
Abstract
Etoricoxib (ETO), Paracetamol (PCM), and two toxic impurities for Paracetamol impurity K (4-aminophenol (PAP)) and impurity E (para-hydroxy acetophenone (PHA)) were separated using a simple and selective HPLC method that was tested for the first time. PCM is a commonly used analgesic and antipyretic medication that has recently been incorporated into COVID-19 supportive treatment. Pharmaceuticals containing PCM in combination with other analgesic-antipyretic drugs like ETO help to improve patient compliance. The studied drugs and impurities were separated on a GL Sciences Inertsil ODS-3 (250 × 4.6) mm, 5.0 µm column, and linear gradient elution was performed using 50 mM potassium dihydrogen phosphate adjusted to pH 4.0 with ortho-phosphoric acid and acetonitrile as mobile phase at 2.0 mL/min flow rate at 25 °C and UV detection at 220 nm. The linearity range was 1.5–30.0 µg/mL for ETO and PCM while 0.5–10.0 µg/mL for PAP and PHA, with correlation coefficients (r) for ETO, PCM, PAP, and PHA of 0.9999, 0.9993, 0.9996, and 0.9998, respectively. The proposed method could be used well for routine analysis in quality control laboratory.
Similar content being viewed by others

Introduction
The global spread of COVID-19 has resulted in an unprecedented disaster. In the early stages, the most common symptom is fever due to the onset of a COVID-19-mediated cytokine storm. NSAIDs are among the most extensively used medicines owing to their efficacy in reducing pain and inflammation and their inclusion in the WHO's Model List of Essential Medicine [1].
Paracetamol (PCM) and Etoricoxib (ETO) are anti-inflammatory, analgesic-antipyretic drugs [2] and approved to treat COVID symptoms. PCM is an N-(4-hydroxyphenyl) acetamide [3]. PCM is a popular over-the-counter analgesic for mild to moderate pain, such as headaches, musculoskeletal discomfort, and fever [4], and has recently been used in the adjunctive treatment of COVID-19 as it acts as an antipyretic for fever resulting from infection with COVID-19 [5,6,7].
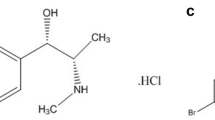
ETO is 5 chloro-6’- methyl-3- [-4- (methyl sulfonyl) phenyl]-2,3’- bipyridine. It is a COX-2-selective NSAID drug with a better COX-1 to COX-2 selectivity ratio than other COX-2-selective NSAIDs [8]. It is also used to treat osteoarthritis, rheumatoid arthritis, gouty arthritis, chronic low back pain, and acute pain [9], and to treat COVID-19 by suppressing cytokine storm [10]. Figure 1A and B show the structures of PCM and ETO.

Structure of a Etoricoxib, b Paracetamol, c 4-aminophenol and d Para-hydroxy acetophenone
During the manufacture of Active Pharmaceutical Ingredients (APIs), some of the starting materials, intermediates, reagents, and reaction by-products end up in the final products as impurities [11].
The presence of impurity in a drug product is mostly a quality issue, as it may compromise the drug's efficacy and may cause potential adverse effects on patients [12].
Each impurity found in a pharmaceutical product must be extensively inspected, both qualitatively and quantitatively, and undergo toxicological testing if necessary [13]. Accordingly, it is necessary to develop a technique for determining pharmaceuticals as well as their impurities at minute levels that may be harmful and have toxic effects.
4-aminophenol (impurity K) (Para aminophenol) is the most common co-existing impurity of PCM in medicinal formulations arising from either degradation or synthesis [14]. 1-(4-hydroxyphenyl) ethanone (impurity E) (para-hydroxy acetophenone) is an impurity of PCM, according to British pharmacopeia [15]. Figure 1C and D show the structures of PAP and PHA.
Several methods, like HPLC [16,17,18,19,20,21,22,23,24,25,26], HPTLC [27], spectrophotometric [28,29,30,31], chemmometric method [32,33,34], and electrochemical methods [35,36,37,38], have been reported for the assay of PCM. Several methods have been reported for the assay of ETO, including HPLC methods [39,40,41,42,43,44], HPTLC [45], spectrophotometric method [46], LC–MS/MS method [47], and ion-selective electrode method [48].
