C before electroporation. Although the transformation efficiency with unmethylated pCons2.1 was improved to values as high as 1 × 104 transformants/μg of unmethylated DNA, the results were not reproducible. It was then decided to add the lysozyme treatment directly to the culture medium, before centrifugation and washing, according to the protocol described in “Methods”. Very reproducible results were then obtained with an optimal lysozyme concentration of 150 μg/ml resulting in a transformation efficiency of 6.5 × 103 transformants/μg of unmethylated DNA, a value in the same range of the transformation efficiency of the sporulating C. acetobutylicum ∆cac1502 ∆cac3535∆upp strain [10].
In a previous study [5], Ch2, a markerless, restrictionless mutant of C. saccharobutylicum was constructed using conjugation to introduce the suicide vectors and the codBA genes and 5-fluorocytosine as a counter selection method. When Ch2 was transformed by electroporation using the unmethylated pMTL84151 replicative plasmid, no transformant could be obtained (Fig. 1) using the classical protocol without lysozyme treatment. On the other hand, when the protocol with the lysozyme treatment (optimized for non-sporulating C. acetobutylicum, i.e., 30 min of lysozyme treatment) was used, a transformation efficiency of 115 transformants/μg of unmethylated DNA was obtained (Fig. 1). After optimizing the incubation time with lysozyme (5 min), the transformation efficiency could be further increased to 255 transformants/μg of unmethylated DNA (Fig. 1). The unmethylated plasmid pMTL84151 was also used to evaluate the transformation efficiency, using the optimized protocol, of the C. saccharobutylicum wild type, ΔhsdR1, Ch1, and Ch2 strains. No transformants could be observed in the wild type and ΔhsdR1 strain. In contrast, the transformation efficiencies of the Ch1 and Ch2 strains using unmethylated pMTL84151 were 58 and 255 transformants/μg of unmethylated DNA, respectively (Table 2).

Effect of lysozyme treatment on the transformation efficiency the C. saccharobutylicum Ch2 (∆hsdR1∆ hsdR2 ∆hsdR3) strain. Lysozyme (at a concentration of 150 μg/ml) was added in the culture medium when the A600 reached a value of 0.6 (see “Methods”). Incubation time varies between 5 and 50 min. The time point at t = 0 min correspond to an experiment without lysozyme added
A generic method for gene knock-out, knock-in, and editing in C. acetobutylicum and C. saccharobutylicum
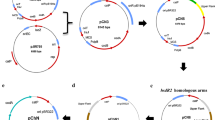
To create the generic method (presented in Fig. 2) for gene modification in both species, two very small shuttle suicide vectors (pCat-upp and pEry-upp) were constructed that carry either a colE1 or a p15A origin of replication functional in E. coli, a upp gene for 5-fluorouracil (5-FU) counterselection and either a catP or a mlsR gene for the selection of single crossing-over integration of the plasmid from thiamphenicol or erythromycin-resistant clones, respectively. Both plasmids have a unique BamHI site for the insertion of the modification cassettes.

General diagram representing the generic method for gene modification by allelic exchange in solventogenic Clostridia using a restrictionless ∆upp strain. The example present the use of the method for markerless gene deletion, but it can also be used for markerless gene editing or DNA insertion. The gene to delete in frame is y. The boxed regions of x and z genes represent approximatively the regions of homology incorporated into the suicide plasmid. a Selection for plasmid integration in 5′ or 3′ using the antibiotic resistance carried by the suicide vector. b Counter selection strategy with the 5-FU/upp system used for the selection of the double crossing-over and the excision of the plasmid
