
Abstract
Background
The development of biological processes that replace the existing petrochemical-based industry is one of the biggest challenges in biotechnology. Aspergillus niger is one of the main industrial producers of lignocellulolytic enzymes, which are used in the conversion of lignocellulosic feedstocks into fermentable sugars. Both the hydrolytic enzymes responsible for lignocellulose depolymerisation and the molecular mechanisms controlling their expression have been well described, but little is known about the transport systems for sugar uptake in A. niger. Understanding the transportome of A. niger is essential to achieve further improvements at strain and process design level. Therefore, this study aims to identify and classify A. niger sugar transporters, using newly developed tools for in silico and in vivo analysis of its membrane-associated proteome.
Results
In the present research work, a hidden Markov model (HMM), that shows a good performance in the identification and segmentation of functionally validated glucose transporters, was constructed. The model (HMMgluT) was used to analyse the A. niger membrane-associated proteome response to high and low glucose concentrations at a low pH. By combining the abundance patterns of the proteins found in the A. niger plasmalemma proteome with their HMMgluT scores, two new putative high-affinity glucose transporters, denoted MstG and MstH, were identified. MstG and MstH were functionally validated and biochemically characterised by heterologous expression in a S. cerevisiae glucose transport null mutant. They were shown to be a high-affinity glucose transporter (K m = 0.5 ± 0.04 mM) and a very high-affinity glucose transporter (K m = 0.06 ± 0.005 mM), respectively.
Conclusions
This study, focusing for the first time on the membrane-associated proteome of the industrially relevant organism A. niger, shows the global response of the transportome to the availability of different glucose concentrations. Analysis of the A. niger transportome with the newly developed HMMgluT showed to be an efficient approach for the identification and classification of new glucose transporters.
Similar content being viewed by others

Background
The development of biological systems for the industrial synthesis of biofuels and chemicals is a main objective of today’s biotechnology. For cost-effective production of valuable products, efficient microbial fermentations of non-food lignocellulosic material are essential. Filamentous fungi, in particular the fungus Aspergillus niger, play a prominent role in this field of biotechnology. A. niger is of significant industrial relevance and has been exploited as a production platform for both organic acids and hydrolytic enzymes [1]. It is an efficient degrader of the major plant cell wall polysaccharides cellulose, hemicellulose and pectin [2], and is one of the main industrial producers of commercial enzymes for plant biomass conversion due to its high enzyme secretory capacity [3]. In the last decades, its versatile arsenal of extracellular enzymes for lignocellulose depolymerisation [4], and the molecular mechanisms controlling their expression, have been well described [5–7]. However, while previous studies have revealed the existence of an array of uptake systems in this fungus [8–10], little is known about the identity and specificity of the transport systems involved in sugar uptake.
Most of the existing knowledge related to monosaccharide uptake in fungi originates from studies in the model yeast Saccharomyces cerevisiae. This yeast is able to transport and metabolise glucose, fructose, mannose and galactose. Transport of these simple sugars is mediated only through facilitated diffusion by the majority of the transporters from the Hxt family, composed of Hxt1–Hxt17, and Gal2. They belong to the sugar porter (SP) family [11], which is the largest subfamily of the major facilitator superfamily (MFS). Hxt1–Hxt4, Hxt6 and Hxt7 have been found to be able to support growth of yeast in glucose on their own, thus being considered the major hexose transporters in yeast. In addition to the Hxt family, three members of the maltose transporter family (Agt1, Mph2 and Mph3) are also able to transport glucose [12]. The individual characterisation of each of these transporters was possible using engineered S. cerevisiae strains, deleted for hxt1–7 [13] and hxt1–17, gal2, agt1, mph2 and mph 3 [12], as hosts for the functional validation and biochemical study of these proteins. These yeast mutant strains, unable to grow on glucose, fructose, mannose and galactose as a single carbon source, have also subsequently been used as tools for the functional characterisation of sugar transporters from other fungal species [14–21].
In contrast to S. cerevisiae, the only functionally validated sugar transporters in A. niger are the recently identified d-galacturonic acid transporter GatA [22, 23], two fructose transporters [10] and the high-affinity sugar/H+ symporter MstA [14]. Furthermore, transcriptional data for the A. niger mstC gene suggests that it encodes a low-affinity glucose transporter [9], but no experimental data supporting its role as a functional sugar transporter is publicly available. MFS proteins display a strong structural conservation [24], and structure-based profile hidden Markov models can be used to identify putative sugar transporters in the A. niger in silico proteome. To obtain a profile hidden Markov model (HMM), a multiple sequence alignment is turned into a position-dependent scoring system with segments of variable conservation levels and length [25]. As a result of the weighted scoring system, HMMs are less sensitive to changes in the non-conserved regions of a given protein family than more traditional methods based on shared primary sequence similarity alone, like e.g. the standard Blast algorithm. These changes include variability of the residues at a certain position as well as insertions and deletions. In this study, a profile hidden Markov model specific for glucose transporters (HMMgluT) was computed based on a structure-based multiple sequence alignment of 42 proteins with a known function related to glucose transport.
Genome information combined with transcriptome analyses of various growth conditions can give a good inventory of A. niger plasma membrane components with hypothetical sugar transporter functions. These data types, however, by definition do not take regulatory events at the posttranscriptional level into account, although these events can influence protein abundances and localisation. An inventory of the A. niger plasma membrane proteome at defined culture conditions can provide a more reliable source of information for the identification of the most important sugar transport components. To date, only few fungal plasmalemma (PM)-enriched proteomes have been reported, and none of them involved an industrially relevant filamentous fungus. The main focus of study has been on S. cerevisiae [26–28] and on several pathogenic fungi [29–31], as their PM represents a cellular component of substantial interest from a diagnostic and therapeutic point of view [29]. Thus, since the PM proteome of A. niger has not been a subject of study yet, it would be a first step towards a better understanding of its dynamics and topology.
Recently, our research group successfully used a shotgun proteomics approach to study protein secretion mechanisms in A. niger, allowing the characterisation of the secretory subproteome of the fungus and its changes in different conditions by using label-free LC–MS/MS [32, 33]. This is a powerful tool to analyse enriched organelle cell fractions, both in qualitative and quantitative terms [34], and thus permits the identification and quantification of the most relevant components of the A. niger cell membranes. In this work, the membrane-associated proteome of A. niger was studied for the identification of new glucose transporters, using newly developed experimental and complementary computational approaches.
Results and discussion
In silico transportome analysis and construction of a hidden Markov model specific for glucose transporters
As an integral part of the membrane, transporters must contain at least one protein domain that is thermodynamically stable in the hydrophobic environment of the phospholipid tails. In eukaryotes, these are typically α-helical structures. Transmembrane proteins can be predicted by applying a transmembrane hidden Markov model (tmHMM) that incorporates the hydrophobicity, charge bias, helix lengths and grammatical constraints of known transmembrane proteins into one model [35]. In addition, a comprehensive list of A. niger sugar transporters can be made by applying hidden Markov models for both the major facilitator superfamily (HMMMFS), and sugar porters (HMMSP). As the largest subfamily of the MFS, the sugar porter (SP) family currently has the most identified members [36], and as such provides a good basis for constructing a specific profile HMM from multiple alignments of extensively characterised members.