Both PCM and ETO were determined by several reported methods, such as HPLC [49,50,51,52,53], HPTLC [54], and spectrophotometric methods [55]. The published methods disregarded the possible impact of PCM impurities such as PHA (hepatotoxic) and PAP (nephrotoxic and teratogenic toxicity), despite the toxicity of these impurities. Also, according to the literature, there is no chromatographic method for determination of PCM and ETO in the presence of PCM impurities.
This work aims to develop, optimize, and validate a simple, sensitive, and selective RP-HPLC technique to be the first method for the simultaneous determination of PCM, ETO, and PCM potential impurities in bulk material and their pharmaceutical formulation.
Experimental
Instrumentation
HPLC SHIMADZU/LC-2030C 3D Plus with vacuum degasser and column GL Sciences Inertsil ODS-3 (250 × 4.6) mm, 5.0 µm. A photodiode Array Detector (PDA), a quaternary pump, a 100 µL (μL) loop, as well as an autosampler injector were employed. The LabSolutions software (v5.90/2017, Shimadzu Corporation, Kyoto, Japan) was used to record and evaluate the data. An electronic balance (Vibra, Japan), a 0.45 µm nylon membrane filter (Millipore, Ireland), an ultrasonic power sonicator (Model 410), and a pH meter (Jenway 3510, UK) were used.
Chemicals and reagents
Pure standard
Pure standard ETO and PCM were donated by SIGMA Pharmaceutical Industries, Cairo, Egypt, and according to the company's analytical certificate, the purity of ETO and PCM was 99.5% and 99.94%, respectively. PAP and PHA were purchased from Sigma-Aldrich, and the purity of PAP and PHA was certified to be 99.73% and 99.61%, respectively.
Reagents
All chemicals and solvents employed in this experiment were of analytical grade. HPLC-grade acetonitrile and methanol were purchased from Sigma-Aldrich, Belgium. Potassium dihydrogen phosphate was supplied by El-NASR Pharmaceutical Chemical Co. (Abu-Zabaal, Cairo, Egypt). Double-distilled water (Otsuka Pharmaceutical Co., Cairo, Egypt) was used. Phosphate buffer (pH = 4) was prepared by dissolving about 6.8 gm of potassium dihydrogen phosphate in 1000 mL of doubled distilled water; the pH was adjusted using orthophosphoric acid (Sigma-Aldrich, Switzerland).
Pharmaceutical dosage form
Intacoxia-P® tablet (Batch no: 5/UA/2017) was purchased from Aagya Biotech Pvt Ltd (Manglaur Roorkee, Uttarakhand, India). Intacoxia-P® tablet is labelled to contain ETO 60 mg and 325 mg of PCM per tablet.
Stock and working standard solutions
In four separate 25 mL volumetric flasks, 25 mg of ETO, PCM, PAP, and PHA were accurately weighed before being dissolved in 15 mL of methanol and sonicated for 10 min. The volume was raised to the mark with methanol, yielding a final concentration of 1.0 mg/mL. Then, from their standard stock solutions, 5, 5, 1.3, and 1.3 mL of ETO, PCM, PAP, and PHA, respectively, were transferred into four separate 50 mL volumetric flasks and the volume was raised to the mark with methanol to obtain standard working solutions of 100, 100, 26, and 26 µg/mL for ETO, PCM, PAP, and PHA, respectively.
Procedure
Chromatographic conditions
The components were separated using a GL Sciences Inertsil ODS-3 column (250 × 4.6) mm, 5.0 µm with a linear gradient elution of 50 mM potassium dihydrogen phosphate adjusted to pH 4.0 with ortho-phosphoric acid (solvent A) and acetonitrile (solvent B). The applied gradient program is shown in Table 1, solvents were filtered through a 0.45 µm Millipore membrane filter and degassed ultrasonically for 15 min before being injected into the HPLC system at a flow rate of 2.0 mL/min. The PDA detector's wavelength was 220 nm, and all chromatographic separations were performed at room temperature (25 ± 2 °C).
