
Abstract
Background
Clostridium saccharobutylicum NCP 262 is a solventogenic bacterium that has been used for the industrial production of acetone, butanol, and ethanol. The lack of a genetic manipulation system for C. saccharobutylicum currently limits (i) the use of metabolic pathway engineering to improve the yield, titer, and productivity of n-butanol production by this microorganism, and (ii) functional genomics studies to better understand its physiology.
Results
In this study, a marker-less deletion system was developed for C. saccharobutylicum using the codBA operon genes from Clostridium ljungdahlii as a counterselection marker. The codB gene encodes a cytosine permease, while codA encodes a cytosine deaminase that converts 5-fluorocytosine to 5-fluorouracil, which is toxic to the cell. To introduce a marker-less genomic modification, we constructed a suicide vector containing: the catP gene for thiamphenicol resistance; the codBA operon genes for counterselection; fused DNA segments both upstream and downstream of the chromosomal deletion target. This vector was introduced into C. saccharobutylicum by tri-parental conjugation. Single crossover integrants are selected on plates supplemented with thiamphenicol and colistin, and, subsequently, double-crossover mutants whose targeted chromosomal sequence has been deleted were identified by counterselection on plates containing 5-fluorocytosine. Using this marker-less deletion system, we constructed the restriction-deficient mutant C. saccharobutylicum ΔhsdR1ΔhsdR2ΔhsdR3, which we named C. saccharobutylicum Ch2. This triple mutant exhibits high transformation efficiency with unmethylated DNA. To demonstrate its applicability to metabolic engineering, the method was first used to delete the xylB gene to study its role in xylose and arabinose metabolism. Furthermore, we also deleted the ptb and buk genes to create a butyrate metabolism-negative mutant of C. saccharobutylicum that produces n-butanol at high yield.
Conclusions
The plasmid vectors and the method introduced here, together with the restriction-deficient strains described in this work, for the first time, allow for efficient marker-less genomic modification of C. saccharobutylicum and, therefore, represent valuable tools for the genetic and metabolic engineering of this industrially important solvent-producing organism.
Similar content being viewed by others

Background
Clostridium saccharobutylicum NCP 262 is a solventogenic strain that has been used in South Africa for the industrial production of acetone, butanol, and ethanol (ABE) by fermentation [1, 2]. C. saccharobutylicum contains the three type I restriction–modification systems (hsdR1: CLSA_RS02150, hsdR2: CLSA_RS14125, and hsdR3: CLSA_RS04425), which might be why it is so difficult to transform. An efficient tri-parental mating system that transfers in vivo methylated DNA [3] by conjugation has, therefore, been developed to prevent DNA restriction and facilitate the genetic engineering of C. saccharobutylicum [4]. Type I restriction–modification (RM) systems consist of three genes, hsdR, hsdM, and hsdS, encoding a restriction enzyme, a methyltransferase, and a specificity subunit, respectively [5]. A restriction-less, marker-less mutant of Clostridium acetobutylicum [6] was previously constructed that greatly facilitates the development of reverse genetic tools for this organism. This mutant will also be useful for functional genomics studies and the efficient genetic and metabolic engineering of C. saccharobutylicum.
To date, most of the knockout mutants of solventogenic clostridia have been constructed by inserting a group II intron [7,8,9] or an antibiotic resistance cassette into, or in place of, the genes of interest [10,11,12,13]. In these cases, persisting DNA sequences such as an intron, an FRT (Flippase Recognition Target), or resistance markers remain in the strain, and are accompanied by polar effects on the expression of downstream genes [14]. Thus, methods that facilitate the generation of marker-less in-frame deletions in solventogenic clostridia are necessary. Moreover, another advantage of such methods is that they can introduce multiple knockouts or insertions, since the number of available resistance markers is not limiting. Typical marker-less deletion systems are two-step methods. First, a non-replicative plasmid containing an antibiotic resistance marker for selecting the allele regions of the target gene is integrated into the bacterial genome by homologous recombination. Then, the vector is excised in a second homologous recombination and selected for using a conditionally lethal counterselection marker present on the plasmid to yield either the wild-type or desired mutant genotype.
Counterselection strategies utilizing the sacB system have been used in several Gram-negative bacteria for this purpose, but do not work satisfactorily in most Gram-positive bacteria [13, 15]. Commonly used approaches for counterselection in Gram-positive bacteria exploit either endogenous toxin/antitoxin systems such as mazE/mazF [16,17,18] or gene-encoding enzymes involved in the purine or pyrimidine metabolism. For example, upp (phosphoribosyltransferase), codA (cytosine deaminase) [19, 20], pyrE/ura5 (orotate phosphoribosyltransferase), and hpt (hypoxanthine phosphoribosyltransferase) have all been used [20,21,22,23,24,25,26]. All these exemplary systems are based on the same selection principle, i.e., that purine or pyrimidine analogs are converted to toxic compounds and that cells can only survive in the presence of the analog when they lack the gene for the converting enzyme. In a previous study by our group, the upp gene was utilized for the counterselection step [27]. The uracil phosphoribosyltransferase encoded by this gene catalyzes the conversion of the pyrimidine analog 5-fluorouracil (5-FU) to 5-fluorouridine-monophosphate [28]. This is then transformed to 5-fluorodesoxyuridine-monophosphate, which elicits a toxic effect by inhibition of thymidylate synthase, thereby blocking DNA repair and replication [29]. Counterselection against this vector was, therefore, performed on media supplemented with 5-FU. In spite of this system’s high efficiency, the requirement for using a Δupp strain limits its application in a variety of solventogenic clostridia used in biotechnology. Cytosine deaminase is an enzyme that participates in pyrimidine salvage metabolism by catalyzing the deamination of cytosine to uracil, but it can also convert the cytosine analog 5-fluorocytosine (5-FC) to 5-FU [30]. A cytosine deaminase system has been used for a negative selection procedure in Streptomyces species and Rhodococcus equi [31], while 5-FC has been used for negative selection conferred by a heterologously expressed E. coli codA gene in mammalian cells and several Gram-positive bacteria [32,33,34,35]. Recent approaches also include the use of the CRISPR/Cas9 systems for counterselection, because the induced double strand breaks in the target gene are lethal in prokaryotes [36,37,38]. In this study, we report the use of the codBA operon genes derived from C. ljungdahlii as counterselection markers in combination with 5-FC as the counterselective compound for the generation of marker-less chromosomal deletions in the Gram-positive species C. saccharobutylicum. This method was used to generate marker-less restriction-deficient mutants of C. saccharobutylicum. In addition, the xylB gene was deleted to study the role of its encoded carbohydrate kinase in xylose and arabinose metabolism and a butyrate metabolism-negative strain that produces n-butanol at high yield was also produced by deletion of the ptb and buk genes.
