
Abstract
To discover multifunctional agents for the treatment of Alzheimer's disease (AD), a new series of 1,2,3-triazole-chromenone derivatives were designed and synthesized based on the multi target-directed ligands approach. The in vitro biological activities included acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibition as well as anti-Aβ aggregation, neuroprotective effects, and metal-chelating properties. The results indicated a highly selective BuChE inhibitory activity with an IC50 value of 21.71 μM for compound 10h as the most potent compound. Besides, compound 10h could inhibit self-induced Aβ1–42 aggregation and AChE-induced Aβ aggregation with 32.6% and 29.4% inhibition values, respectively. The Lineweaver–Burk plot and molecular modeling study showed that compound 10h targeted both the catalytic active site (CAS) and peripheral anionic site (PAS) of BuChE. It should be noted that compound 10h was able to chelate biometals. Thus, the designed scaffold could be considered as multifunctional agents in AD drug discovery developments.

Similar content being viewed by others


Introduction
Dementia is one of the noteworthy problems in the public health management as over 80% of dementia cases are suffering from Alzheimer’s disease (AD). Currently, available therapies provide temporary symptomatic relief but do not target the distractive neuropathology. Therefore, a new treatment to delay or halt disease progression has remained as an urgent medical need.
The pathophysiological processes in AD have still remained unclear to this day. However, alongside its complexity, several neurodegenerative processes could be identified which include (I) aggregation of insoluble amyloid beta (Aβ) plaques mostly trigger from sequential cleavage of amyloid precursor protein (APP) by the aspartyl protease β-site APP cleaving enzyme-1 (BACE1) and γ-secretase, (II) neurofibrillary tangles (NFTs) form through hyperphosphorylation of tau proteins, (III) biometals dysfunction, and (IV) oxidative stress which in return results in synapse loss and death of neuronal cells in the brain [1]. Also, different hallmarks have been recognized including the loss of cholinergic neurons, reduction of the neurotransmitter acetylcholine (ACh), and increased expression of inflammatory mediators [2,3,4].
Based on the approved theory for AD, the loss of cholinergic neurons causes reduction of ACh. As a result, inhibition of the acetylcholinesterase (AChE) raises the level of ACh and improves cognitive performance at the early stage of AD. The critical point is that the AChE level decreases with the progression of AD, subsequently, AChE inhibition seems to be ineffective during the progression of AD [5, 6]. Interestingly, the level of butyrylcholinesterase (BuChE) remains unchanged or even increases at the late stage of disease [7]. BuChE can hydrolyze ACh and thereby, compensates the reduction of AChE activity [8]. A recent experiment with AChE knockout mice supported this hypothesis [9]. Results from further studies were in accordance with the role of BuChE in AD brains and showed a positive correlation between selective BuChE inhibition and improved cognitive performance and memory [10, 11].
BuChE has 65% amino acid sequence similarity to AChE along with mostly similar functions [12,13,14]. AChE and BuChE active sites have five subsites, including catalytic active site (CAS), peripheral anionic site (PAS), acyl binding pocket, oxyanion hole, and anionic subsite [15]. One of the structural differences between AChE and BuChE can be associated with acyl pocket size. More specifically, smaller residue such as Leu286 and Val288 of BuChE acyl pocket provide a larger site in BuChE while aromatic and bulky Phe295 and Phe297 residues of AChE acyl pocket afford smaller space in AChE. Mentioned structural differences contribute to the design of selective inhibitors [5, 8, 16].
Moreover, produced Aβ peptides can aggregate into Aβ plaques which initiate pathogenic cascade leading to neuronal loss and dementia. Inhibition of the accumulation of Aβ peptide in the brain could be another therapeutic strategy against the development of AD [2]. The metal chelatory potential of compounds has also been demonstrated to exert beneficial effects via decreasing plaque aggregation [17, 18].
Results and discussion
Design
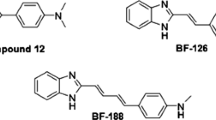
Because of the multifactorial and sporadic nature of AD, the modern approach “multi-target-one disease” could be efficient to develop effective agents to act simultaneously at different targets. Selective BuChE inhibitors could be a promising target for the treatment of AD at the moderate and advanced stages of the disease [19]. Closer looks at X-ray crystallography of BuChE depicted that it usually tolerates bigger scaffolds than AChE as the active site of BuChE is approximately 200 Å larger than AChE. Analysis of the potent BuChE inhibitors revealed that the N-containing ring could be effective for the interactions with ChE active site (Fig. 1). Molecular docking evaluation of compound A depicted that coumarin moiety interacted with Trp231 and Phe329 residues of CAS pocket and benzyl pyridinium moiety interacted with Trp82 of BuChE PAS pocket [20]. According to the interaction mode of compound B, it can be understood that 1,2,3-triazole-aryl moiety led to the formation of hydrophobic interactions with amino acids of PAS and chromenone ring oriented towards CAS pocket [21]. In the case of compound C, diphenyl fit into the BuChE CAS pocket while the benzyl triazine pendant group showed H-bond interaction with the PAS of BuChE [22, 23]. As appeared in compounds B and C, increasing bulkiness and length of drug candidate could increase the selectivity of BuChE over AChE. As can be seen in compound D, the presence of relatively spacious moieties can affect the interaction of the designed compounds with that amino acid within the active site of BuChE. As a result, introducing a dimethylamino propenone entity into our system increased the length and volume of the designed scaffold which might be a good strategy to increase selectivity towards BuChE [24]. Besides, this part could also exhibit metal-chelating potential [25].