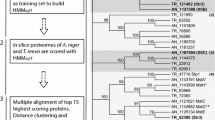
The experimental steps and complementary bioinformatics pipeline to identify and validate A. niger glucose transporters are depicted in Fig. 1. Initial in silico analysis of the theoretical A. niger proteome with the precomputed HMMMFS and HMMSP, obtained from the Pfam database [37], showed that more than 250 of the proteins predicted in A. niger ATCC1015 have a conserved domain architecture related to sugar transport (Table 1). Very similar results were obtained for the A. niger CBS 513.88 strain (not shown). This high abundance of sugar transporters makes A. niger a versatile host for the bioconversion of lignocellulosic biomass to products of interest, especially also in comparison to S. cerevisiae, which cannot metabolise as many sugars and consequently has fewer sugar transporters [11]. Although the HMMSP captures the conserved domain architecture of potential sugar porters, it cannot discriminate between different sugar substrates, and was thus considered to be too broad for the purpose of this study. Therefore, a list of 42 biochemically characterised glucose transporters from 10 different organisms was obtained from the UniProt database [38], and a profile HMM specific for glucose transporters (HMMgluT) was built from these sequences (see alignment in Additional file 1). The functions of proteins are often more conserved in their tertiary structure than in their primary amino acid sequence, as residues that are not crucial for the function of the protein will be subject to evolutionary changes over time. The typical structure of known glucose transporters comprises 12 transmembrane helix domains divided into 2 groups of 6 helixes [39, 40], and the HMMgluT was computed from a structure-based multiple sequence alignment incorporating transmembrane helix predictions, rather than using an alignment algorithm based on the primary amino acid sequence alone. Appropriate threshold values, above which a hit with the HMMgluT has to score in order to be considered a true positive, can be calculated by evaluating the properties of the HMMgluT. Receiver operating characteristic (ROC) curves are instrumental in assigning the best threshold values, since they display the trade-off between sensitivity and specificity. In order to obtain ROC curves, the HMMgluT was first validated using a 10 × 3-fold cross-validation approach (Fig. 2). Threshold values with the best trade-off between the true and false-positive rates of the prediction were determined from the resulting ROC curve. In this study, two thresholds were used. The first is d min, which is the point on the curve that has the minimal distance to [0 1] in the 2-dimensional x–y plane. Another way to determine the best trade-off point is by calculating the point in which the Matthews correlation coefficient is maximal (MCCmax), as the prediction is regarded as better the closer the MCC value is to 1, whereas a value of 0 is regarded as no better than a random guess. To compare the performance of this approach to a more traditional approach, in which Blast is used to identify homologous proteins, each of the 42 verified glucose transporters was used as query in a separate Blast search against the same dataset used to evaluate the HMMgluT. The resulting ROC curves for each of the methods is depicted in Additional file 2 and shows that, for the datasets used, an approach using HMMs outperforms Blast in discriminating glucose transporters from other sugar transporters.

Bioinformatic and proteomic profiling of the A. niger transportome. The flow chart outlines the complementary approach taken to identify putative glucose transporters. Experimental validation of the transporters identified was carried out in S. cerevisiae

Receiver operating characteristic (ROC) curve for the HMMgluT transporter model. Plotted is the mean of 30 runs of a 10 × 3-fold cross-validation. The confidence interval is shown in grey. Calculated inclusion thresholds, d min (white square) and MCCmax (black circle), are indicated
By using the previously calculated thresholds as lower limits for the HMMgluT score, the list of putative glucose transporters was effectively narrowed down to 19 sugar porters when the d min threshold was applied, and to only 5 sugar porters, including MstA, when the MCCmax threshold was applied (Table 1). Note that MstA was truly ‘discovered’ by the HMMgluT, since it was not a priori included in the training set. The average sensitivity, specificity, accuracy and inferred HMMgluT scoring values at the two threshold points, d min and MCCmax, are noted in Table 2. As expected, the specificity at the MCCmax threshold is very high, but it comes at the cost of having a lower sensitivity. The d min threshold on the other hand is less strict, thus allowing for a higher false-positive rate in order to not neglect any true positives.
The A. niger plasmalemma proteome in response to different glucose concentrations at low pH
A way to verify both the HMMgluT and the predicted glucose transporter candidates is by using an experimental setup, in which the abundance of transmembrane proteins in relevant conditions can be compared with their HMMgluT scores. In this complementary work, the focus was put on the study of the A. niger transportome in the presence of different concentrations of glucose at a pH which is both physiologically and biotechnologically relevant for this fungus. Mycelium of A. niger N400 was pre-grown for 18 h in minimal medium with 100 mM sorbitol as sole carbon source and subsequently transferred in equal amounts to controlled fermenters containing minimal medium with the following carbon source compositions: 100 mM sorbitol (reference condition), 100 mM sorbitol plus 1 mM glucose (low-glucose condition) and 100 mM sorbitol plus 60 mM glucose (high-glucose condition). The initial pH of these cultures was set at pH 4.0, which corresponded with the final pH measured at the pre-growth stage. Immediately after inoculation the pH of the fermenter cultures started to drop, reaching pH 3.5, in all cases, after 50–60 min. For the remainder of the experiment this was kept constant at pH 3.5. High-resolution analysis of the sugar content in the culture medium sampled 2 h after inoculation showed that there was sorbitol consumption in the reference condition; sorbitol and glucose consumption in the low-glucose condition; and glucose but no sorbitol consumption in the high-glucose condition. This result confirmed, as reported [41], that the organism strongly favours consumption of good carbon sources like glucose, over poorer carbon sources, such as sorbitol, even when the latter is also present at a high concentration (100 mM). The 2-h time-point was selected for membrane-associated protein analysis in all three conditions.
Isolation of fractions enriched for cell membranes was performed using a protocol similar to the one developed by Oliveira and co-workers [32]. This protocol involves the previously described workflow: cell disruption, crude organelle separation, and subsequent enrichment [42]. After several differential centrifugation steps, a pellet containing crude low-density organelles (P3) was obtained and subjected to density gradient centrifugation, yielding a set of five fractions (P3A–P3E). The PM marker vanadate-sensitive H+ ATPase and the mitochondrial membrane cytochrome c oxidase activities were then measured in the initial cell-free extract, the P3 pellet and the P3A to P3E fractions derived from it. Compared to the cell-free extract, the P3 pellet was shown to be 2.4–3.2 times enriched in plasma membranes. No further enhanced PM enrichment was found in the P3A to P3E fractions; however, cytochrome c oxidase activity was higher in the P3A to P3E fractions when compared to P3. Since mitochondrial membranes were not the main focus of this research, the P3 pellets, considered to be more optimal for the analysis of plasmalemma proteins, were further processed and subjected to shotgun proteomics analysis (detailed information regarding subcellular fractionation, marker enzyme assays and sample preparation for LC–MS/MS can be found in the “Methods”).