Results
Generation of the ΔhsdR1 strain, the first marker-less C. saccharobutylicum strain that is transformable without prior in vivo plasmid methylation
The genome of the biotechnologically important solventogenic Clostridium saccharobutylicum NCP 262 contains three operons coding for genes of presumed type I RM systems belonging to the families A and C. The first RM system (RM1) consists of three genes, hsdR1, hsdM1, and hsdS1, encoding the restriction, methylation, and specificity subunits, respectively. Similarly, the second (RM2) and third RM (RM3) systems are composed of the hsdR2, hsdM2, and hsdS2 and the hsdR3, hsdM3, and hsdS3 genes, respectively. The previous work in our laboratory aimed at determining the importance of RM1 and RM2 in the restriction of exogenous DNA introduced into C. saccharobutylicum, resulted in the generation of the hsdR1::int ClosTron mutant. This strain was used to prevent exogenous DNA from degradation by both restriction systems by introducing (by conjugation) recombinant DNA that had been previously methylated in vivo for protection against degradation by RM2 [4]. Furthermore, we constructed a vector suitable for counterselection in C. saccharobutylicum using the codBA operon genes from E. coli K12 that encode a cytosine transporter (codB) and a cytosine deaminase (codA). These two genes have been successfully used by us as a counterselection marker in combination with 5-FC as the counterselective compound in the Gram-positive bacterium Bacillus licheniformis [34]. The hsdR1::int gene was deleted using a suicide vector carrying the replacement cassette, which was constructed in two steps. First, the pCN3 vector was produced by replacing the bla, ermC, and the pre genes from pKVM4 by the catP gene from pJIR750 (Fig. 1a). Then, an upstream and a downstream flanking region of the target hsdR1 gene were amplified (each region about 1 kb), fused, and inserted into pCN3 in place of the Gram-positive pE194ts replicon to yield the suicide vector pCN6 (Fig. 1b). After in vivo methylation using E. coli, Top 10 containing pJL2, pCN6 was introduced into the C. saccharobutylicum hsdR1::int strain by tri-parental conjugation. The transconjugants were plated on 2×YTG plates supplemented with 15 μg/ml thiamphenicol and 10 μg/ml colistin (for selection against E. coli cells used in tri-parental mating) and incubated overnight at 37 °C under anaerobic conditions. PCR showed that clones resistant to thiamphenicol were the result of homologous recombination of pCN6 with either the upstream or the downstream region of hsdR1 on the C. saccharobutylicum hsdR1::int strain chromosome. Colonies were streaked on MES-MM plates containing 0.01% yeast extract and 60–600 μg/ml of 5-FC, to select clones that have lost the codBA operon genes after a second crossover. However, after overnight incubation at 37 °C, all the colonies obtained were still resistant to thiamphenicol when tested by replica plating. Furthermore, colony PCR analysis showed that the catP gene was still present and that the colonies contained a mix of single integrants comprising cells of the hsdR1::int strain and ΔhsdR1 mutants. This suggested that the 5-FC selection did not function optimally, perhaps, because the codBA operon was not well expressed. To isolate a ΔhsdR1 mutant, a colony, giving, after PCR, a high amount of amplification product specific for ΔhsdR1, was picked and plated on MES-MM plates containing 0.01% yeast extract, and around 400 colonies were replica plated on the same medium supplemented with 5 μg/ml erythromycin. Among these, two clones were erythromycin-sensitive and, when analyzed by PCR, were shown to be ΔhsdR1 mutants (Fig. 2a).