The recipient strain should be restrictionless, but should also carry a upp deletion for counterselection using 5-FU. Such a strain was already constructed for C. acetobutylicum [10]. However, the Ch2 mutant of C. saccharobutylicum still had a functional upp gene. The pCat-upp-Dupp plasmid was then constructed by inserting in pCat-upp the upp deletion cassette containing two 1-kbp regions flanking the upp gene on the chromosome of C. saccharobutylicum. When Ch2 was transformed with 200 μg of this plasmid using the optimized electroporation protocol presented above, no clones resistant to thiamphenicol could be obtained. As such clones would result from a RecA-dependent crossing-over between the homologous regions of the plasmid and the chromosome, and as it is well known that RecA is more efficient on single-stranded DNA, the pCat-upp-Dupp plasmid was first denatured at 95 °C for 5 min and rapidly cooled on ice before electroporation. Applying this DNA pretreatment, approximately 10 thiamphenicol colonies were obtained. PCR analysis of the different clones showed (Fig. 3b) that integration was obtained both in the upstream and downstream regions of upp. Two clones with an integration in each homologous arm were grown in 2×YTG, and appropriate dilutions were plated on MES-MM (0.01% yeast extract) with 5-FU at 1 mM. To select integrants having excised and lost pCat-upp-Dupp, 5-FU-resistant clones were replica plated on both MES-MM (0.01% yeast extract) + 5-FU at 1 mM and 2xYTG with thiamphenicol at 15 µg/ml. To identify mutants that lost pCat-upp-Dupp and possessed a markerless upp deletion, clones resistant to 5-FU and sensitive to thiamphenicol (at 25 μg/ml) were checked by PCR analysis (with primers Upp-check-F and Upp-check-R located outside of the upp deletion cassette). All the 5-FU-resistant, thiamphenicol-sensitive clones showed that upp was deleted when analyzed by PCR (Fig. 3c). The fermentation profiles of one of the C. saccharobutylicum ∆hsdR1∆hsdR2∆hsdR3∆upp clones were evaluated in batch fermentation performed without pH regulation in MES-MM (0.001% yeast extract) medium. Solvent and acid formation by C. saccharobutylicum ∆hsdR1∆hsdR2∆hsdR3∆upp was similar to that of the wild-type strain (Table 3), indicating that no physiological modifications were introduced during the construction of the mutant.

Markerless deletion of the upp gene in the C. saccharobutylicum Ch2 (∆hsdR1∆ hsdR2 ∆hsdR3) strain. a Map of pCat-upp-Dupp and chromosomal region around upp. b Insertion of pCat-upp-Dupp in 5′ and 3′ of upp. Clones were characterized using the upp-check-F and check-catp-F primers. c Excision of the plasmid by a second crossing-over using 5-FU/upp as a counter selection tool and isolation of the C. saccharobutylicum ∆hsdR1∆ hsdR2 ∆hsdR3 ∆upp strain. All the 5FU-resistant thiamphenicol-sensitive clones had a upp deletion as demonstrated by PCR using upp-check-F and upp-check-R primers
Gene deletion and editing in C. acetobutylicum using the generic method
The alsD gene (CA_C2967) encodes an acetolactate decarboxylase involved in the last step of acetoin formation [24]. To delete alsD, the alsD deletion cassette was cloned into the BamHI site of the pCat-upp to generate the plasmid pCat-upp-alsD. The plasmid pCat-upp-alsD was used to transform the C. acetobutylicum ∆cac1502 ∆cac3535∆upp strain by electroporation without previous in vivo methylation, and pCat-upp-alsD integrants were selected on RCA plates with thiamphenicol at 20 µg/ml. Two colonies were cultured for 24 h in liquid SM–glucose medium and then subcultured in liquid 2xYTG medium without antibiotic. Appropriate dilutions were plated on RCA with 5-FU at 1 mM. To select integrants having excised and lost pCat-upp-alsD, 5-FU-resistant clones were replica plated on both RCA + 5-FU and RCA with thiamphenicol at 40 µg/ml. To identify mutants possessing a markerless alsD deletion, clones resistant to 5-FU and sensitive to thiamphenicol were checked by PCR analysis (with primers alsd-0 and alsd-5 located outside of the alsD deletion cassette and primers alsd-F and alsd-R located inside alsD). Approximately half of the clones had an alsD deletion, and half had a wild-type genotype for alsD. The C. acetobutylicum ∆cac1502 ∆cac3535∆upp∆alsD strain was isolated. The fermentation profile of this strain was compared to that of the C. acetobutylicum ∆cac1502 ∆cac3535∆upp control strain during batch fermentation at pH 4.8 (Fig. 4). Surprisingly, the production of acetoin was only slightly decreased, indicating that either acetolactate can be chemically decarboxylated in vivo [25] or that Adc, the acetoacetate decarboxylase involved in the last step of acetone formation (15), can also decarboxylate acetolactate.