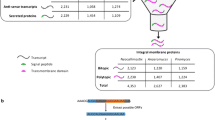
The LC–MS/MS spectra obtained were processed as described in the “Methods”. In total, 833 proteins were identified, of which 432 were present in all three conditions, 72 proteins were found only in the reference condition, 34 only in the low-glucose condition, and 106 proteins were found exclusively in the high-glucose condition. Of the proteins identified, almost 30 % had one or more predicted tmHMM domains, indicative of integral membrane proteins (Additional file 3). The relative abundances of the proteins containing at least one tmHMM domain, which add up to 15.28, 17.50 and 15.45 % of the total protein isolated in the reference, low- and high-glucose conditions, respectively, can be found in Additional file 4. Proteins associated with the mitochondrial membrane were most abundant, followed by the proteins associated with the plasma membrane. However, the plasma membrane-associated fraction consists of a higher number of different proteins than the mitochondrial membrane-associated fraction, with 86 and 35 identified proteins, respectively. Proteins associated with the membrane-bound endoplasmic reticulum (ER) constitute the third most abundant group of the list, with a total of 48 different proteins identified. Finally, proteins linked to other membrane-associated organelles, comprising the endomembrane system and membrane-bound organelles, such as the Golgi apparatus, vacuoles and lysosomes, account for <1 % of the total protein isolated (Additional file 5). In all three conditions, approximately one-third of the proteins that have one or more tmHMM domains are annotated as proteins with a transporter function. This fraction, denoted as the A. niger transportome, accounted for 3.9, 4.4 and 4.1 % of the total protein isolated in the reference, low- and high-glucose conditions, respectively. Mitochondrial carrier proteins are prevalent in all three conditions, followed by ATPases. Proteins with the MFS architecture, which are the main interest in this study, comprise the third most abundant group of the isolated A. niger transportome. Proteins (putatively) involved in the secretory pathway and amino acid transport were found in relative abundances of <0.5 % of the total protein isolated. Other transporters, comprising (putative) oligopeptide transporters, formate/nitrite transporters, ammonium transporters, iron permeases, Na+/solute symporters, ABC transporters, inorganic phosphate transporters, nucleotide-sugar transporters, major intrinsic proteins and the translocation protein Sec62, accounted for ≤0.01 % of the total protein isolated, respectively.
Performance of the HMMgluT for the identification of candidate glucose transporters
The A. niger transportome was further analysed for the presence of glucose transporters. Table 3 summarises the results of the bioinformatics analysis, which was performed in the same way as previously for the entire A. niger in silico proteome, i.e. the data were first queried with the hidden Markov models specific for proteins containing a MFS or SP domain. As already observed for the A. niger in silico proteome, a search with the HMMSP lowered the number of proteins that scored above the inclusion threshold in comparison to the search performed with the less specific HMMMFS. Querying the transportome with the newly developed HMMgluT, using the previously calculated thresholds, lowered the number of putative, albeit highly probable, glucose transporters to 4, 3, and 2 promising hits in the reference, low- and high-glucose conditions, respectively.
The relative abundances of the identified MFS proteins that scored above the default inclusion threshold of HMMgluT, HMMSP and HMMMFS, ordered by their HMMgluT scores, can be found in Table 4. High HMM scores corresponded to MFS porters that have been related to glucose uptake in previous studies, or novel MFS porters that, in the present study, showed abundance patterns that point towards a possible role as glucose transporter. In Fig. 3a, the proteins found in the three conditions are highlighted on the HMM curves for the glucose and general sugar transporter model depicted. The colour coding corresponds to their relative abundances in the three conditions (see “Methods” for details). The HMM curves as such show the scores of all proteins in the theoretical A. niger ATCC1015 proteome that hit above the default inclusion threshold of both HMMgluT and HMMSP. The majority of proteins that were more abundant in the low-glucose condition relative to both the reference and high-glucose condition cluster closer to the d min and MCCmax thresholds, whereas the proteins that were more abundant in the high-glucose condition scored overall lower on the HMMgluT. This indicates that the model is better at detecting high-affinity glucose transporters, which might be due to the sequences that were selected to build the HMMgluT (Additional file 1). A close-up of the top scoring proteins is depicted in Fig. 3b. The highest score was obtained with protein ID 1121621, which is the putative low-affinity glucose transporter MstC. Studies on its transcriptional regulation during the exponential growth phase in batch fermentation and in chemostat cultures, carried out at different dilution rates, have been performed by Jørgensen et al. [9]. The mstC transcripts were only detected during the batch fermentation phase, indicating that mstC expression is associated with higher glucose concentrations or with specific growth rates. In the present work, the protein abundance of the MstC protein was found to be similar in all the three conditions studied. This result, together with the previous findings of Jørgensen et al., suggests that the presence of MstC is independent of the glucose concentration. The second best hit, with protein ID 1142882, and the fourth best hit, with protein ID 1143598, are yet-uncharacterised transport proteins, henceforth denoted as MstG and MstH, respectively. Their high HMMgluT scores and overall abundance pattern in the three experimental conditions strongly indicate a possible role for them as glucose transporters. MstG and MstH were found with relatively high abundance levels only in the two non-carbon catabolite-repressing conditions studied, being higher in abundance when low glucose concentrations were present. This type of abundance pattern fits that of high-affinity glucose transporters described in S. cerevisiae and Aspergillus species [14, 43, 44], that are preferably expressed in the presence of low concentrations of glucose, poor carbon (de-repressing) sources and also in starvation conditions. Another protein with protein ID 1125134, scoring in between MstG and MstH in the HMMgluT was not observed in the three studied conditions. It is denoted as MstE (An03g02190) in the UniProt database, and has been found to be expressed in germinating spores [45]. The fifth highest scoring hit, with protein ID 1143191, is MstA. The mstA coding gene is transcriptionally controlled by the carbon catabolite repressor CreA and the environmental pH regulator PacC. Its transcript levels were found to be higher during carbon starvation at pH 6.0 [14]. Van Kuyk et al. also observed low expression levels at pH 4.0 and 8.0, and in the presence of repressing glucose concentrations at pH 6.0 [14]. In the present study, MstA was detected in very low abundance; however, only in the de-repressing reference condition, supporting previous results that suggested a limited role of this transporter when the environmental pH is low. The combined results of the specific protein abundance patterns and the high HMMgluT scores indicate that MstG and MstH could have a role as high/mid-affinity glucose transporters, therefore both transporters were selected for further characterisation.

HMMSP and HMMgluT scores of MFS porter proteins. a HMM scores for the theoretical ATCC1015 proteome, with HMMgluT = black dotted line and HMMSP = orange dotted line. Horizontal lines indicate the HMMgluT thresholds d min (dashed) and MCCmax (solid). MFS porter proteins found in the three conditions applied are indicated with filled circles and colour-coded according to their relative abundance patterns. b Close-up of the proteins that scored above the d min (dashed line) and MCCmax (solid line) thresholds. The galacturonic acid transporter GatA (indicated with a grey triangle and not found in the conditions applied) is added as a reference. With HMMgluT, the Mst transporters are well separated from GatA, whereas the more general HMMSP does not discriminate the putative glucose transporters from the galacturonic acid transporter
Transcriptional analysis of mstG and mstH in the presence of different carbon sources
In order to obtain additional insights in the regulation and possible biological role of MstG and MstH, transcriptional levels were analysed in mycelium samples from cultures containing: minimal medium with 100 mM sorbitol (used as a reference condition) and minimal medium plus glucose, fructose or mannose at high (55 mM) or low (5 mM) concentration. Samples taken 2 h after mycelium transfer were processed and RT-qPCR analysis was performed. Both genes were found to be expressed in all the studied conditions, but their expression levels were clearly influenced by the different culture conditions (Fig. 4). The relative transcript levels of both genes in the reference, low-glucose and high-glucose conditions showed a pattern comparable to what was observed at proteomic level; mstG and mstH were upregulated in the presence of a low glucose concentration and downregulated when the glucose concentration was high. However, mstG and mstH expression patterns showed to be divergent in the presence of high and low concentrations of fructose and mannose. With these carbon sources mstG expression levels were similar to those observed with a low concentration of glucose. In the case of mstH, as observed in the presence of glucose 55 mM, high concentrations of fructose (55 mM) and mannose (55 mM) also led to a decrease of its transcript levels when compared to the reference condition. While these results do not provide conclusive evidence that MstG is indeed a high-affinity glucose transporter they are not in disagreement with its possible role either. In the case of mstH, only the glucose 5 mM condition enhanced its expression, suggesting that MstH could have a specific role in A. niger when low concentrations of the monosaccharide are available.