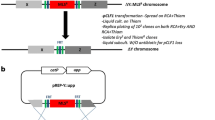
Schematic representation of deletion vector construction. a pCN3, a shuttle vector for C. saccharobutylicum NCP262 in which the antibiotic cassette of pKVM4 is replaced by the catP gene from pJIR750. b pCN6, a suicide vector to delete the hsdR1 gene, where the pE194ts replicon is replaced by hsdR1 homologous arms. c pCN8, where the homologous arms of pCN6 are replaced by those hsdR2. d pChN1, a deletion vector for the hsdR2 where the codBA operon genes of pCN8 are replaced by those from C. ljungdahlii. e pChN, a deletion vector cassette produced by removing the hsdR2 homologous arms from pChN1

Gene replacement via allelic exchange at the hsdR1, hsdR2, hsdR3, xylB, and ptb–buk loci. PCR confirmation of the different double-crossover deletion mutants using external primers annealing to the chromosome upstream and downstream of each deletion cassette. Strains (a) ΔhsdR1. b ΔhsdR1 ΔhsdR2. c ΔhsdR1 ΔhsdR2 ΔhsdR3. d ΔhsdR1 ΔhsdR2 ΔxylB. e ΔhsdR1 ΔhsdR2 Δptb Δbuk. ΔhsdR1: 2141 bp (a, b, c, d, e), WT of hsdR1: 5553 bp (a), catP gene: 622 bp (a, b, c, d, e). ΔhsdR2: 2064 bp (b, c, d, e), WT of hsdR2: 5259 bp (b) ΔhsdR3: 2078 bp (c), WT of hsdR3: 5010 bp (c). ΔxylB: 2081 bp (d), WT of xylB: 3549 bp (d). Δptb Δ buk: 2042 bp (e), and WT of ptb–buk: 4026 bp (e)
Construction of a C. saccharobutylicum ΔhsdR1ΔhsdR2 strain using the codB–codA genes from C. ljungdahlii
Since 5-FC counterselection was suboptimal, we assumed that the codBA operon genes from E. coli were not sufficiently well expressed in C. saccharobutylicum, and consequently, we decided to construct a new suicide vector, pChN1, using the codBA operon genes from Clostridium ljungdahlii to delete hsdR2. First, upstream and downstream flanking regions of the target hsdR2 gene were amplified (each region about 1 kb), fused, and inserted into pCN6 in place of the hsdR1 deletion cassette to yield pCN8 (Fig. 1c). Then, the codBA operon genes from E. coli were replaced by their clostridial orthologs (CLJU_RS09415 and CLJU_RS09420) from C. ljungdahlii (Fig. 1d). After in vivo methylation against HsdR2 restriction using pJL2, pChN1 was introduced into the C. saccharobutylicum ΔhsdR1 strain by tri-parental conjugation as described by Lesiak et al. [4]. The transconjugants were then plated on 2×YTG supplemented with 15 μg/ml thiamphenicol and 10 μg/ml colistin (for selection against E. coli cells in the conjugation mix) and incubated overnight at 37 °C under anaerobic conditions. PCR showed that the clones resistant to thiamphenicol were the result of homologous recombination of pChN1 with either the upstream or the downstream region of hsdR2 on the chromosome of the C. saccharobutylicum ΔhsdR1 strain. Colonies were then streaked and grown overnight on MES-MM plates supplemented with 0.001% yeast extract and 500 μg/ml of 5-FC to select for clones that had lost the codBA operon genes by a second crossover (Fig. 2b).
The colonies were then replica plated on the same medium and on MES-MM plates containing 0.001% yeast extract and 15 μg/ml of thiamphenicol. Twenty colonies that did not grow on the thiamphenicol plate were analyzed by PCR for hsdR2 deletion. About half (9 of 20) possessed the desired genotype (i.e., deletion of hsdR2), while the remainder were wild type. This demonstrates that the codBA operon genes from C. ljungdahlii were functionally expressed in C. saccharobutylicum and that they can be used in combination with 5-FC for counterselection. The resulting C. saccharobutylicum ΔhsdR1ΔhsdR2 strain, which we named C. saccharobutylicum Ch1, was further used to construct a restriction-deficient strain by deletion of the hsdR3 gene.
Construction of C. saccharobutylicum ΔhsdR1ΔhsdR2ΔhsdR3, a restriction-minus strain that can be subjected to iterative genome modification without marker limitations
Based on the success of the hsdR2 deletion using the pChN1 deletion vector and the codBA operon genes from C. ljungdahlii for counterselection, we used pChN1 as a backbone to construct a generic deletion vector, pChN, lacking homologous arms (Fig. 1e). About 1 kb of the upstream and downstream flanking regions of the target hsdR3 gene were amplified, fused, and inserted into pChN to produce the pChN2 plasmid. This plasmid was introduced into the C. saccharobutylicum Ch1 strain by tri-parental conjugation [4] without prior in vivo methylation. A clone with a deletion in hsdR3 was selected, as described above for hsdR2 (Fig. 2c), to produce the C. saccharobutylicum ΔhsdR1ΔhsdR2ΔhsdR3 strain, which we named C. saccharobutylicum Ch2.
The unmethylated plasmid pMTL84151 was used to evaluate the conjugation efficiency of the C. saccharobutylicum wild type, ΔhsdR1, Ch1 and Ch2 strains. As reported previously [4], no transconjugants could be observed in the wild-type strain without prior in vivo methylation of the plasmid. In contrast, the conjugation efficiencies of the Ch1 and Ch2 strains using unmethylated pMTL84151 were twofold and tenfold higher, respectively, than the ΔhsdR1 strain (Table 1).
The fermentation profiles of the different strains were evaluated in batch fermentation performed without pH regulation in MS medium. Solvent and acid formation by C. saccharobutylicum Ch1 were similar to the wild-type strain (Table 2), indicating that no physiological modifications were introduced during the construction of the mutants.
Application of 5-FC counterselection using the pChN plasmid in C. saccharobutylicum Ch1 to study the role of the xylB carbohydrate kinase gene in xylose and arabinose metabolism
Clostridium saccharobutylicum possesses an operon, CLSA_RS15825-CLSA_RS15800, containing six genes potentially involved in xylose metabolism and predicted to code for (1) carbohydrate kinase (xylB), (2) ROK family transcriptional regulator, (3) fructose-6-phosphate aldolase, (4) transketolase, (5) DUF4867 family protein, and (6) l-fucose isomerase, with a promoter region-mapped upstream of the CLSA_RS15825 gene. Since the triple-restriction-minus strain was not available at the time of these experiments, the Ch1 double mutant was used as the parental strain. To delete the xylB gene from C. saccharobutylicum Ch1, pChN3 was constructed from pChN. About 1 kb each of the upstream and downstream flanking regions of the xylB gene was amplified, fused, and inserted into pChN to produce the pChN3 plasmid. This plasmid was then introduced into C. saccharobutylicum Ch1 by tri-parental conjugation [4] without prior in vivo methylation. Strains with a deletion in the xylB gene were selected as described above for hsdR2 (Fig. 2d). Growth of C. saccharobutylicum Ch1 and C. saccharobutylicum Ch1 ΔxylB on MES-MM liquid cultures supplemented with 0.001% yeast extract or with d-glucose, d-xylose or l-arabinose as sole carbon sources was evaluated. While C. saccharobutylicum Ch1 grew on all three carbon sources (Fig. 3a), C. saccharobutylicum Ch1 ΔxylB only grew on glucose and arabinose but not on xylose (Fig. 3b). This demonstrates that XylB is specifically required for xylose but not for arabinose metabolism.