Solvent, acetoin and acid production by C. acetobutylicum ∆cac1502 ∆cac3535 ∆upp and C. acetobutylicum ∆cac1502 ∆cac3535 ∆upp ∆alsD in batch culture at pH4.8 in SM medium. Cultures were ran for 72 h
In a project aiming to improve the isopropanol tolerance of C. acetobutylicum using an adaptive laboratory evolution (ALE) approach, three individual clones (IPT4, IPT7, and IPT10) able to grow at isopropanol concentrations higher than 40 g/l were isolated (Fig. 5). When the genomes of these three strains were sequenced, 26 mutations present in the three strains were identified. Among all the mutated genes, two retained our attention: CA_C 0437 and CA_C3368, which encode a phosphatase that catalyzes the dephosphorylation of Spo0A [26] and a putative permease, respectively. The mutation in each gene is translated at the protein level to C1151A and G506A mutations. To evaluate the effect of these mutations on isopropanol tolerance, the genome-editing method presented above was used to introduce each of the two mutations in the genome of C. acetobutylicum ∆cac1502 ∆cac3535∆upp. For this purpose, two editing cassettes were created by directly amplifying a two kbp region centered around the point mutations in CA_C 0437 and CA_C3368 from the genome of the evolved strains and directly cloning them in pCat-upp to yield pCat-upp-CAC0437* and pCat-upp-CAC3368*.

Isopropanol tolerance of various mutants of C. acetobutylicum ∆cac1502 ∆cac3535 ∆upp
Each plasmid was transformed by electroporation in the C. acetobutylicum ∆cac1502 ∆cac3535∆upp strain and integrants were selected by their resistance to thiamphenicol. The generic method described in Fig. 2 was then used to select for the second crossing-over. Clones with the proper mutations were identified by a mismatch amplification mutation assay PCR (MAMA PCR) [27], and validation was finally performed by sequencing the region corresponding to the editing cassette plus 1 kbp on each side. The C. acetobutylicum ∆cac1502 ∆cac3535∆upp::cac0437* and C. acetobutylicum ∆cac1502 ∆cac3535∆upp::cac3368* were obtained and then characterized for their tolerance to isopropanol. The tolerance of both edited strains was not significantly different from the control strain (Fig. 5), indicating that those two mutations are either not involved in isopropanol tolerance or alone are not able to significantly participate in the isopropanol tolerance of C. acetobutylicum. Using the generic method described in this manuscript, a reverse strategy is currently under way, i.e., the editing back to wild type of each of the 26 mutations identified in one of the isopropanol tolerant strains and analysis of the isopropanol tolerance of the strains obtained.
Use of the gene-editing method to assess the effect of the Spo0A G179S mutation on the control of sporulation and solvent formation in C. acetobutylicum and C. saccharobutylicum
During the selection process of the C. acetobutylicum ∆cac1502 ∆cac3535∆upp∆pSOL strain, a mutant not producing solvent but still having the pSOL1 plasmid was identified and isolated. When the genome of this mutant was sequenced, a point mutation in the spo0A gene was identified, translating to the G179S mutation at the protein level. The mutated glycine residue is in a very conserved region of the Spo0A protein in all Firmicutes [28], IIHEIGVPAHIKGY, in which the lysine residue was shown to be involved in DNA binding to the Spo0A box [29].
This mutant was still able to sporulate, although at a lower frequency (Fig. 6), but after classical heat shock (70 °C for 10 min), no colony forming units were obtained for the G179S Spo0A mutant, while 4 × 105 CFU/ml were obtained for the control strain (Table 4). Analysis of the product profile of the mutant showed that it no longer produced solvents, and only acetic and butyric acid accumulated in the fermentation broth (Table 5).

Sporulation of C. acetobutylicum ∆cac1502 ∆cac3535 ∆upp and C. saccharobutylicum ∆hsdR1∆ hsdR2 ∆hsdR3 ∆upp with and without the mutation in Spo0A (G179S and G172S, respectively)
Using the gene-editing method, the same mutation in spo0A (translating to the G172S mutation at the protein level, as this protein is 7 amino acid residues shorter in N-terminal than the corresponding C. acetobutylicim protein) was introduced in the C. saccharobutylicum ∆hsdR1∆hsdR2∆hsdR3∆upp strain. This mutant was still able to sporulate (Fig. 6), but similar to the C. acetobutylicum G179S Spo0A mutant, it no longer produced solvent (Table 3), and the spores were thermally sensitive (Table 4). A tdcR knock-out mutant of C. difficile was previously shown to also produce heat-sensitive spores, which was associated with a lower expression of the SigE- and SigF-dependent sporulation genes [30].