Construction of calibration graphs
Into 10 mL volumetric flasks, aliquots of standard working solution were diluted with the mobile phase (50:50 v/v, phosphate buffer: acetonitrile) to achieve concentrations ranging from 1.5 to 30 μg/mL for ETO and PCM and 0.5‒10 μg/mL for PAP and PHA. Diluted standard solutions of varying concentrations were subsequently injected by auto-sampler (80 µL volume) in triplicates into the HPLC system and chromatographed under the above-mentioned chromatographic conditions. Using a PDA detector, chromatographic peaks were obtained at 220 nm.
Analysis of tablet formulation
Ten tablets were weighed to determine the mean weight, then they were finely ground. The weight of the crushed powder corresponding to 18.5 mg of ETO and 100 mg of PCM was accurately weighed out and dissolved in 50 mL of the mobile phase (50:50 v/v, phosphate buffer: acetonitrile) in a 100 mL volumetric flask. After 15 min of sonication, the volume was completed to the mark with the same solvent. After that, the solution was filtered via a 0.45 µm membrane filter, yielding an initial stock solution claimed to contain 0.18 mg/mL of ETO and 1.0 mg/mL of PCM. The obtained solution was further diluted using the mobile phase to reach the final concentration 1.8 µg/mL and 10 µg/mL for ETO and PCM respectively, then injected in triplicate. The separation was accomplished using the chromatographic conditions described above. The concentrations of the listed drugs were determined using the calculated regression equations. We applied a standard addition technique to further evaluate the suggested method's accuracy.
Method validation
The proposed method was validated in accordance to ICH guidelines [56].
Results and discussion
The current work intends to develop and validate the first chromatographic method for determining PCM and ETO simultaneously as well as PCM toxic impurities.
Method development and optimization
The chromatographic settings were optimized to produce a good resolution of the investigated components with sharp symmetric peaks in a short run time.
Following an examination of different solvent compositions, methanol was chosen for preparing the stock and working standard solutions since all standards displayed acceptable solubility at the examined concentrations. Components of interest were separated using the suggested HPLC gradient elution method combined with UV detection at 220 nm. Figure 2 shows typical chromatograms of well-defined symmetrical peaks for PAP, PCM, PHA, and ETO mixtures.

HPLC chromatogram of 4-aminophenol (10 μg/mL, tR: 1.728 min), paracetamol (30 μg/mL, tR: 3.413 min), para-hydroxy acetophenone (10 μg/mL, tR: 5.003 min), and Etoricoxib (30 μg/mL, tR: 7.275 min) using a linear gradient elution of solvent A (50 mM of potassium dihydrogen phosphate, adjusted to pH 4.0 with ortho-phosphoric acid) and solvent B (acetonitrile) as a mobile phase at a flow rate of 2.0 mL/min at 220 nm
Various experimental factors which affect separation were studied, including:
Choosing a suitable wavelength
We adjusted the PDA detector at several wavelengths to find the optimal one regarding the sensitivity and peak shape of the components under study, and all compounds had reasonable UV absorption at 220 nm. As a result, the 220 nm wavelength was selected for the investigation and quantification of ETO, PCM, and impurities of PCM.
Selection of the column
The HPLC column was chosen after trying several packing materials. C8, and C18 columns such as GL Sciences Inertsil ODS-3 column (250 × 4.6) mm, 5.0 µm and Kinetex C8 (4.6 × 100 mm, 5 µm; Phenomenex, USA), were tried. Employing a C8 column, some of the components (polar components like PAP and PCM) were retained in the column with a long separation time, which was most likely due to the high polarity of the C8 column. The separation was enhanced and the best results with excellent sharp peaks were obtained by utilizing a GL Sciences Inertsil ODS-3 column (250 × 4.6) mm, 5.0 µm instead of C8 columns. Furthermore, the influence of column temperatures ranging from 25 to 40 °C was examined. However, no enhancement in terms of analysis time was found upon increasing column temperature due to the low viscosity of the mobile phase. As a result, no significant variations in retention times were seen over the temperature range investigated. Finally, the temperature was kept at 25 °C.