Transcriptional analysis of mstG and mstH. Mycelium of the A. niger strain N400 was precultured on minimal medium with sorbitol 100 mM and thereafter transferred to minimal medium with the following carbon sources: sorbitol 100 mM, glucose 5 mM, glucose 55 mM, fructose 5 mM, fructose 55 mM, mannose 5 mM and mannose 55 mM. Samples from the seven culture conditions were taken 2 h after mycelium transfer, and expression analyses of mstG and mstH were performed by qPCR using a histone-like gene (gene ID 207921) transcript for normalisation. Results are given as relative transcript rations in logarithmic scale (lg). The values provided in the figures are means of two biological replicates. Transcript levels always refer to the reference sample (sorbitol 100 mM), indicated with an asterisk
Functional validation of the A. niger sugar transporters MstG and MstH
As discussed above, the high HMMgluT scores, the protein abundance patterns and transcriptional analyses of the MFS porters MstG and MstH point towards a role in glucose uptake. In order to test their functionality, the engineered S. cerevisiae strain EBY.VW.4000 [12], a glucose transporter null mutant unable to grow on glucose, mannose, galactose or fructose as carbon source, was chosen as host for functional complementation analysis. The yeast strain was transformed with the 2μ expression plasmid p426HXT7-6His-mstG or p426HXT7-6His-mstH, containing the respective cDNA under control of the constitutive promoter HXT7p and the terminator CYC1t. Single colony transformants were isolated from minimal medium agar plates containing 2 % maltose and the ability of both genes to restore growth of the EBY.VW.4000 transformant strain in the presence of different monosaccharides was studied. Tenfold serial dilutions of exponentially growing cells from at least two different transformants expressing each gene were spotted on different minimal medium plates supplemented with 1 % (w/v) of the following carbon sources: glucose, galactose, fructose, mannose, sucrose and maltose (Fig. 5). The mstG transformants were able to grow on glucose, galactose, mannose and sucrose as single carbon sources, whereas mstH transformants grew on glucose, sucrose, mannose, galactose and, in contrast to mstG strains also on fructose. MstH transformants also showed better growth on mannose and sucrose, but poorer growth on galactose. Regarding the sucrose transport by both transporters, since the EBY.VW4000 strain encodes an extracellular invertase [18], it is unknown if the transported substrate was sucrose or glucose in the case of MstG, and sucrose, glucose or fructose in the case of MstH. These results indicated that MstG and MstH are functional sugar transporters with the ability to transport a variety of substrates.

MstG and MstH functional analysis. Growth of strain EBY.VW4000 containing the mstG gene (mstG +), mstH gene (mstH +), or harbouring the empty expression vector p426HXT7-6His (EV) in minimal medium agar plates containing the following sugars at a final concentration of 1 % (w/v): maltose, glucose, sucrose, mannose, fructose and galactose. Agar plates were incubated at 30 °C for 96 h. All transformants expressing each gene showed the same growth pattern; therefore, the figure depicts only one transformant per transporter as representative
In order to have better insights about the affinity for glucose of both transporters, the growth rates of the transformants expressing MstG and MstH were studied in the presence of different glucose concentrations. Additional file 6 shows growth curves and glucose consumption figures of the transformant strains grown on minimal medium with glucose 2.5, 10 and 50 mM. Both transporters showed to be functional in all glucose concentrations. However, when comparing individual growth curves and glucose uptake, some differences could be observed. The growth rate of the MstG transformant during the exponential growth phase was comparable in the three different conditions, while in case of the MstH transformant, at the highest glucose concentration the growth rate was reduced. Accordingly, the glucose consumption rate of the MstG transformant in the 50 mM condition was much faster, being able to deplete the monosaccharide after around 40 h of growth, whereas in the same time-span the MstH transformant was only able to consume a 60 % of the total amount suggesting that MstG and MstH are glucose transporters with different affinities for the sugar.
To determine MstG and MstH transport characteristics, (14C) glucose transport assays were performed with the transformant strains as described [46]. Initial glucose uptake rates at various substrate concentrations were fitted to the Michaelis–Menten model with non-competitive substrate inhibition and used to estimate the appropriate kinetic parameters as described [47]. MstG was confirmed to transport glucose with apparent K m values of 0.5 ± 0.04 mM, and V max of 5.8 ± 0.04 nmol min−1 mg DW−1 (Fig. 6). Its affinity for glucose was lower than the one reported for A. niger MstA with a K m value of 0.025 ± 0.01 mM. However, MstG can still be classified as a high-affinity glucose transporter, since its K m value is lower than the one reported for the S. cerevisiae high-affinity hexose transporter HXT6 (K m value 1.4 ± 0.1 mM) [47]. Initially MstH kinetics were determined using the same range of substrate concentrations as for MstG. The results obtained suggested that MstH had a much higher affinity for glucose than MstG. Therefore, the assay was optimised and repeated using a micro-molar range of glucose concentrations. MstH was confirmed to transport glucose with apparent K m values of 0.06 ± 0.005 mM and a V max of 1.3 ± 0.2 nmol min−1 mg DW−1 (Fig. 6), similar to what has been reported for A. niger MstA.

Uptake of 14C-labelled glucose by the yeast hexose transporter null mutant EBY.VW4000 expressing mstG or mstH as a function of glucose concentration. a, b Diamonds represent measured uptake rates of 14C labelled glucose (n = 3) as a result of expression of mstG (a) or mstH (b). Black lines represent least-squares-fitted Michaelis–Menten model (mstG; K m = 0.5 mM; V max = 5.8 nmol min−1 mg DW−1; mstH: K m = 0.06 mM, and a V max = 1.3 nmol min−1 mg DW−1). Insert results for mstH using micro-molar substrate concentrations. c Uptake rate of 1 mM 14C-labelled glucose by EBY.VW4000 expressing mstG (filled boxes) or mstH (open boxes) in the presence of a tenfold excess of competing sugars
Since MstG and MstH were able to restore growth of the hexose non-transporting EBY.VW4000 on other monosaccharides as well, their substrate specificity was evaluated by determining glucose relative transport levels in the presence of a tenfold excess of various competing sugars (Fig. 6). Both transporters clearly showed higher specificities for glucose. However, besides glucose, galactose, fructose and xylose were able to inhibit MstG glucose transport by 39, 29 and 25 %, respectively; whereas, in the MstH transformant mannose and fructose reduced the uptake of labelled glucose by 70 and 50 %, respectively. A. niger MstA also showed a preference for glucose, while being able to transport other monosaccharides as well [14]. Similarly, substrate specificity of Aspergillus nidulans hexose transporters HxtB and HxtC have been assayed [20], and a reduction of glucose uptake of 70–80 % was observed for galactose, fructose and mannose. In accordance to the functioning of many glucose transporters from other filamentous fungi, the function of MstG and MstH was found to be energy dependent via proton symport. This was confirmed by measurements of glucose initial uptake rate in the presence of CCCP that resulted in a 60 ± 8 % (n = 3) and 64 ± 6 % (n = 3) reduction of labelled glucose uptake by MstG and MstH, respectively.