Conclusions
The restrictionless, markerless generic method for genome modification in C. acetobutylicum and C. saccharobutylicum is a simple and useful tool for research groups involved in functional genomic studies and for further metabolic engineering of these two industrially important strains. As a demonstration of the efficiency of the method, we deleted the alsD gene in C. acetobutylicum to better understand how acetoin is produced in this microorganism. Furthermore, using this method we successfully edited genes to better characterize how C. acetobutylicum can develop isopropanol tolerance through adaptive laboratory evolution. Finally, we identified a mutation (G179S) in the Spo0A protein that abolishes solvent formation in both microorganisms while still allowing sporulation, although the spores produced were heat sensitive. Compared to the CRISPR/Cas9 method, that due to the large size of the cas9 gene imposes the use of replicative, this method allows the use of suicide vectors avoiding the step of plasmid curing that can be troublesome.
In the future, with the combined use of the pCat-upp and pEry-upp vectors developed in this study, it should be possible to simultaneously inactivate two genes in case each of the single knock-out mutants is not viable, while the double knock-out mutant is viable.
Methods
Bacterial strain, plasmids, and oligonucleotides
The bacterial strain and plasmids used in this study are listed in Table 6. The specific oligonucleotides used for PCR amplification were synthesized by Eurogentec (Table 7).
Culture and growth conditions
Clostridium acetobutylicum and C. saccharobutylicum were maintained as spores in (SM) and MES-MM (0.001% yeast extract) synthetic media, respectively, as previously described [31,32,33]. Spores were activated by heat treatment at 70 °C for 10 min. All C. acetobutylicum and C. saccharobutylicum strains were grown in anaerobic conditions at 37 °C in SM or MES-MM (0.001% yeast extract), in Clostridium growth medium (CGM) [34] in 2xYTG [35], or in reinforced clostridial medium (RCM) (Fluka). Solid media were obtained by adding 1.5% agar to the liquid media. Media were supplemented, when required, with the appropriate antibiotic in the following concentrations: for C. acetobutylicum and C. saccharobutylicum, erythromycin at 40 µg/ml and thiamphenicol between 15 and 25 µg/ml; for E. coli, erythromycin at 200 µg/ml, and chloramphenicol at 30 µg/ml. 5-Fluorouracil (5-FU) was purchased from Sigma, and stock solutions were prepared in DMSO.
Selection of isopropanol tolerant C. acetobutylicum mutant strains
An isopropanol tolerant population was selected using an Adaptive Laboratory Evolution (ALE) strategy using serial subcultures in SM–glucose medium with increasing concentration of isopropanol up to 5% W/V. Individual colonies were then on SM–glucose plates containing 4% W/V isopropanol. 10 clones were then evaluated for their isopropanol tolerance in liquid culture and the three best ones were sent for genome resequencing.
Analytical methods
Cell growth was monitored by measuring optical density at 600 nm (OD600). Solvent and acid production as well as glucose consumption in cell-free supernatant samples were determined based on high-performance liquid chromatography (HPLC) [36] using H2SO4 at 0.5 mM, as mobile phase.
DNA manipulation techniques
Total genomic DNA from C. acetobutylicum and C. saccharobutylicum were isolated as previously described [35]. Plasmid DNA was extracted from E. coli with the QIAprep kit (Qiagen, France). Pfu DNA Polymerase (Roche) was used to generate PCR products for cloning, and Taq Polymerase (New England BioLabs) was used for screening colonies by PCR with standard PCR protocols employed for all reactions. DNA restriction and cloning were performed according to standard procedures [37]. Restriction enzymes and Quick T4 DNA ligase were obtained from New England BioLabs (Beverly, MA) and were used according to the manufacturer’s instructions. DNA fragments were purified from agarose gels with the QIAquick gel purification kit (Qiagen, France).