Selection of mobile phase
The examined substances show a significant difference in lipophilicity (log P) of 0.47, 0.51, 1.23, and 2.79 for PAP, PCM, PHA, and ETO, respectively.
At the beginning of the study, isocratic elution was used to separate the four components using varied ratios of water/methanol, water/ethanol, and water/acetonitrile as mobile phases.
In these trials, there was either inadequate separation or overlapping peaks, particularly PCM and PAP peaks, as their polarity is nearly the same, while ETO took more time to be separated. So, we shifted to gradient elution with the same mobile phases, acetonitrile was more suitable for the separation of the studied components than methanol and ethanol, where acetonitrile is less polar than methanol and ethanol. In addition, the UV cut-off of acetonitrile (190 nm) is less than methanol (210 nm) and ethanol (210 nm).
Buffer was tried instead of water and pH was varied from 2 to 8, using phosphate buffer, pH 4.0 resulted in an obvious improvement giving sharp peak shapes and excellent separation of the mixture simultaneously with symmetric peaks in short run time.
Finally, a linear gradient elution using 50 mM potassium dihydrogen phosphate adjusted to pH 4.0 with ortho-phosphoric acid (solvent A) and acetonitrile (solvent B) was conducted as described in Table 1.
The optimal resolution, unambiguous baseline separation with adequate retention times, and symmetric peaks of the investigated drugs were obtained. UV detection was carried out at 220 nm. A good resolution was achieved with retention times at tR of 1.728 ± 0.01, 3.413 ± 0.01, 5.003 ± 0.01, 7.275 ± 0.01 min for PAP, PCM, PHA, and ETO, respectively, as shown in Fig. 2.
Effect of flow rate
Different flow rates of the mobile phase were tried, ranging from 1.0 to 2.0 mL/min, to separate the analytes’ peaks from the mobile phase. The best flow rate for the effective elution of the drugs was 2.0 mL/min, giving better peak shape and a shorter retention time for all analytes while keeping acceptable peak resolution.
System suitability testing
To assess the performance of the operating system for the required separation, Table 2 shows the results of system suitability parameters, and satisfactory results were obtained according to USP pharmacopoeia [57], demonstrating perfect baseline separation of the separated peaks and high selectivity of the suggested method.
Robustness
The robustness of the HPLC technique was evaluated by analyzing the influence of slight modifications on the chromatographic conditions, such as the percentage of buffer ± 1% in the mobile phase components, the flow rate of the mobile phase (2.0 ± 0.1 mL/min), and pH (4 ± 0.2). Even minor changes to the perfect conditions had no discernible effect on retention times, tailing factor, and resolution of the examined components, which was proved by the low %RSD values, indicating the reliability of the proposed method during routine use (Table 3).
Method validation
The proposed method was validated in accordance to ICH guidelines [56], as shown in Table 4.
The suggested method's calibration curves were generated to illustrate the relationship between the mean peak area and the corresponding concentration in range of 1.5–30 µg/mL for ETO and PCM and 0.5–10 µg/mL for PAP and PHA, as shown in Fig. 3. The regression equations have been computed and results are shown in Table 4.

Calibration curve of a Etoricoxib, b paracetamol, c 4-aminophenol and d para-hydroxy acetophenone
The accuracy of the proposed method was assessed across the required range by analyzing three different concentrations of pure samples in triplicate, and the mean of the percentage recoveries ± SD was calculated (100.21 ± 0.973, 100.58 ± 0.858, 98.17 ± 1.362, and 99.32 ± 1.163) for ETO, PCM, PAP, and PHA, respectively, confirming accuracy of the method as shown in Table 4.
Repeatability and intermediate precision were also investigated, three different concentration levels within the specified range, either within the same day or on three successive days to investigate intra-day and inter-day precision, respectively. As proven by the low %RSD values, the suggested analytical procedure yielded data with acceptable precision (Table 4).