Growth of C. saccharobutylicum Ch1 (a) and C. saccharobutylicum Ch1 ΔxylB (b) on different carbon sources. Cells were grown in 30 ml of MES-MM supplemented with 0.001% yeast extract and 40 g/l d-glucose (black circle), 40 g/l l-Arabinose (black square) or 40 g/l d-xylose (white up-pointing triangle)
Application of 5-FC counterselection using the pChN plasmid for metabolic engineering using the C. saccharobutylicum Ch1 strain: deletion of the ptb–buk operon to create a strain with increased n-butanol production
The ptb and buk genes were targeted for deletion to test the applicability of 5-FC counterselection using the pChN plasmids to the metabolic engineering of C. saccharobutylicum. The ptb and buk genes, which encode a phosphotransbutyrylase and a butyrate kinase, respectively, have been targets for gene inactivation in C. acetobutylicum, because the butyrate synthesis pathway competes with the butanol synthesis pathway [39], since the consumption of butyryl-CoA for butyrate formation reduces n-butanol yield. The pChN4 vector was, therefore, constructed to delete the ptb–buk operon from the C. saccharobutylicum Ch1 mutant. About 1 kb of sequence upstream and a downstream of the target ptb–buk operon were amplified, fused, and inserted into pChN to produce the pChN4 plasmid, which was then introduced into the C. saccharobutylicum Ch1 strain by tri-parental conjugation [4] without prior in vivo methylation. Clones with a deletion of the ptb–buk operon were selected as described above for hsdR2 (Fig. 2e).
The fermentation profile of the C. saccharobutylicum Ch1∆ptb–buk strain was compared to that of the C. saccharobutylicum Ch1 control strain in batch fermentation performed without pH regulation in MS medium. The formation of butyrate was highly decreased in the mutant strain and the yield of n-butanol on glucose increased from 0.155 to 0.215 g/g (Table 2).
Discussion
A simple and efficient method to introduce targeted mutations without leaving behind marker remnants in the chromosome was established for Clostridium saccharobutylicum.
This method needs: (i) a suitable conjugative suicide shuttle vector; (ii) a deletion cassette containing fused upstream and downstream flanking regions of the target gene; (iii) an efficient counterselection marker, namely the codBA operon genes from Clostridium ljungdahlii. The codBA operon genes encode a cytosine permease and a cytosine deaminase facilitate the conversion 5-FC to 5-FU, which is toxic to the cell. The initial attempts to use the codBA operon genes from E. coli were unsuccessful, probably because their expression was not codon-optimized for C. saccharobutylicum and was, therefore, too low [40]. Other studies have relied on the use of E. coli codA alone. However, we have demonstrated before that the additional expression of the gene codB, which encodes a cytosine transporter that can presumably transport the cytosine analog 5-FC, enhances the counterselection [34].
The use of suicide plasmids requires high transformation or conjugation efficiencies. This was achieved by employing tri-parental conjugation of C. saccharobutylicum with the E. coli strains [4] and use of C. saccharobutylicum strains with deleted restriction systems. Integration by single crossover was then easily selected for by the thiamphenicol resistance of the clones. The construction of the deletion cassette for the codBA operon deletion system for C. saccharobutylicum was achieved by fusion PCR based on the SLiCE method. The codBA operon genes are located on the pChN plasmid, outside of the deletion cassette. This allows for the positive selection of clones that have lost the plasmid and the integrated deletion cassette via a double recombination event. Once a deletion cassette is integrated into the chromosome, a clean in-frame deletion of the targeted gene can be obtained, thus avoiding polar effects in operon structures. Such strategies were previously applied to construct marker-less gene deletions in E. coli [41, 42], Clostridium difficile [19], Bacillus licheniformis [35], Gluconobacter oxydans [34], and many other organisms.
In this study, genes encoding the three type I restriction enzymes of C. saccharobutylicum, HsdR1, HsdR2, and HsdR3 (hsdR1: CLSA_RS02150, hsdR2: CLSA_RS14125, and hsdR3: CLSA_RS04425, respectively), were deleted to produce a restriction-deficient strain. The conjugation efficiencies of the C. saccharobutylicum Ch1 and C. saccharobutylicum Ch2-recipient strains using an unmethylated pMTL84151 plasmid, were twofold and tenfold higher than for C. saccharobutylicum ΔhsdR1. The C. saccharobutylicum Ch2 strain should be especially useful for future genetic engineering efforts, e.g., for mariner transposon mutagenesis using a suicide vector introduced by conjugation or for the development of a protocol for the transformation of plasmids by electroporation [43]. The C. saccharobutylicum Ch1 strain and the codBA-based counterselection method described here were successfully used to investigate the role of the putative xylB gene in xylose and arabinose metabolism. This work demonstrated that xylB encodes a xylulokinase that is essential for the utilization of xylose as a carbon source in C. saccharobutylicum.