Conclusions
In this study, two complementary approaches were used to both increase the understanding of the A. niger membrane-associated proteome, and to identify novel glucose transporters. In a purely in silico approach, a hidden Markov model (HMMgluT) was constructed for the identification of glucose transporters. HMMgluT performed well in the identification and segmentation of functionally validated glucose transporters. In a complementary in vivo approach, defined culture conditions were applied to study the response of the A. niger membrane-associated proteome to different glucose concentrations. To the best of our knowledge, this is the first study on the membrane-associated proteome of the industrially relevant fungus A. niger. The study provided a better understanding of the membrane composition and topology, especially with respect to proteins with a putative transporter function; the A. niger transportome. Analysis of the A. niger in vivo transportome with HMMgluT was shown to be effective; by combining the abundance patterns of the proteins identified in the experimental conditions with their respective HMMgluT scores, two new putative glucose transporters, MstG and MstH, were identified. They were functionally validated in an engineered yeast strain with a monosaccharide transporter null-background and confirmed to be high-affinity transporters for glucose.
Methods
In silico transportome analysis and construction of a hidden Markov model specific for glucose transporters
The A. niger ATCC1015 and S. cerevisiae CEN.PK proteomes plus their annotations, which were used for the in silico analysis, were obtained from the JGI database [48]. For A. niger ATCC1015, only the best model proteome was used. Transmembrane helix domains were predicted with a standalone version of the TMHMM tool from the TMHMM website [35]. Hidden Markov models for the major facilitator superfamily (HMMMFS) and sugar porter (HMMSP) domains were obtained from the Pfam database [37].
The protein sequences used to build the HMMgluT were obtained by entering the search term “sugar transporter” in the uniprot database and downloading the resultant xml and canonical fasta files. The xml file was parsed for proteins that were not experimentally verified to have the biological function assigned to them. The remaining proteins were divided into two separate datasets; the first dataset contained only the proteins with an experimentally verified GO term related to glucose transport (GO:0005355 and GO:0015758); the second dataset contained the remaining proteins (core dataset). The protein sequences of the experimentally verified glucose transporters were aligned by accessing the PRALINE structural alignment tool [49] via the SOAP interface and using the following settings: BLOSUM62, PSI-BLAST pre-profile processing (Homology extended alignment, 3 iterations), structural features: DSSP-defined secondary structure search, PSIPRED and TMHMM, fasta outputfile. The aligned fasta output was converted to the Stockholm format, and the HMMgluT was built from the resultant output.sto file using the “hmmbuild” command from HMMER v3.0 [50]. A. niger MstA was excluded from the multiple alignment.
For the threefold cross-validation, the initial dataset, containing the 42 verified glucose transporters, was randomly partitioned into three equally sized subsets. The model was then built from every combination of two subsets, while the validation of the model was carried out on the core dataset plus the third subset of the glucose transporters not used for the model building process. This allowed for the determination of false and true negatives in comparison to false and true positives, and thus gave an estimate of the prediction power of the model. The threefold cross-validation was repeated ten times, each time with different random subsets of the verified glucose transporters. Two thresholds were determined from the average ROC curve resulting from the 10 × 3-fold cross-validation of the HMMgluT:
1. Minimal distance to the point [0 1] (d min):
With s n = sensitivity (TPR) and sp = specificity (TNR).
Another metric to calculate the optimal trade-off point between true and false predictions is the Matthews correlation coefficient. The Matthews correlation coefficient is essentially a one number representation of the confusion matrix, and is thus suitable to compare the outcome of different model predictions. This coefficient is calculated as follows:
2. Matthews correlation coefficient (MCC):
Here, a value closer to 1 is clearly a better prediction, and thus the inferred HMMgluT score at the highest MCC value calculated along the ROC curve, MCCmax, was set as second threshold.
Similarly, the Blast approach was evaluated by using each of the 42 verified glucose transporters as separate query for a local Blastp search against the core dataset plus the other verified glucose transporters, allowing for the determination of true and false-positive rates (see Additional file 2).
Strains and growth conditions
Escherichia coli DH5α (endA1, hsdR17, gyrA96, thi-1, relA1, supE44, recA1, Δ lacU169 (Φ80 lacZΔM15)), grown at 37 °C, was used for cloning experiments and plasmid propagation. Luria broth (LB) was used as growth medium (1 % w/v tryptone, 0.5 % w/v yeast extract, 1 % w/v NaCl) with or without 100 μg mL−1 ampicillin.
The wild-type strain A. niger N400 (CBS 120.49), used for the plasma membrane proteomics analysis, was grown at 30 °C on complete medium plates for spores generation and maintenance [51]. Mycelial biomass of the N400 strain was obtained after 18 h of growth in liquid cultures containing minimal medium [51], including 5 g L−1 of yeast extract, with 100 mM sorbitol as carbon source. Equal amounts of water-rinsed mycelium were transferred to 1-L benchtop fermenters (Sartorius) with 750 mL of minimal medium containing 4.50 g NaNO3, 1.13 g KH2PO4, 0.38 g KCl, 0.38 g MgSO4·7 H2O and 750 µL of Vishniac solution [51, 52]. Three different conditions, varying on the carbon source composition, were studied: sorbitol 100 mM (reference condition), sorbitol 100 mM plus glucose 1 mM (low-glucose condition) and sorbitol 100 mM plus glucose 60 mM (high-glucose condition). Two biological replicates per condition were studied. Fermenters were stirred at 1000 rpm and aerated with filtered air (0.6 L min−1), keeping oxygen levels over 60 %. The initial pH, set at 4.0, was allowed to drop until pH 3.5 and kept constant afterwards by sodium hydroxide addition.
Mycelium samples for RT-qPCR analysis were obtained from a mycelium transfer experiment similar to the one described above, using minimal medium with the following carbon source compositions: 100 mM sorbitol (reference condition), glucose 5 mM, glucose 55 mM, fructose 5 mM, fructose 55 mM, mannose 5 mM and mannose 55 mM. Two biological replicates per condition were studied as well. Cultures were performed in Erlenmeyer flasks, stirred at 200 rpm and the initial pH of the medium in all conditions was set at 4.0. Two hours after inoculation mycelium samples were taken and quickly washed, dried with a single-use towel, snap-frozen with liquid nitrogen and stored at −80 °C until further processing.
The S. cerevisiae strain EBY.VW4000 (CEN.PK2-1C hxt13Δ::loxP; hxt15::ΔloxP; hxt16Δ::loxP; hxt14Δ::loxP; hxt12Δ::loxP; hxt9Δ::loxP; hxt11Δ::loxP; hxt10Δ::loxP; hxt8Δ::loxP; hxt514Δ::loxP; hxt2Δ::loxP; hxt367Δ::loxP; gal2Δ; stl1Δ::loxP; agt1::loxP; ydl247wΔ::loxP; yjr160cΔ::loxP), used for the functional complementation experiments and the characterisation of glucose transporters [12], was grown at 30 °C and maintained in solid complete medium containing 10 g L−1 of yeast extract, 20 g L−1 of peptone and 20 g L−1 of maltose. The EBY.VW4000-derived strains obtained in the present study were grown in liquid minimal medium containing 6.7 g L−1 of yeast nitrogen base with ammonium sulphate (Difco), 20 g L−1 of maltose, supplemented with leucine (30 mg L−1), tryptophan (20 mg L−1) and histidine (20 mg L−1).
A. niger membrane-associated protein purification and quality control analysis
Aspergillus niger mycelium samples (2–3 g, press-dried), washed with a 20 mM HEPES buffer pH 7.6 containing 150 mM NaCl, and resuspended in the same solution containing 1 % (v/v) protease inhibitor cocktail for yeast and fungi (Sigma), were mechanically disrupted using a French press (8000 psi). Cell-free extracts were centrifuged for 5 min at low speed (500g), in order to remove unbroken cells and pellet debris. The supernatants were then centrifuged during 20 min at medium speed (5000g), to pellet and remove remaining heavy organelles. The remaining supernatants were centrifuged for 120 min at high speed (~85,000g), to pellet light organelles (P3).