Limit of detection (LOD) and limit of quantitation (LOQ) were calculated for ETO, PCM, PAP, and PHA based on the standard deviation of the intercept (SD) and the slope obtained from the calibration curves of each component. The low LOD values (0.304, 0.397, 0.113, and 0.062) were obtained, demonstrating the proposed method's great sensitivity for ETO, PCM, PAP, and PHA, respectively, as shown in Table 4.
Assay of pharmaceuticals dosage form
The proposed method successfully determined ETO and PCM in Intacoxia-P tablets. The standard addition technique was employed to validate the proposed method for determining ETO and PCM selectively in the presence of formulation excipients and additives, and good results were obtained (Table 5; Fig. 4).

HPLC chromatogram of sample solution (dosage form)
Statistical comparison
Results obtained from the analysis of pure ETO and PCM were statistically compared with those obtained by the reported HPLC method [49]. The comparison revealed that the calculated F and student’s t-test values are less than the tabulated ones, revealing no significant difference between the proposed and reported method as shown in Table 6.
The proposed method was compared to the other published methods [49,50,51,52,53] and the findings showed that the proposed method was more sensitive than the published ones. The proposed method excels the published ones as it separates and quantifies PCM toxic impurities as well as PCM and ETO mixtures, which was not published before, as shown in Table 7.
Conclusion
A validated, robust, precise, accurate, and selective gradient RP-HPLC method was used to determine PCM and ETO in pharmaceutical preparations without interference from PCM impurities. The methodology was validated in agreement with the ICH recommendations. The results show that using a linear gradient system with respect to the mobile phase allows the separation of studied drugs and impurities with high resolution and relatively short analysis time. The proposed method was shown to be suitable for use in quality control laboratories for determining PCM in pure form or pharmaceutical dosage forms with ETO.
Availability of data and materials
Datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Shah S, Mehta V. Controversies and advances in non-steroidal anti-inflammatory drug (NSAID) analgesia in chronic pain management. Postgrad Med J. 2012;88(1036):73–8.
Patrignani P, Capone ML, Tacconelli S. Clinical pharmacology of etoricoxib: a novel selectiveCOX2 inhibitor. Expert Opin Pharmacother. 2003;4(2):265–84.
Merck NJ. In The Merck index. Royal Society of Chemistry: London, p. 47.
Gordon RM: Pain/Burning Sensation on Back. Case Studies in Geriatric Primary Care & Multimorbidity Management-E-Book 2019. p. 316.
Pandolfi S, Simonetti V, Ricevuti G, Chirumbolo S. Paracetamol in the home treatment of early COVID-19 symptoms: A possible foe rather than a friend for elderly patients? J Med Virol. 2021;93(10):5704.
Pergolizzi JV, Varrassi G, Magnusson P, LeQuang JA, Paladini A, Taylor R, Wollmuth C, Breve F, Christo P. COVID-19 and NSAIDS: a narrative review of knowns and unknowns. Pain Ther. 2020;9:353–8.
Casalino G, Monaco G, Di Sarro PP, David A, Scialdone A. Coronavirus disease 2019 presenting with conjunctivitis as the first symptom. Eye. 2020;34(7):1235–6.
Cochrane DJ, Jarvis B, Keating GM. Etoricoxib. Drugs. 2002;62(18):2637–51.
Dallob A, Hawkey CJ, Greenberg H, Wight N, De Schepper P, Waldman S, Wong P, DeTora L, Gertz B, Agrawal N. Characterization of etoricoxib, a novel, selective COX-2 inhibitor. J Clin Pharmacol. 2003;43(6):573–85.
Wang R. Etoricoxib may inhibit cytokine storm to treat COVID-19. Med Hypotheses. 2021;150: 110557.
Bolt HM, Foth H, Hengstler JG, Degen GH. Carcinogenicity categorization of chemicals—new aspects to be considered in a European perspective. Toxicol Lett. 2004;151(1):29–41.
de Oliveira Melo SR, Homem-de-Mello M, Silveira D, Simeoni LA. Advice on degradation products in pharmaceuticals: a toxicological evaluation. PDA J Pharm Sci Technol. 2014;68(3):221–38.
Bundgaard H, De Weck A. The role of amino-reactive impurities in acetylsalicylic acid allergy. Int Arch Allergy Immunol. 1975;49(1–2):119–24.