Furthermore, the described method was successfully used for metabolic engineering by creating a butyrate metabolism-minus strain that produces n-butanol at high yield. A similar strain was previously described for C. acetobutylicum [39]. A more detailed characterization of the C. saccharobutylicum Ch1 ΔptbΔbuk growing in both batch and continuous culture is currently in progress in our laboratory.
Conclusion
The restriction-deficient and marker-less genomic mutants constructed in this study, as well as the associated gene deletion method, will provide, to our scientific community, the simple and convenient tools for the genetic engineering of C. saccharobutylicum that can be used for future metabolic engineering of this industrially important strain to enhance the production of chemicals and biofuels.
Methods
Bacterial strains, culture and growth conditions, plasmids/oligonucleotides, and tests for 5-FU and 5-FC sensitivity
The bacterial strains and plasmids used in this study are listed in Table 3. Oligonucleotides were obtained from Eurofins MWG GmbH (Ebersberg, Germany) and are listed in Table 4. C. saccharobutylicum strains were grown under anaerobic conditions at 37 °C in CGM [44], 2×YTG [4], or MES-MM and MS media with a d-glucose concentration of 50 g/l [45]. Solid media were produced by adding 1.5% agar to the liquid media. Media were supplemented, when required, with the appropriate antibiotic at the following concentrations: erythromycin at 5 μg/ml and thiamphenicol at 15 μg/ml for C. saccharobutylicum; kanamycin at 50 μg/ml, chloramphenicol at 25 μg/ml and colistin at 10 μg/ml for E. coli. Growth curves in batch cultures were generated in 30 ml modified MES-MM medium supplemented with 0.001% yeast extract and 40 g/l d-glucose (GOPOD Format, K-GLUC, Megazyme, Ireland), or 40 g/l d-xylose (K-XYLOSE, Megazyme, Ireland) or 40 g/l l-arabinose (K-ARGA, Megazyme, Ireland) for 3 days. 5-FU was purchased from Sigma-Aldrich (Steinheim, Germany) and 5-FC from TCI Europe N.V. (Zwijndrecht, Belgium). Both were prepared in water as stock solutions of 10 mg/ml. Minimal inhibitory concentrations of 5-FC and 5-FU were determined in MES-MM [45] supplemented with 1%, 0.1%, 0.01%, or 0.001% yeast extract (see Additional file 1).
DNA manipulation techniques
Routine molecular biological procedures were performed using the standard protocols [48]. NucleoSpin® Plasmid EasyPure kit (Macherey–Nagel, Germany) was used for plasmid preparation. Genomic DNA from C. saccharobutylicum was extracted with an Epicenter MasterPure DNA purification kit (Madison, USA) and DNA purification was performed with a NucleoSpin® PCR clean-UP Gel extraction kit (Macherey–Nagel, Düren, Germany). Cloning was via the SLiCE method, which utilizes easily obtained bacterial cell extracts to assemble multiple DNA fragments into recombinant DNA molecules in a single in vitro recombination reaction [49]. PCR was performed according to the manuals provided for enzymes from Thermo Scientific (Schwerte, Germany). Phire Green Hot Start II DNA polymerase was used for analytical reactions and Phusion High-Fidelity DNA polymerase for amplifications requiring proofreading. TakaRa Bio (Otsu, Shiga, Japan) PrimeSTAR® GXL DNA polymerase was used for the amplification of products ≥ 30 kb in length. Colony PCR [50] was used to screen for mutants or to confirm the integration of a deletion vector into the genome.
Construction of deletion vectors
PCR primers used in the production of all constructs are listed in Table 4. The pCN3 shuttle vector for C. saccharobutylicum and E. coli was constructed by replacing the bla and ermC resistance cassettes of pKVM4 [35] with the catP gene from pJIR750 [51] (Fig. 1a). The backbone was amplified using pKVM4 as a template. The catP gene fragment was amplified using catp_FpJIR_IV and catp_RpJIR_IV primers and pJIR750 as a template. Cloning was performed using the SLiCE method.
To construct the pCN6 suicide vector for the deletion of the hsdR1 gene of C. saccharobutylicum, the pE194ts Gram-positive origin of replication in pCN3 was replaced by a fragment consisting of fused upstream and downstream flanking regions of the hsdR1 gene (Fig. 1b). The upstream and downstream flanking regions were amplified using chromosomal DNA from C. saccharobutylicum wild type as a template, while the backbone was amplified using pCN3 as a template. Cloning was performed using the SLiCE method. Plasmid integration by single crossover was detected using HsdR1_check_F and Catp_FpJIR_IV primers for 5′ integration and HsdR1_check_R and check_pre_R primers for 3′ integration. After selecting clones that had lost the integrated plasmid containing the codBA operon genes via a second crossover event, loss was confirmed using colony PCR. The presence or absence of catP was confirmed by PCR.
For construction of the pChN1 suicide vector for deletion of the hsdR2 gene of C. saccharobutylicum, approximately 1 kb of the flanking regions upstream and downstream of the hsdR2 gene were amplified using chromosomal DNA of C. saccharobutylicum wild type as a template, fused, and then inserted into pCN6 in place of the hsdR1 deletion cassette to produce pCN8 (Fig. 1c). The backbone used was the same as for pCN6. The codBA operon genes from E. coli were then replaced by the clostridial orthologs (CLJU_RS09415 and CLJU_RS09420) from Clostridium ljungdahlii, which were amplified with CLcodBA_F2 and R2 primers and using the chromosomal DNA of wild-type C. ljungdahlii as a template. The backbone was amplified using pCN8 as a template and cloning was performed using the SLiCE method (Fig. 1d). Plasmid integration by single crossover was detected using HsdR2_check_F and pCLcodBA_F_IV primers for 5′ integration and HsdR2_check_R and Check_catp_F primers for 3′ integration. After selecting clones that had lost the codBA operon genes via a second crossover event, loss was confirmed using colony PCR. The presence or absence of catP was confirmed by PCR.
pChN is a generic vector containing the codBA operon genes from C. ljungdahlii but lacking any homologous arms for a target gene (Fig. 1e). Since pChN1 was successfully used to delete hsdR2, we used pChN1 as a template to PCR-amplify the pChN fragment using the pChN_backbone_F and pChN_backbone_R primers. Ligation was performed using the SLiCE method.