Transformation protocol
Transformations of C. acetobutylicum and C. saccharobutylicum were conducted by electroporation according to the following protocol. A 10% inoculum of C. acetobutylicum or C. saccharobutylicum was grown in CGM up to A600 of 0.6. This culture was used to inoculate a serum bottle with 50 ml of 2×YTG. When the culture reaches A600 of 0.6, 100 µl of 8% NH4OH is added to the cultures before putting it on ice. In the normal protocol, developed for sporulating C. acetobutylicum, cells were then harvested by centrifugation at 4500g and 4 °C for 10 min and the culture resuspended in 10 ml of ice cold 0.5 M sucrose, 10 mM MES, pH6 (EPB). After a second centrifugation under the same conditions, the pellet is resuspended in 400 µl of EPB. Cells were chilled on ice for 1 min in a sterile electrotransformation vessel (0.4 cm electrode gap × 1.0 cm) and plasmid DNA (5–200 μg) dialysed against EPB buffer was added to the suspension keeping the total volume constant at 0.6 ml. A 1.8 kV discharge was applied to the suspension from a 25 μF capacitor and a resistance in parallel of 200 Ω using the Gene Pulser (Bio-Rad Laboratories, Richmond, CA). The cells were immediately transferred to 10 ml of prewarmed 2×YTG and incubated overnight at 30 °C prior to plating on 2×YTG with 20 μg/ml and 15 μg/ml thiamphenicol for C. acetobutylicum and C. saccharobutylicum, respectively.
For the poorly transformable strains, i.e., non-sporulating C. acetobutylicum and C. saccharobutylicum, a lysozyme (from chicken egg white, 7000 U/mg, Sigma-Aldrich) treatment (final concentration ranging from 15 to 1500 μg/ml) for 5 to 30 min was introduced immediately after cooling on ice the culture. This lysozyme pretreatment was optimized for both C. acetobutylicum ∆cac1502 ∆cac3535∆upp∆pSOL (a restrictionless non-sporulating strain) and C. saccharobutylicum Ch2 (a restrictionless sporulating strain).
Construction of pCat-upp
This plasmid contains a colE1 origin of replication functional in E. coli, a catP gene conferring resistance to thiamphenicol and chloramphenicol, the upp gene (encoding the uracil phosphoribosyl-transferase of C. acetobutylicum) and a unique BamHI site for the cloning of the replacement cassette. This plasmid was constructed by PCR (Phusion) amplification of a 2845 bp fragment on the pCons::UPP plasmid DNA using oligonucleotides pcat-Upp-F and BamHI-pCat-Upp-R. This fragment was digested by BamHI and ligated. The pCat-upp plasmid (2829 bp) was obtained.
Construction of pEry-upp
This plasmid contains a p15A origin of replication functional in E. coli, an mlsR gene conferring resistance to erythromycin, a upp gene and a unique BamHI site for the cloning of the replacement cassette. This plasmid was constructed in five steps.
-
1.
PCR (Phusion) amplification of the p15A replication origin (P15A fragment) on the plasmid pAN1, with the primers p15A-F and p15A-R.
-
2.
PCR (Phusion) amplification of the MLSR (EryR) cassette (EryUpp fragment) on the pSOS95-Upp plasmid with the primers eryUpp-F and eryUpp-R.
-
3.
PCR (Phusion) amplification of the adhE2 terminator (Teradhe2 fragment) on Clostridium acetobutylicum genomic DNA with the primers upp-Teradhe2-F and teradhe2-R.
-
4.
PCR fusion (Phusion) of the “EryUpp” and “Term-B” fragments using the primers eryUpp-F and termadhe2-R to get the “EryUpp- Teradhe2” fragment.
-
5.
Digestion by BamHI and SalI of the “P15A” with “EryUpp- Teradhe2″ fragments and ligation to get the pEry-Upp plasmid (2582 bp).
Construction of pCat-upp-Dupp
Two DNA fragments surrounding the upp-encoding gene (CLSA_RS02460) were PCR amplified with the Phusion DNA polymerase with total DNA from C. saccharobutylicum as template and two specific couples of oligonucleotides as primers. With the couples of primers Upp-Csa-1–Upp-Csa-2 and Upp-Csa-3–Upp-Csa-4, 1045 bp and 1047 bp DNA fragments were, respectively, obtained. Both primers Upp-Csa-1 and Upp-Csa-4 introduce a BamHI site, while primers Upp-Csa-2 and Upp-Csa-3 have complementary 5′ extended sequences. DNA fragments Upp-Csa-1–Upp-Csa-2 and Upp-Csa-3–cac-4 were joined in a PCR fusion experiment with primers Upp-Csa-1 and Upp-Csa-4 and the resulting fragment was cloned in the pCR4-TOPO-Blunt vector to yield pTOPO:upp. The upp replacement cassette obtained after BamHI digestion of the resulting plasmid was cloned, at the BamHI, site into pCat-upp to yield the pCat-upp-Dupp plasmid.