Dejaegher B, Bloomfield MS, Smeyers-Verbeke J, Vander Heyden Y. Validation of a fluorimetric assay for 4-aminophenol in paracetamol formulations. Talanta. 2008;75(1):258–65.
British Pharmacopeia, vol. II, 6th edition edn. London: Stationary Office Medicines and Health Care Products Regulatory Agencies; 2009.
Salih ME, Aqel A, Abdulkhair BY, Alothman ZA, Abdulaziz MA, Badjah-Hadj-Ahmed AY. Simultaneous determination of paracetamol and chlorzoxazone in their combined pharmaceutical formulations by reversed-phase capillary liquid chromatography using a polymethacrylate monolithic column. J Chromatogr Sci. 2018;56(9):819–27.
Ibrahim H, Hamdy AM, Merey HA, Saad AS. Simultaneous determination of paracetamol, propyphenazone and caffeine in presence of paracetamol impurities using dual-mode gradient HPLC and TLC densitometry methods. J Chromatogr Sci. 2021;59(2):140–7.
Borahan T, Unutkan T, Şahin A, Bakırdere S. A rapid and sensitive reversed phase-HPLC method for simultaneous determination of ibuprofen and paracetamol in drug samples and their behaviors in simulated gastric conditions. J Sep Sci. 2019;42(3):678–83.
Pereira FJ, Rodríguez-Cordero A, López R, Robles LC, Aller AJ. Development and validation of an RP-HPLC-PDA method for determination of paracetamol, caffeine and tramadol hydrochloride in pharmaceutical formulations. Pharmaceuticals. 2021;14(5):466.
Ibrahim H, Hamdy AM, Merey HA, Saad AS. Dual-mode gradient HPLC and TLC densitometry methods for the simultaneous determination of paracetamol and methionine in the presence of paracetamol impurities. J AOAC Int. 2021;104(4):975–82.
Farid NF, Abdelhamid NS, Naguib IA, Anwar BH, Magdy MA. Quantitative determination of anti-migraine quaternary mixture in presence of p-aminophenol and 4-chloroacetanilide. J Chromatogr Sci. 2022;60(6):538–44.
El-Yazbi AF, Guirguis KM, Bedair MM, Belal TS. Simultaneous quantitation of paracetamol and lornoxicam in the presence of five related substances and toxic impurities by a Selective HPLC–DAD Method. J AOAC Int. 2022;105(4):972–8.
El-Yazbi AF, Guirguis KM, Bedair MM, Belal TS. Validated specific HPLC-DAD method for simultaneous estimation of paracetamol and chlorzoxazone in the presence of five of their degradation products and toxic impurities. Drug Dev Ind Pharm. 2020;46(11):1853–61.
Abdelwahab NS, Abdelrahman MM, Boshra JM, Taha AA. Different stability-indicating chromatographic methods for specific determination of paracetamol, dantrolene sodium, their toxic impurities and degradation products. Biomed Chromatogr. 2019;33(9): e4598.
Călinescu O, Badea IA, Vlădescu L, Meltzer V, Pincu E. HPLC separation of acetaminophen and its impurities using a mixed-mode reversed-phase/cation exchange stationary phase. J Chromatogr Sci. 2012;50(4):335–42.
Belal T, Awad T, Clark CR. Stability-indicating simultaneous determination of paracetamol and three of its related substances using a direct GC/MS method. J AOAC Int. 2009;92(6):1622–30.
Abdelaleem EA, Naguib IA, Hassan ES, Ali NW. HPTLC and RP-HPLC methods for simultaneous determination of Paracetamol and Pamabrom in presence of their potential impurities. J Pharm Biomed Anal. 2015;114:22–7.
Pasha C. Determination of paracetamol in pharmaceutical samples by spectrophotometric method. Eclética Química. 2020;45(3):37–46.
Farid JF, Mostafa NM, Fayez YM, Essam HM, ElTanany BM. Chemometric quality assessment of Paracetamol and Phenylephrine Hydrochloride with Paracetamol impurities; comparative UV-spectrophotometric implementation of four predictive models. Spectrochim Acta Part A Mol Biomol Spectrosc. 2022;265: 120308.