For construction of the pChN2 suicide vector for deletion of the hsdR3 gene, approximately 1 kb flanking regions upstream and downstream of hsdR3 were amplified using chromosomal DNA of wild-type C. saccharobutylicum as a template, fused, and inserted into pChN (Fig. 1e) to produce pChN2. The backbone was amplified using pChN as a template and cloning was performed using the SLiCE method. Plasmid integration via single crossover was detected by PCR using HsdR3_check_F and catp_FpJIR_IV primers for 5′ integration and HsdR3_check_R and codBA_CL_R primers for 3′ integration. After selecting clones that had lost the codBA operon genes via a second crossover event, loss was confirmed by colony PCR. The presence or absence of catP was confirmed by PCR.
For construction of the pChN3 suicide vector for deletion of the xylB gene, approximately 1 kb flanking regions up- and downstream of xylB were amplified using chromosomal DNA of wild-type C. saccharobutylicum as template, fused, and inserted into pChN (Fig. 1e). The backbone was amplified with pChN as a template and cloning was performed using the SLiCE method. Plasmid chromosomal integration via single crossover was detected by PCR using xylB_check_F and catp_FpJIR_IV for 5′ integration and xylB_check_R and codBA_CL_R primers for 3′ integration. After selecting clones that had lost the codBA operon genes via a second crossover, loss was confirmed by colony PCR. The presence or absence of catP was confirmed by PCR.
For construction of the pChN4 suicide vector for the deletion of the buk and ptb genes, an approximately 1 kb region upstream of ptb and a second approximately 1 kb region downstream of buk were amplified using chromosomal DNA of wild-type C. saccharobutylicum as a template, fused, and then inserted into pChN (Fig. 1e) The backbone was amplified using pChN as a template. Cloning was performed using the SLiCE method. Plasmid integration by single crossover was detected by PCR using PTB_check_F3 and catp_FpJIR_IV primer for 5′ integration and BUK_check_R2 and PTB_F_IV primers for 3′ integration. After selecting clones that had lost the codBA operon genes via a second crossover event, loss was confirmed by colony PCR. The presence or absence of catP was confirmed by PCR.
Tri-parental conjugation
To conjugate pChN plasmids into C. saccharobutylicum, we modified the tri-parental conjugation protocol [4] as follows. C. saccharobutylicum-recipient cells in Hungate tube-containing anaerobic 2×YTG medium were heat-shocked at 70 °C for 5 min and then incubated at 37 °C, overnight. Donor cells containing the deletion vector in LB medium-containing chloramphenicol at 25 μg/ml and helper E. coli CA434 cells in LB medium-containing 50 μg/ml kanamycin were grown aerobically at 37 °C overnight. Cultures of recipient, donor, and helper cells were then inoculated to an OD600 of 0.1–0.2 and grown to an OD600 of 1 in the respective media described above. One ml each of the donor cells and helper cells were then mixed in the same Eppendorf tube and centrifuged at 6000 rpm at room temperature for 5 min. After washing the cells with 1 ml of phosphate-buffered saline (PBS), the pellet was transferred to an anaerobic chamber. Pellets were resuspended in 200 μl of recipient culture and six drops (about 25 μl per drop) were transferred to 2×YTG plates lacking any antibiotics and incubated overnight at 37 °C. Under anaerobic chamber, the cell mixture was collected from the surface of the agar plate, resuspended in 400 μl of PBS, and plated on 2×YTG plates supplemented with 15 μg/ml thiamphenicol and 10 μg/ml colistin and incubated at 37 °C.
General procedure for the construction of chromosomal deletion strains of Clostridium saccharobutylicum using codBA operon-based counterselection
The general outline for the deletion method is given below, using the deletion of the hsdR2 gene from C. saccharobutylicum (Fig. 4) as an example. First, a deletion vector containing about 1 kb fused flanking regions from the genomic locus targeted for deletion was constructed. The suicide deletion vector (pChN1 for deletion of hsdR2) was methylated by propagation in E. coli Top10-containing pJL2 and then introduced into the recipient C. saccharobutylicum ΔhsdR1 by tri-parental conjugation, and with E. coli CA434 as a helper strain. Transconjugants are transferred to 2×YTG plates containing 15 μg/ml thiamphenicol for pChN1 selection and 10 μg/ml colistin for elimination of E. coli. Since the suicide vector has no functional Gram-positive origin of replication, overnight growth at 37 °C yielded clones with the deletion plasmid integrated into the chromosomal target locus via homologous recombination. Colonies were then picked and streaked on the same medium. The presence of the catP gene and integration was confirmed by colony PCR. For counterselection, colonies were streaked on MES-MM supplemented with 0.001% yeast extract containing 500 µg/ml 5-FC, which selected against the vector-encoded codBA operon genes. After incubation at 37 °C overnight, only cells that had lost the integrated vector via a second homologous recombination formed colonies. The presence of the expected mutation in the resulting colonies was finally tested by PCR and confirmed by sequencing.