P3 pellets were resuspended using a Dounce homogenizer in 1 mL of a 20 mM HEPES buffer pH 7.6 containing 250 mM sucrose. P3 suspensions were overlayed in a discontinuous sucrose density gradient, prepared by layering successive decreasing sucrose densities solutions (6 × 1 mL), with concentrations ranging from 1.20 to 0.70 M, upon one another. Sucrose density gradients were centrifuged (~100,000g, 60 min) to isolate different membrane-associated fractions from P3 pellet. Five fractions were obtained (P3A, P3B, P3C, P3D and P3E).
Sample preparation for LC–MS/MS
The protein content of enriched plasma membrane-associated fractions was determined using the BCA protein assay. Membrane proteins were solubilised by mixing volumes of each fraction containing 25 μg of protein with equal volumes of a 2× solution of 20 mM HEPES pH 7.6 containing 1 M 6-aminocaproic acid and 10 g L−1 of n-dodecyl-beta-d-maltoside. Cell membrane–detergent mixes were incubated in a thermoblock (ThermoMixer) for 1 h at 20 °C and vigorous stirring (1000 rpm). Afterwards, samples were sonicated in a water bath for 15 min, and finally they were centrifuged at 22,000g for 30 min. Supernatants containing solubilised proteins were concentrated using MMicrocon YM-10 columns (cutoff, 10 kDa; Millipore, Eschborn, Germany) and loaded into a 12 % SDS–polyacrylamide gel, which was run until the loaded samples entered into the gel. The gel was stained according to the manufacturer’s instructions using Page Blue staining (Fermentas) and rinsed with ultrapure water. Each sample-gel lane was cut into one slice (approx. 1 cm2), carefully sliced into smaller pieces of about 1 mm3 and transferred into microcentrifuge tubes. Samples were destained and equilibrated through three washing steps using the following solutions: 50 mM ammonium bicarbonate (ABC) (incubated 5 min), ABC/acetonitrile (1:1, v/v) (incubated 5 min) and neat acetonitrile (incubated 5 min). These washing steps were successively repeated two times. The gel samples were then swelled in 10 mM dithiothreitol (DTT) for 20 min at 56 °C to reduce protein disulfide bonds. Subsequently, the DTT solutions were removed and samples were alkylated with 50 mM 2-chloroacetamide in ABC, for 20 min, at room temperature, in the dark. The 2-chloroacetamide solutions were removed, and samples were again washed twice with: neat acetonitrile (incubated 5 min), ABC (incubated 5 min) and neat acetonitrile (incubated 5 min). Approximately 150 μL of digestion buffer, containing sequencing grade modified trypsin (12.5 ng μL−1) (Promega, Madison, WI, USA) in ABC, was added to each sample, making sure that all gel pieces were kept wet during digestion (adding, if necessary, additional ABC solution). Protein samples were digested overnight at 37 °C. Peptide digestion products were extracted by adding 50 μL of 2 % trifluoroacetic acid (TFA), followed by an incubation step in a thermoblock (ThermoMixer) for 20 min, at room temperature and vigorous stirring (1400 rpm). Gel pieces were then subjected to 20 s sonication in a water bath, centrifuged and supernatants were transferred to new tubes. The peptide extraction step was then repeated once by washing the gel pieces with buffer B (80 % acetonitrile, 0.1 % formic acid) followed by the mentioned incubation and sonication steps. Supernatants from both extractions were pooled and samples were placed in a vacuum centrifuge for acetonitrile evaporation until 20–40 μL were left. Finally, samples were acidified by addition of TFA (1:1, v/v) and peptide clean-up procedure, prior to LC–MS/MS analysis, was performed using the “STop And Go Extraction” procedure as described before [53].
Mass spectrometric measurements
LC–MS/MS analysis was performed at Radboud Proteomics Centre as described previously [54]. Measurements were performed by nanoflow reversed-phase C18 liquid chromatography (EASY nLC, Thermo Scientific) coupled online to a 7-Tesla linear ion trap Fourier-Transform ion cyclotron resonance mass spectrometer (LTQ FT Ultra, Thermo Scientific).
Proteomics data analysis
The LC–MS/MS spectra obtained from the proteomics experiment were identified and quantified using the maxQuant software [55]. The peptides were mapped against the annotated A. niger ATCC1015 in silico proteome obtained from the JGI database (http://genome.jgi-psf.org/Aspni7/Aspni7.home.html) using the default settings of the maxQuant version 1.3.0.5 [variable modifications: oxidation (M) and acetylation (protein N-term); enzyme used: trypsin/P; fixed modifications carbamidomethyl (cys)], except for the variables affecting the label-free quantification. For this, the multiplicity was set to 1, and the parameters for label-free quantification as well as the iBAQ and peak property calculations were selected. Only proteins with two or more unique peptide hits were considered for further analysis. Protein localisation was determined using the softberry protComp prediction server (http://linux1.softberry.com). The relative abundances of the identified proteins are represented as follows:
where A L is the relative abundance of the protein in the low-glucose condition; A H, relative abundance of the protein in the high-glucose condition; A S, relative abundance of the protein in the reference (sorbitol) condition.
Transcriptional analysis of mstG and mstH genes
Mycelium samples were disrupted with glass beads in a Fastprep-24 instrument, and RNA extraction was performed by a Maxwell 16 instrument using the Maxwell 16 LEV simplyRNA kit (Promega). Reverse transcription and qPCR analysis were performed following the protocols and instruments described in Mach-Aigner et al. [56]. The previously described histone-like gene “hist” transcript (gene ID 207921) was used as reference for normalisation of the expression data [56]. The following sequences belong to the primers used for qPCR analysis in this study: hist-FW, ACAATGACTGGCCGTGGAAAGG; hist-RV, ATACGCTTGACACCACCACGAC; mstG-FW, CGGTGGTGGTATGGCTTTCT; mstG-RV, GTTCTCAGGCACACCGTACA; mstH-FW, GCCATCATGATCGGTCTGTTTGTC, mstH-RV, ACTGATGGTTCCGGTGTCATATCC.
Construction of S. cerevisiae EBY.WV4000 transformants expressing A. niger mstG and mstH genes and glucose uptake assays
The coding sequence of the genes mstG and mstH, digested with SpeI and XhoI were cloned on the S. cerevisiae expression vector p426HXT7-6His, previously linearised with SpeI and XhoI under the control of the constitutive promoter HXT7p and the terminator CYC1t. Transformation of S. cerevisiae EBY.WV4000 with p426HXT7-6His (empty vector), p426HXT7-6His_mstG and p426HXT7-6His_mstH was performed as described [57].
Uptake assays were performed as described with minor adjustments [46]. 5 mL of Synthetic Complete medium without uracil (SC-ura; 0.67 % YNB Difco + dropout supplement mix without uracil Sigma-Aldrich) with 2 % maltose as a carbon source was inoculated from plate with EBY.VW4000 with pRS426H7 (control) and EBY.VW4000 with pRS426H7_mstG or pRS426H7_mstH and incubated overnight (30 °C, 225 rpm) as pre-inoculum. Pre-inoculum was transferred to 500 mL SC-ura with 2 % maltose and incubated for 24 h. Cells were harvested by centrifugation (4000g, 10 min) and washed with 50 mL ice-cold MQ. Cells were then resuspended in 2 mL ice-cold SC-ura without carbon source, divided in 40-μL aliquots and kept on ice.