Kokilambigai K, Lakshmi K. Utilization of green analytical chemistry principles for the simultaneous estimation of paracetamol, aceclofenac and thiocolchicoside by UV spectrophotometry. Green Chem Lett Rev. 2021;14(1):99–107.
Youssef SH. Hegazy MA-M, Mohamed D, Badawey AM: Analysis of paracetamol, pseudoephedrine and cetirizine in Allercet Cold® capsules using spectrophotometric techniques. Chem Cent J. 2018;12(1):1–14.
Palur K, Archakam SC, Koganti B. Chemometric assisted UV spectrophotometric and RP-HPLC methods for simultaneous determination of paracetamol, diphenhydramine, caffeine and phenylephrine in tablet dosage form. Spectrochim Acta Part A Mol Biomol Spectrosc. 2020;243: 118801.
Uddin M, Mondol A, Karim M, Jahan R, Rana A. Chemometrics assisted spectrophotometric method for simultaneous determination of paracetamol and caffeine in pharmaceutical formulations. Bangladesh J Sci Indus Res. 2019;54(3):215–22.
Saad AS, AbouAlAlamein AM, Galal MM, Zaazaa HE. Traditional versus advanced chemometric models for the impurity profiling of paracetamol and chlorzoxazone: Application to pure and pharmaceutical dosage forms. Spectrochim Acta Part A Mol Biomol Spectrosc. 2018;205:376–80.
Niedziałkowski P, Cebula Z, Malinowska N, Białobrzeska W, Sobaszek M, Ficek M, Bogdanowicz R, Anand JS, Ossowski T. Comparison of the paracetamol electrochemical determination using boron-doped diamond electrode and boron-doped carbon nanowalls. Biosens Bioelectron. 2019;126:308–14.
Ulusoy Hİ, Yılmaz E, Soylak M. Magnetic solid phase extraction of trace paracetamol and caffeine in synthetic urine and wastewater samples by a using core shell hybrid material consisting of graphene oxide/multiwalled carbon nanotube/Fe3O4/SiO2. Microchem J. 2019;145:843–51.
Wong A, Santos AM, Fatibello-Filho O. Simultaneous determination of paracetamol and levofloxacin using a glassy carbon electrode modified with carbon black, silver nanoparticles and PEDOT: PSS film. Sens Actuators, B Chem. 2018;255:2264–73.
Amare M. Voltammetric determination of paracetamol in tablet formulation using Fe (III) doped zeolite-graphite composite modified GCE. Heliyon. 2019;5(5): e01663.
Gika HG, Theodoridou A, Michopoulos F, Theodoridis G, Diza E, Settas L, Nikolaidis P, Smith C, Wilson ID. Determination of two COX-2 inhibitors in serum and synovial fluid of patients with inflammatory arthritis by ultra performance liquid chromatography–inductively coupled plasma mass spectroscopy and quadrupole time-of-flight mass spectrometry. J Pharm Biomed Anal. 2009;49(3):579–86.
Patel H, Suhagia B, Shah S, Rathod I. Determination of etoricoxib in pharmaceutical formulations by HPLC method. Indian J Pharm Sci. 2007;69(5):703.
Singh B, Santhakumar R, Bala I, Prasad SB, Verma S. Development and Validation of RP-HPLC method for the dissolution and assay of etoricoxib in pharmaceutical dosage forms. Int J Pharm Qual Assurance. 2015;6(01):1–7.
Andraws G, Trefi S. Ionisable substances chromatography: A new approach for the determination of Ketoprofen, Etoricoxib, and Diclofenac sodium in pharmaceuticals using ion–pair HPLC. Heliyon. 2020;6(8): e04613.
Topalli S, Chandrashekhar T, Annapurna MM. Validated RP-HPLC method for the assay of etoricoxib (a non-steroidal anti-inflammatory drug) in pharmaceutical dosage forms. E-J Chem. 2012;9(2):832–8.