General diagram representing gene replacement via allelic exchange at the target gene. a C. saccharobutylicum NCP262 genomic regions surrounding CLSA_RS14125 (hsdR2). The deletion vector pChN1, containing approximately 1 kbp of upstream and downstream sequences of hsdR2 and the codBA operon from C. ljungdahlii. b Counterselection strategy with the 5-FC/codBA system resulting in a marker-less deletion mutant lacking CLSA_RS14125 (hsdR2) between the two flanking regions
Analytical methods
Cell growth was monitored by measuring optical density at 600 nm (OD600). Solvent and acid production as well as glucose consumption in cell-free supernatant samples were determined based on high-performance liquid chromatography (HPLC) [52] equipped with refractive index and UV detectors. The separation was obtained with an Aminex HPX-87H (Bio-Rad, Chemical Division, Richmond, USA) column (300 by 7.8 mm). The operating conditions were as follows: temperature, 17 °C; mobile phase, H2SO4 (0.25 mM); flow rate, 0.5 ml/min [52].
Abbreviations
- 5-FU:
-
5-fluorouracil
- 5-FC:
-
5-fluorocytosine
- CGM:
-
Clostridium growth medium
- 2×YTG:
-
2× yeast tryptone glucose medium
- PCR:
-
polymerase chain reaction
- MES-MM:
-
MES-based mineral medium
- RM system:
-
restriction modification system
- catP :
-
thiamphenicol resistance gene
- MS:
-
synthetic mineral medium
- SLiCE:
-
seamless ligation cloning extract
References
Ni Y, Xia Z, Wang Y, Sun Z. Continuous butanol fermentation from inexpensive sugar-based feedstocks by Clostridium saccharobutylicum DSM 13864. Bioresour Technol. 2013;129:680–5.
Poehlein A, Hartwich K, Krabben P, Ehrenreich A, Liebl W, Durre P, et al. Complete genome sequence of the solvent producer Clostridium saccharobutylicum NCP262 (DSM 13864). Genome Announc. 2013;1:00997–1013.
Mermelstein LD, Papoutsakis ET. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol. 1993;59:1077–81.
Lesiak JM, Liebl W, Ehrenreich A. Development of an in vivo methylation system for the solventogen Clostridium saccharobutylicum NCP 262 and analysis of two endonuclease mutants. J Biotechnol. 2014;188:97–9.
Murray NE. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol Mol Biol Rev. 2000;64(2):412–34.
Croux C, Nguyen NP, Lee J, Raynaud C, Saint-Prix F, Gonzalez-Pajuelo M, et al. Construction of a restriction-less, marker-less mutant useful for functional genomic and metabolic engineering of the biofuel producer Clostridium acetobutylicum. Biotechnol Biofuels. 2016;9:23.
Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods. 2007;70:452–64.
Jang YS, Lee JY, Lee J, Park JH, Im JA, Eom MH, et al. Enhanced butanol production obtained by reinforcing the direct butanol-forming route in Clostridium acetobutylicum. MBio. 2012;3:e00314–412.
Dong H, Zhang Y, Dai Z, Li Y. Engineering clostridium strain to accept unmethylated DNA. PLoS ONE. 2010;5:e9038.
Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN. Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology. 1996;142:2079–86.
Harris LM, Welker NE, Papoutsakis ET. Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol. 2002;184:3586–97.
Hölscher T, Görisch H. Knockout and overexpression of pyrroloquinoline quinone biosynthetic genes in Gluconobacter oxydans 621H. J Bacteriol. 2006;188:7668–76.
Hölscher T, Weinert-Sepalage D, Görisch H. Identification of membrane-bound quinoprotein inositol dehydrogenase in Gluconobacter oxydans ATCC 621H. Microbiology. 2007;153:499–506.
Yoo M, Croux C, Meynial-Salles I, Soucaille P. Elucidation of the roles of adhE1 and adhE2 in the primary metabolism of Clostridium acetobutylicum by combining in-frame gene deletion and a quantitative system-scale approach. Biotechnol Biofuels. 2016;9:92.
Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI. Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J Bacteriol. 1985;164:918–21.
Morimoto T, Ara K, Ozaki K, Ogasawara N. A new simple method to introduce marker-less deletions in the Bacillus subtilis genome. Genes Genet Syst. 2009;84:315–8.
Zhang XZ, Yan X, Cui ZL, Hong Q, Li SP. mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis. Nucleic Acids Res. 2006;34:e71.
Al-Hinai MA, Fast AG, Papoutsakis ET. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling marker-less chromosomal gene deletions and DNA integration. Appl Environ Microbiol. 2012;78:8112–21.
Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol. 2012;78:4683–90.
Ehsaan M, Kuit W, Zhang Y, Cartman ST, Heap JT, Winzer K, Minton NP. Mutant generation by allelic exchange and genome resequencing of the biobutanol organism Clostridium acetobutylicum ATCC 824. Biotechnol Biofuels. 2016;9:4.
Boeke JD, La Croute F, Fink GR. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet (MGG). 1984;197:345–6.
Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–75.
Fabret C, Ehrlich SD, Noirot P. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol. 2002;46:25–36.
Keller KL, Bender KS, Wall JD. Development of a marker-less genetic exchange system for Desulfovibrio vulgaris Hildenborough and its use in generating a strain with increased transformation efficiency. Appl Environ Microbiol. 2009;75:7682–91.
Pritchett MA, Zhang JK, Metcalf WW. Development of a marker-less genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl Environ Microbiol. 2004;70:1425–33.
Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, et al. Versatile genetic tool box for the crenarchaeote Sulfolobus acidocaldarius. Front Microbiol. 2012;3:214.
Peters B, Junker A, Brauer K, Muhlthaler B, Kostner D, Mientus M, et al. Deletion of pyruvate decarboxylase by a new method for efficient marker-less gene deletions in Gluconobacter oxydans. Appl Microbiol Biotechnol. 2013;97:2521–30.