Aliquots were incubated for 5 min at 30 °C before uptake assay was started. To start the reaction, 10 μL of a five times concentrated labelled glucose solution (d-[U-14C]-glucose, Campro Scientific) was added. After exactly 10 s the reaction was stopped by the addition of 1 mL of 100 mM LiCl and vacuum filtration (0.45 μm HV filters, 1225 sampling manifold, Millipore), with subsequent washing with 2 × 5 mL of ice-cold 100 mM LiCl. After 5 min of drying in the vacuum manifold, the filters were transferred to scintillation vials with 7.5 mL scintillation liquid (Ultima Gold, Perkin Elmer) and activity was counted (Packard Tricarb 1600TR). All reactions were performed in triplicates. For each reaction, a negative control assay without incubation was performed.
To determine transport kinetics, uptake of labelled glucose with a range of concentrations from 1 μM to 10 mM was measured in SC-ura at pH 5.0. Glucose solutions with an activity of approximately 700–70,000 Bq were used. To determine kinetic parameters K m and V max, the data were fitted to the following Michaelis–Menten model with substrate inhibition using the least-squares method.
To determine substrate specificity, the uptake of 1 mM of labelled glucose was measured in the presence of 10 mM of a competing carbon source. d-glucose, d-sorbitol, d-xylose, d-mannose, l-rhamnose, l-arabinose, d-fructose and d-galactose were selected as competing carbon sources.
To determine if the transporter functions via proton symport, activity was measured while uncoupling the proton gradient by carbonyl cyanide m-chlorophenyl hydrazine (CCCP). This inhibitor is dissolved in DMSO and added before temperature equilibration of the cells. The uptake rate in the presence of 250 μM of CCCP and 2 % DMSO was compared with the uptake rate in the presence of only 2 % DMSO.
Abbreviations
- HMM:
-
hidden Markov model
- MFS:
-
major facilitator superfamily
- SP:
-
sugar porter
- Hxt:
-
hexose transporter
- PM:
-
plasmalemma
- tmHMM:
-
transmembrane hidden Markov model
- ROC:
-
receiver operating characteristic
- MCC:
-
Matthews correlation coefficient
- d :
-
distance
- ER:
-
endoplasmic reticulum
- Sorb:
-
sorbitol
- Glu:
-
glucose
- Fruc:
-
fructose
- Man:
-
mannose
- CCCP:
-
carbonyl cyanide m-chlorophenyl hydrazine
- GO:
-
gene ontology
- LB:
-
Luria broth
- ABC:
-
ammonium bicarbonate
- TmD:
-
transmembrane domains
- protID:
-
protein identity
- rel. ab:
-
relative abundance
- sd:
-
standard deviation
- O.D.:
-
optical density
References
Andersen MR, Salazar MP, Schaap PJ, Van De Vondervoort PJI, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, Van Dijck PWM, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, Van Ooyen AJJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A et al (2011) Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21:885–897
De Souza WR, Maitan-alfenas GP, de Gouvêa PF, Brown NA, Savoldi M, Battaglia E, Goldman MHS, De Vries RP, Goldman GH (2013) The influence of Aspergillus niger transcription factors AraR and XlnR in the gene expression during growth in d-xylose, l-arabinose and steam-exploded sugarcane bagasse. Fungal Genet Biol 60:29–45
Tamayo-Ramos J, Orejas M (2014) Enhanced glycosyl hydrolase production in Aspergillus nidulans using transcription factor engineering approaches. Biotechnol Biofuels 7:103
De Vries RP, Visser J (2001) Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol Mol Biol Rev 65:497–522
Van Peij NN, Gielkens MM, de Vries RP, Visser J, de Graaff LH (1998) The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol 64:3615–3619
Battaglia E, Hansen SF, Leendertse A, Madrid S, Mulder H, Nikolaev I, de Vries RP (2011) Regulation of pentose utilisation by AraR, but not XlnR, differs in Aspergillus nidulans and Aspergillus niger. Appl Microbiol Biotechnol 91:387–397
Gruben BS, Zhou M, Wiebenga A, Ballering J, Overkamp KM, Punt PJ, De Vries RP (2014) Aspergillus niger RhaR, a regulator involved in l-rhamnose release and catabolism. Appl Microbiol Biotechnol 98:5531–5540
Torres NV, RiolCimas JM, Wolschek M, Kubicek CP (1996) Glucose transport by Aspergillus niger: the low affinity carrier is only formed during growth on high glucose concentrations. Appl Microbiol Biotechnol 44:790–794
Jørgensen TR, vanKuyk PA, Poulsen BR, Ruijter GJG, Visser J, Iversen JJL (2007) Glucose uptake and growth of glucose-limited chemostat cultures of Aspergillus niger and a disruptant lacking MstA, a high-affinity glucose transporter. Microbiology 153:1963–1973
Coelho MA, Gonçalves C, Sampaio JP, Gonçalves P (2013) Extensive intra-kingdom horizontal gene transfer converging on a fungal fructose transporter gene. PLoS Genet 9:e1003587
Leandro MJ, Fonseca C, Gonçalves P (2009) Hexose and pentose transport in ascomycetous yeasts: an overview. FEMS Yeast Res 9:511–525
Wieczorke R, Krampe S, Weierstall T, Freidel K, Hollenberg CP, Boles E (1999) Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett 464:123–128
Reifenberger E, Freidel K, Ciriacy M (1995) Identification of novel HXT genes in Saccharomyces cerevisiae reveals the impact of individual hexose transporters on glycolytic flux. Mol Microbiol 16:157–167
Vankuyk PA, Diderich JA, MacCabe AP, Hererro O, Ruijter GJG, Visser J (2004) Aspergillus niger mstA encodes a high-affinity sugar/H+ symporter which is regulated in response to extracellular pH. Biochem J 379(Pt 2):375–383
Polidori E, Ceccaroli P, Saltarelli R, Guescini M, Menotta M, Agostini D, Palma F, Stocchi V (2007) Hexose uptake in the plant symbiotic ascomycete Tuber borchii Vittadini: biochemical features and expression pattern of the transporter TBHXT1. Fungal Genet Biol 44:187–198
Saloheimo A, Rauta J, Stasyk OV, Sibirny AA, Penttilä M, Ruohonen L (2007) Xylose transport studies with xylose-utilizing Saccharomyces cerevisiae strains expressing heterologous and homologous permeases. Appl Microbiol Biotechnol 74:1041–1052
Du J, Li S, Zhao H (2010) Discovery and characterization of novel d-xylose-specific transporters from Neurospora crassa and Pichia stipitis. Mol BioSyst 6:2150–2156
Wahl R, Wippel K, Goos S, Kämper J, Sauer N (2010) A novel high-affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis. PLoS Biol 8:e1000303
Leandro MJ, Sychrová H, Prista C, Loureiro-Dias MC (2013) ZrFsy1, a high-affinity fructose/H+ symporter from fructophilic yeast Zygosaccharomyces rouxii. PLoS One 8:e68165
Dos Reis TF, Menino JF, Bom VLP, Brown NA, Colabardini AC, Savoldi M, Goldman MHS, Rodrigues F, Goldman GH (2013) Identification of glucose transporters in Aspergillus nidulans. PLoS One 8:e81412
Colabardini AC, Nicolas L, Ries A, Brown NA, Fernanda T, Savoldi M, Goldman MHS, Menino JF, Rodrigues F, Goldman GH, Ries LNA, Dos Reis TF (2014) Functional characterization of a xylose transporter in Aspergillus nidulans. Biotechnol Biofuels 7:46
Sloothaak J, Schilders M, Schaap PJ, de Graaff LH (2014) Overexpression of the Aspergillus niger GatA transporter leads to preferential use of d-galacturonic acid over d-xylose. AMB Express 4:66