Construction of pCat-upp-alsd
Two DNA fragments surrounding the alsD encoding gene (CAC2967) were PCR amplified with the Phusion DNA polymerase with total DNA from C. acetobutylicum as template and two specific couples of oligonucleotides as primers. With the couples of primers Alsd-Cac-1– Alsd-Cac-2 and Alsd-Cac-3–Alsd-Cac-4, 1010 bp and 1011 bp DNA fragments were, respectively, obtained. Both primers Alsd-Cac-1 and Alsd-Cac-4 introduce a BglI site, while primers Alsd-Cac-2 and Alsd-Cac-3 have complementary 5′ extended sequences that introduced an in frame deletion of alsD. DNA fragments Alsd-Cac-1– Alsd-Cac-2 and Alsd-Cac-3–cac-4 were joined in a PCR fusion experiment with primers cac-1 and cac-4 and the resulting fragment was cloned in the pCR4-TOPO-Blunt vector to yield pTOPO:alsD. The alsD replacement cassette obtained after BglI digestion of the resulting plasmid was cloned, at the BamHI, site into pCat-upp to yield the pCat-upp-alsd plasmid.
Construction of pCat-upp-spo0A*Csa
Two DNA fragments surrounding the point mutation introduced in the spo0A-encoding gene (CLSARS02460) were PCR amplified with the Phusion DNA polymerase with total DNA from C. saccharobutylicum as template and two specific couples of oligonucleotides as primers. With the couples of primers spo0A*-Csa-1–spo0A*-Csa-2 and spo0A*-Csa-3–spo0A*-Csa-4, 797 bp, and 1204 bp DNA fragments were, respectively, obtained. Both primers spo0A*-Csa-1 and spo0A*-Csa-4 introduce a BamHI site, while primers spo0A*-Csa-2 and spo0A*-Csa-3 have complementary 5′ extended sequences which introduce the point mutation. DNA fragments spo0A*-Csa-1–spo0A*-Csa-2 and spo0A*–spo0A*-3–spo0A*-4 were joined in a PCR fusion experiment with primers spo0A*-1 and spo0A*-4 and the resulting fragment was cloned in the pCR4-TOPO-Blunt vector to yield pTOPO: spo0A*-Csa. The spo0A replacement cassette obtained after BamHI digestion of the resulting plasmid was cloned, at the BamHI, site into pCat-upp to yield the pCat-upp-spo0A*-Csa plasmid.
Construction of pCat-upp-cac0437* and pCat-upp-cac3368*
Cassettes containing the desired mutations surrounded by 1 kb upstream and downstream were PCR amplified with the Phusion DNA polymerase using total DNA from an isolated evolved isopropanol tolerant C. acetobutylicum strain (IPT4) as template and a specific couple of oligonucleotides as primers. For the CAC0437 PCR, the primers CAC0437_Bam_F and CAC0437_Bam_R were used to introduce a BamH1 site, whereas for the CAC3368 PCR, the primers CAC3368_BglII_F and CAC3368_BglII_R were used to introduce a BglII site. The resulting fragments were cloned into the pCR4-TOPO-Blunt vector to generate pTOPO::CAC0437 C1151A and pTOPO::CAC3368 G506A, respectively. The CAC0437 C1151A fragment obtained after BamH1 digestion and the CAC3368 G506A fragment obtained after BglII digestion were cloned at the BamHI site into pCat-upp to generate the pCat-upp-CAC0437* and the pCat-upp-CAC3368* plasmids, respectively.
Mismatch amplification mutation assay (MAMA PCR)
Primers for MAMA PCR were designed as described in publication [27] from Cha et al. Briefly, in each PCR, a forward MAMA primer and a reverse primer were used in a PCR reaction to detect the desired mutation. The PCR fragment was only generated from the wild-type gene and not from the gene with the mutation at the location covered by the mismatch position on the MAMA primer. For the CAC0437 C1151A mutation detection, the CAC0437_MAMA WT_F and CAC0437_ext_R primers were used. For the CAC3368 G506A mutation detection, the CAC3368_MAMA WT_F and the CAC3368_MAMA _R were used.
Mutants’ characterization
For each mutant strain, two clones of were systematically selected and their deletion cassettes sequenced after integration into the chromosome by double crossing-over.