Martinussen J, Hammer K. Cloning and characterization of upp, a gene encoding uracil phosphoribosyltransferase from Lactococcus lactis. J Bacteriol. 1994;176:6457–63.
Neuhard J. Pyrimidine nucleotide metabolism and pathways of thymidine triphosphate biosynthesis in Salmonella typhimurium. J Bacteriol. 1968;96:1519–27.
Danielsen S, Kilstrup M, Barilla K, Jochimsen B, Neuhard J. Characterization of the Escherichia coli codBA operon encoding cytosine permease and cytosine deaminase. Mol Microbiol. 1992;6:1335–44.
Dubeau MP, Ghinet MG, Jacques PE, Clermont N, Beaulieu C, Brzezinski R. Cytosine deaminase as a negative selection marker for gene disruption and replacement in the genus Streptomyces and other Actinobacteria. Appl Environ Microbiol. 2009;75:1211–4.
Maier AG, Braks JAM, Waters AP, Cowman AF. Negative selection using yeast cytosine deaminase/uracil phosphoribosyl transferase in Plasmodium falciparum for targeted gene deletion by double crossover recombination. Mol Biochem Parasitol. 2006;150:118–21.
Mullen CA, Kilstrup M, Blaese RM. Transfer of the bacterial gene for cytosine deaminase to mammalian-cells confers lethal sensitivity to 5-fluorocytosine—a negative selection system. Proc Natl Acad Sci USA. 1992;89:33–7.
Kostner D, Peters B, Mientus M, Liebl W, Ehrenreich A. Importance of codB for new codA-based marker-less gene deletion in Gluconobacter strains. Appl Microbiol Biotechnol. 2013;97:8341–9.
Kostner D, Rachinger M, Liebl W, Ehrenreich A. Marker-less deletion of putative alanine dehydrogenase genes in Bacillus licheniformis using a codBA-based counterselection technique. Microbiology. 2017;163:1532–9.
Li Q, Chen J, Minton NP, Zhang Y, Wen Z, Liu J, et al. CRISPR-based genome editing and expression control systems in Clostridium acetobutylicum and Clostridium beijerinckii. Biotechnol J. 2016;11:961–72.
Wang Y, Zhang ZT, Seo SO, Lynn P, Lu T, Jin YS, et al. Bacterial genome editing with CRISPR-Cas9: deletion, integration, single nucleotide modification, and desirable “clean” mutant selection in Clostridium beijerinckii as an example. ACS Synth Biol. 2016;5:721–32.
Wasels F, Jean-Marie J, Collas F, López-Contreras AM, Lopes Ferreira N. A two-plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum. J Microbiol Methods. 2017;140:5–11.
Yoo M, Croux C, Meynial-Salles I, Soucaille P. Metabolic flexibility of a butyrate pathway mutant of Clostridium acetobutylicum. Metab Eng. 2017;40:138–47.
Sharp PM, Emery LR, Zeng K. Forces that influence the evolution of codon bias. Philos Trans R Soc Lond B Biol Sci. 2010;365:1203–12.
Pósfai G, Kolisnychenko V, Bereczki Z, Blattner FR. Marker-less gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999;27:4409–15.
Yu BJ, Kang KH, Lee JH, Sung BH, Kim MS, Kim SC. Rapid and efficient construction of marker-less deletions in the Escherichia coli genome. Nucleic Acids Res. 2008;36:e84.
Minton NP, Ehsaan M, Humphreys CM, Little GT, Baker J, Henstra AM, et al. A roadmap for gene system development in Clostridium. Anaerobe. 2016;41:104–12.
Wiesenborn DP, Rudolph FB, Papoutsakis ET. Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl Environ Microbiol. 1988;54:2717–22.
Monot F, Martin JR, Petitdemange H, Gay R. Acetone and butanol production by Clostridium acetobutylicum in a synthetic medium. Appl Environ Microbiol. 1982;44:1318–24.
Heap JT, Pennington OJ, Cartman ST, Minton NP. A modular system for Clostridium shuttle plasmids. J Microbiol Meth. 2009;78:79–85.
Purdy D, O’Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, Minton NP. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol. 2002;46(2):439–52.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Woodbury: Cold Spring Harbor Laboratory Press; 1989.
Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40:e55.
Güssow D, Clackson T. Direct clone characterization from plaques and colonies by the polymerase chain reaction. Nucleic Acids Res. 1989;17:4000.
Bannam TL, Rood JI. Clostridium perfringens-Escherichia coli shuttle vectors that carry single antibiotic resistance determinants. Plasmid. 1993;29:233–5.
Dusséaux S, Croux C, Soucaille P, Meynial-Salles I. Metabolic engineering of Clostridium acetobutylicum ATCC 824 for the high-yield production of a biofuel composed of an isopropanol/butanol/ethanol mixture. Metab Eng. 2003;18:1–8.
Authors’ contributions
CNH, AE, and WL conceived the study; CNH performed all the experimental work. CHN drafted the manuscript together with AE and WL. All authors read and approved the final manuscript.
Acknowledgements
We thank Helga Gaenge for the technical support.
Competing interests
The authors declare that they have no competing interests
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Financial support from the European Commission (Collaborative FP7-KBBE Project Valor Plus, grant agreement No. 613802) is gratefully acknowledged.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1.
Minimal inhibitory concentration.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huang, CN., Liebl, W. & Ehrenreich, A. Restriction-deficient mutants and marker-less genomic modification for metabolic engineering of the solvent producer Clostridium saccharobutylicum. Biotechnol Biofuels 11, 264 (2018). https://doi.org/10.1186/s13068-018-1260-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-018-1260-3