Martens-Uzunova ES, Schaap PJ (2008) An evolutionary conserved d-galacturonic acid metabolic pathway operates across filamentous fungi capable of pectin degradation. Fungal Genet Biol 45:1449–1457
Vardy E, Arkin IT, Gottschalk KE, Kaback HR, Schuldiner S (2004) Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci 13:1832–1840
Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:755–763
Kim H, Melén K, Osterberg M, von Heijne G (2006) A global topology map of the Saccharomyces cerevisiae membrane proteome. Proc Natl Acad Sci USA 103:11142–11147
Delom F, Szponarski W, Sommerer N, Boyer JC, Bruneau JM, Rossignol M, Gibrat R (2006) The plasma membrane proteome of Saccharomyces cerevisiae and its response to the antifungal calcofluor. Proteomics 6:3029–3039
Szopinska A, Degand H, Hochstenbach J-F, Nader J, Morsomme P (2011) Rapid response of the yeast plasma membrane proteome to salt stress. Mol Cell Proteom 10(M111):009589
Cabezón V, Llama-Palacios A, Nombela C, Monteoliva L, Gil C (2009) Analysis of Candida albicans plasma membrane proteome. Proteomics 9:4770–4786
Ouyang H, Luo Y, Zhang L, Li Y, Jin C (2010) Proteome analysis of Aspergillus fumigatus total membrane proteins identifies proteins associated with the glycoconjugates and cell wall biosynthesis using 2D LC–MS/MS. Mol Biotechnol 44:177–189
Rogers PD, Vermitsky JP, Edlind TD, Hilliard GM (2006) Proteomic analysis of experimentally induced azole resistance in Candida glabrata. J Antimicrob Chemother 58:434–438
De Ferreira Oliveira JMP, Van Passel MWJ, Schaap PJ, De Graaff LH (2010) Shotgun proteomics of Aspergillus niger microsomes upon d-xylose inductions. Appl Environ Microbiol 76:4421–4429
Ferreira de Oliveira JMP, van Passel MWJ, Schaap PJ, de Graaff LH (2011) Proteomic analysis of the secretory response of Aspergillus niger to d-maltose and d-xylose. PLoS One 6(6):e20865. doi:10.1371/journal.pone.0020865
Patel VJ, Thalassinos K, Slade SE, Connolly JB, Crombie A, Murrell JC, Scrivens JH (2009) A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J Proteome Res 8:3752–3759
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Pao SS, Paulsen IT, Saier MH (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucleic Acids Res 40(Database issue):D290–D301
UniProt Consortium (2014) UniProt: a hub for protein information. Nucleic Acids Res 43(Database issue):D204–D212
Zeng H, Parthasarathy R, Rampal AL, Jung CY (1996) Proposed structure of putative glucose channel in GLUT1 facilitative glucose transporter. Biophys J 70:14–21
Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125
Strauss J, Horvath HK, Abdallah BM, Kindermann J, Mach RL, Kubicek CP (1999) The function of CreA, the carbon catabolite repressor of Aspergillus nidulans, is regulated at the transcriptional and post-transcriptional level. Mol Microbiol 32:169–178
De Oliveira JMPF, de Graaff LH (2011) Proteomics of industrial fungi: trends and insights for biotechnology. Appl Microbiol Biotechnol 89:225–237
Ozcan S, Johnston M (1999) Function and regulation of yeast hexose transporters. Microbiol Mol Biol Rev 63:554–569
Forment JV, Flipphi M, Ventura L, González R, Ramón D, MacCabe AP (2014) High-affinity glucose transport in Aspergillus nidulans is mediated by the products of two related but differentially expressed genes. PLoS One 9:e94662
Van Leeuwen MR, Krijgsheld P, Wyatt TT, Golovina EA, Menke H, Dekker A, Stark J, Stam H, Bleichrodt R, Wösten HAB, Dijksterhuis J (2013) The effect of natamycin on the transcriptome of conidia of Aspergillus niger. Stud Mycol 74:71–85
Walsh MC, Smits HP, Scholte M, van Dam K (1994) Affinity of glucose transport in Saccharomyces cerevisiae is modulated during growth on glucose. J Bacteriol 176:953–958
Maier A, Völker B, Boles E, Fuhrmann GF (2002) Characterisation of glucose transport in Saccharomyces cerevisiae with plasma membrane vesicles (countertransport) and intact cells (initial uptake) with single Hxt1, Hxt2, Hxt3, Hxt4, Hxt6, Hxt7 or Gal2 transporters. FEMS Yeast Res 2:539–550
Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, Smirnova T, Grigoriev IV, Dubchak I (2014) The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res 42((Database issue)):D26–D31
Simossis VA, Heringa J (2005) PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res 33(Web Server issue):W289–W294
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39(Web Server issue):W29–W37
Pontecorvo G, Roper JA, Chemmons LM, Macdonald KD, Bufton AWJ (1953) The genetics of Aspergillus nidulans. Adv Genet 5:141–238
Vishniac W, Santer M (1957) The thiobacilli. Bacteriol Rev 21:195–213
Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75:663–670
Rajala N, Hensen F, Wessels HJCT, Ives D, Gloerich J, Spelbrink JN (2015) Whole cell formaldehyde cross-linking simplifies purification of mitochondrial nucleoids and associated proteins involved in mitochondrial gene expression. PLoS One 10:e0116726
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372
Mach-Aigner AR, Omony J, Jovanovic B, van Boxtel AJB, de Graaff LH (2012) d-Xylose concentration-dependent hydrolase expression profiles and the function of CreA and XlnR in Aspergillus niger. Appl Environ Microbiol 78:3145–3155
Gietz RD, Woods RA (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96
Authors’ contributions
LHG, PJS and JATR conceived and designed the study. PJS, VAPMS and JATR supervised and coordinated the study. JS, DIO and JATR developed methodologies, and collected and analysed the data. JS and JATR performed the laboratory experiments and DIO performed the in silico analysis. DIO, JS, PJS and JATR drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work has been carried out on the basis of a Grant in the framework of the BE-BASIC program F01.011 Transport processes in the production of organic acids by Aspergillus niger. We would like to thank Dr Eckhard Boles for providing us the S. cerevisiae strain EBY.VW4000 and the plasmid p426HXT7-6His and Adrie Westphal for his suggestions on the enzyme kinetics.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
J Sloothaak, DI Odoni contributed equally to this work
Additional files
13068_2015_317_MOESM3_ESM.pdf
Additional file 3. Number of proteins identified in the 3 different conditions studied in the A. niger membrane-associated proteome analysis.
13068_2015_317_MOESM4_ESM.pdf
Additional file 4. Relative abundances of proteins from the A. niger membrane-associated proteome with at least one tmHMM domain.
13068_2015_317_MOESM5_ESM.pdf
Additional file 5. Relative abundances of identified A. niger proteins grouped according to their location within the cell.
13068_2015_317_MOESM6_ESM.pdf
Additional file 6. Growth of S. cerevisiae EBY.VW4000 expressing the A. niger mstG and mstH genes, at different glucose concentrations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sloothaak, J., Odoni, D.I., de Graaff, L.H. et al. Aspergillus niger membrane-associated proteome analysis for the identification of glucose transporters. Biotechnol Biofuels 8, 150 (2015). https://doi.org/10.1186/s13068-015-0317-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-015-0317-9