Abbreviations
- 5-FU:
-
5-fluorouracil
- CGM:
-
Clostridium growth medium
- DMSO:
-
dimethyl sulfoxide
- FLP:
-
flippase
- FRT:
-
flippase recognition target
- MLS r :
-
the macrolide lincosamide streptogramin B resistance gene
- PCR:
-
polymerase chain reaction
- EPB:
-
electroporation buffer
- RBS:
-
ribosome binding site
- RCM:
-
reinforced clostridial medium
- SM:
-
synthetic medium
- MES-MM:
-
synthetic medium supplemented with MES
- Th R :
-
thiamphenicol resistance gene
- UPRTase:
-
uracil phosphoribosyl-transferase
References
Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, et al. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol. 2001;183:4823–38.
Poehlein A, Solano JDM, Flitsch SK, Krabben P, Winzer K, Reid SJ, Jones DT, et al. Microbial solvent formation revisited by comparative genome analysis. Biotechnol Biofuels. 2017;10:017–0742.
Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN. Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology. 1996;142:2079–86.
Green EM, Bennett GN. Inactivation of an aldehyde/alcohol dehydrogenase gene from Clostridium acetobutylicum ATCC 824. Appl Biochem Biotechnol. 1996;57–58:213–21.
Huang CN, Liebl W, Ehrenreich A. Restriction-deficient mutants and marker-less genomic modification for metabolic engineering of the solvent producer Clostridium saccharobutylicum. Biotechnol Biofuels. 2018;11:264.
Harris LM, Welker NE, Papoutsakis ET. Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol. 2002;184:3586–97.
Al-Hinai MA, Fast AG, Papoutsakis ET. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl Environ Microbiol. 2012;78:8112–21.
Liu CC, Qi L, Yanofsky C, Arkin AP. Regulation of transcription by unnatural amino acids. Nat Biotechnol. 2011;29:164–8.
Heap JT, Ehsaan M, Cooksley CM, Ng YK, Cartman ST, Winzer K, Minton NP. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res. 2012;40:e59.
Croux C, Nguyen NP, Lee J, Raynaud C, Saint-Prix F, Gonzalez-Pajuelo M, et al. Construction of a restriction-less, marker-less mutant useful for functional genomic and metabolic engineering of the biofuel producer Clostridium acetobutylicum. Biotechnol Biofuels. 2016;9:23.
Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods. 2007;70:452–64.
Shao L, Hu S, Yang Y, Gu Y, Chen J, Yang Y, Jiang W, Yang S. Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res. 2007;17:963–5.
Lesiak JM, Liebl W, Ehrenreich A. Development of an in vivo methylation system for the solventogen Clostridium saccharobutylicum NCP 262 and analysis of two endonuclease mutants. J Biotechnol. 2014;188:97–9.
Mermelstein LD, Papoutsakis ET. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol. 1993;59:1077–81.
Yoo M, Croux C, Meynial-Salles I, Soucaille P. Elucidation of the roles of adhE1 and adhE2 in the primary metabolism of Clostridium acetobutylicum by combining in-frame gene deletion and a quantitative system-scale approach. Biotechnol Biofuels. 2016;9:92.
Sektas M, Szybalski W. Tightly controlled two-stage expression vectors employing the Flp/FRT-mediated inversion of cloned genes. Mol Biotechnol. 1998;9(1):17–24.
Kato J, Hashimoto M. Construction of long chromosomal deletion mutants of Escherichia coli and minimization of the genome. Methods Mol Biol. 2008;416:279–93.
Ehsaan M, Kuit W, Zhang Y, Cartman ST, Heap JT, Winzer K, Minton NP. Mutant generation by allelic exchange and genome resequencing of the biobutanol organism Clostridium acetobutylicum ATCC 824. Biotechnol Biofuels. 2016;9:4.
Dong H, Zhang Y, Dai Z, Li Y. Engineering Clostridium strain to accept unmethylated DNA. PLoS ONE. 2010;5:e9038.
Sabathe F, Croux C, Cornillot E, Soucaille P. amyP, a reporter gene to study strain degeneration in Clostridium acetobutylicum ATCC 824. FEMS Microbiol Lett. 2002;210:93–8.
Powell IB, Achen MG, Hillier AJ, Davidson BE. A simple and rapid method for genetic transformation of lactic streptococci by electroporation. Appl Environ Microbiol. 1988;54:655–60.