Gangane P, Bagde S, Mujbaile S, Niranjane K, Gangane P. Development and Validation of HPLC assay method for etoricoxib in bulk drug and tablet formulation. Indian J Nat Sci. 2014;4(24):1565–625.
Rajmane VS, Gandhi SV, Patil UP, Sengar MR. High-performance thin-layer chromatographic determination of etoricoxib and thiocolchicoside in combined tablet dosage form. J AOAC Int. 2010;93(3):783–6.
Singh S, Mishra A, Verma A, Ghosh AK, Mishra AK. A simple Ultraviolet spectrophotometric method for the determination of etoricoxib in dosage formulations. J Advan Pharma Technol Res. 2012;3(4):237.
Jalakam SP, Waghmode J, Pawar P, Mane G. Development of Simple and Rapid LC-MS/MS Method for Determination of Etoricoxib in Human Plasma and its Application to Bioequivalence Study. Biomirror. 2016;7:67.
Rassi S. Novel PVC membrane selective electrode for the determination of etoricoxib in pharmaceutical preparations. Jordan Journal of Chemistry (JJC). 2011;6(4):423–37.
Narajji C, Karvekar MD. Method development and validation for simultaneous estimation of Paracetamol and Etoricoxib in pharmaceutical dosage form by RP-HPLC method. Der Pharma Chem. 2011;3:7–12.
Pattan S, Jamdar S, Godge R, Dighe N, Daithankar A, Nirmal S, Pai M. RP-HPLC method for simultaneous estimation of paracetamol and etoricoxib from bulk and tablets. J Chem Pharm Res. 2009;1(1):329–35.
Gupta KR, Likhar AD, Wadodkar SG. Application of stability indicating HPLC Method for quantitative determination of etoricoxib and paracetamol in pharmaceutical dosage form. Eurasian J Anal Chem. 2011;5(3):218–26.
Zaveri M, Khandhar A. Quantitative determination of Etoricoxib and Paracetamol in pharmaceutical dosage form and in-vitro comparison by reversed-phase high performance liquid chromatography (RP-HPLC). Asian J Pharm Res Health Care. 2010;2:4.
Rao KP, Ramana GV. Cost effective isocratic RP-HPLC method for simultaneous determination of Etoricoxib and Paracetamol in pure and in tablet formulation. J Advan Stud Agric Biol Environ Sci. 2014;1(2):201–9.
Chaube PH, Gandhi SV, Deshpande PB, Kulkarni VG. High performance thin layer chromatographic analysis of paracetamol and etoricoxib in spiked human plasma. Res J Pharm Technol. 2011;4(8):1303–6.
Patel AB, Vaghasiya E, Vyas AJ, Patel AI, Patel NK. Spectrophotometric first order derivative method for simultaneous determination of etoricoxib and paracetamol in tablet dosage form. J Med Chem Sci. 2020;3(3):300–7.
ICH, ICH Topic: Validation of analytical procedures: text and methodology. In: Q2 (R1): 2005. p. 1–17.
The United States Pharmacopoeia (USP32), National Formulary (NF27), Asian ed The United State Pharmacopoeia Convention, 2009.
Acknowledgements
The authors express their gratitude to SIGMA Pharmaceutical Industries for donating us the pure Etoricoxib and Paracetamol samples.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This work wasn't funded by any third party.
Author information
Authors and Affiliations
Contributions
MAA: data curation, validation, investigation, resources, writing—original draft. SAA: validation, formal analysis, investigation, data curation, supervision, writing—review and editing, visualization. SSE-M: validation, formal analysis, investigation, data curation, supervision, writing—review and editing, visualization. MRE: conceptualization, methodology, validation, formal analysis, investigation, data curation, supervision, review and editing, visualization. HEZ: conceptualization, methodology, validation, formal analysis, investigation, data curation, supervision, review and editing, visualization. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Abdel Rahman, M.A., Elghobashy, M.R., Zaazaa, H.E. et al. Validated HPLC–PDA methodology utilized for simultaneous determination of Etoricoxib and Paracetamol in the presence of Paracetamol toxic impurities. BMC Chemistry 16, 108 (2022). https://doi.org/10.1186/s13065-022-00904-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-022-00904-z