McDonald IR, Riley PW, Sharp RJ, McCarthy AJ. Factors affecting the electroporation of Bacillus subtilis. J Appl Bacteriol. 1995;79:213–8.
Wei M-Q, Rush CM, Norman JM, Hafner LM, Epping RJ, Timms P. An improved method for the transformation of Lactobacillus strains using electroporation. Microbiol Methods. 1995;21:97–109.
Yoo M, Bestel-Corre G, Croux C, Riviere A, Meynial-Salles I, Soucaille P. A quantitative system-scale characterization of the metabolism of Clostridium acetobutylicum. MBio. 2015;6:e01808–15.
Rondags E, Germain P, Marc IJLL. Cinétiques de décarboxylation oxydative extra-et intracellulaire d’α-acétolactate par un Lactococcus lactis ssp. Lait. 1998;78:135–43.
Steiner E, Dago AE, Young DI, Heap JT, Minton NP, Hoch JA, Young M. Multiple orphan histidine kinases interact directly with Spo0A to control the initiation of endospore formation in Clostridium acetobutylicum. Mol Microbiol. 2011;80:641–54.
Cha RS, Zarbl H, Keohavong P, Thilly WG. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl. 1992;2:14–20.
Wörner K, Szurmant H, Chiang C, Hoch JA. Phosphorylation and functional analysis of the sporulation initiation factor Spo0A from Clostridium botulinum. Mol Microbiol. 2006;59:1000–12.
Zhao H, Msadek T, Zapf J, Hoch JA, Varughese KI. DNA complexed structure of the key transcription factor initiating development in sporulating bacteria. Structure. 2002;10:1041–50.
Girinathan BP, Monot M, Boyle D, McAllister KN, Sorg JA, Dupuy B, Govind R. Effect of tcdR mutation on sporulation in the epidemic Clostridium difficile strain R20291. mSphere. 2017;2:e00383-16.
Vasconcelos I, Girbal L, Soucaille P. Regulation of carbon and electron flow in Clostridium acetobutylicum grown in chemostat culture at neutral pH on mixtures of glucose and glycerol. J Bacteriol. 1994;176:1443–50.
Peguin S, Goma G, Delorme P, Soucaille P. Metabolic flexibility of Clostridium acetobutylicum in response to methyl viologen addition. Appl Microbiol Biotechnol. 1994;42:611–6.
Monot F, Martin JR, Petitdemange H, Gay R. Acetone and butanol production by Clostridium acetobutylicum in a synthetic medium. Appl Environ Microbiol. 1982;44:1318–24.
Wiesenborn DP, Rudolph FB, Papoutsakis ET. Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl Environ Microbiol. 1988;54:2717–22.
Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Biotechnology. 1992;10:190–5.
Dusseaux S, Croux C, Soucaille P, Meynial-Salles I. Metabolic engineering of Clostridium acetobutylicum ATCC 824 for the high-yield production of a biofuel composed of an isopropanol/butanol/ethanol mixture. Metab Eng. 2013;18:1–8.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Woodbury: Cold Spring Harbor Laboratory Press; 1989.
Authors’ contributions
AE, WL, IMS, and PS conceived the study; AT performed the initial construction of the pCAT-UPP vector; and AT and NPTN optimized the method for efficiently transforming C. acetobutylicum. CNH optimized the method for efficiently transforming C. saccharobutylicum. MY constructed the C. acetobutylicum ∆alsD strain and perform the cultures in fermentors. CF performed all the other deletions and gene editing of C. acetobutylicum. TWS perform the shake flask experiments. CNH performed all the deletions and gene editing of C. saccharobutylicum. PS drafted the manuscript and supervised the work. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Jonathan Humphreys for help in the whole genome resequencing data analysis.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All the data analyzed in this study are included in this manuscript.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was financially supported by the European Community’s Seventh Framework Program “CLOSTNET” (PEOPLE-ITN-2008-237942) (to TN) and “KBBE Valor Plus” (Grant agreement No. 613802). This work was partially financed by Total Raffinage Chimie as part of a collaborative program with Total.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Foulquier, C., Huang, CN., Nguyen, NPT. et al. An efficient method for markerless mutant generation by allelic exchange in Clostridium acetobutylicum and Clostridium saccharobutylicum using suicide vectors. Biotechnol Biofuels 12, 31 (2019). https://doi.org/10.1186/s13068-019-1364-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-019-1364-4