Designed hybrids investigated as selective BuChE inhibitors with anti-Aβ aggregatory, neuroprotective, and metal chelating properties
In the case of anti-Aβ plaques aggregation, it is important to keep the potential moiety in the structure to inhibit the aggregation of the toxic peptide.
Coumarin structures as active natural compounds may simultaneously possess anti-oxidative [2, 26], neuroprotective [27] anti-ChE [20] and anti-Aβ aggregative properties [28, 29]. Coumarins pharmacophore owing to the presence of polar elements in the structure (Fig. 1, compound E and F) might help to inhibit amyloid fibril formation through the interaction with the polar surface of Aβ [30,31,32]. Hence, coumarins could serve as a potent framework for the prevention of Aβ1-42 aggregation [28]. In addition to the inhibition of BuChE and Aβ plaques aggregation, an inhibitor that can tackle toxicity of Aβ peptide, ROS and RNS could be effective for a longer period of AD progression. Recently, iminochromene ring was characterized as a potent neuroprotective agent. In this project, the iminochromene group of compound G was bioisosterically replaced with chromenone moiety to evaluate the possible neuroprotectivity [33]. Hence, in the present work, a molecular hybridization and bioisosteric replacement approaches were used to design and synthesize multi-target agents with anti-BuChE, anti-Aβ aggregation, neuroprotective, and metal chelating properties.
Synthesis of 1,2,3-triazole-dimethylaminoacryloyl-chromenones
Synthesis of the tilted compounds 10a-m was conducted according to the steps shown in Scheme 1. Desired starting material, 2-hydroxy-4-(prop-2-yn-1-yloxy)benzaldehyde (3) was exactly prepared according to the literature [34]. Then, the reaction of compound 3 and excess amounts of ethyl acetoacetate (4) in ethanol at room temperature overnight afforded 3-acetyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (5). Reaction of compound 5 and dimethylformamid-dimethylacetal (DMA-DMF) in 1,4-dioxane under reflux conditions for 6 h led to the formation of (E)-3-(3-(dimethylamino)acryloyl)-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (7). Finally, click reaction [35] of compound 7 and in situ prepared azides 9 in the presence of triethylamine and CuSO4.5H2O in the mixture of water and tert-butyl alcohol at room temperature for 24 h gave the corresponding products 10a-m.

The synthetic route for the preparation of compounds 10a-m
AChE and BuChE inhibitory activity
In vitro anti-AChE and anti-BuChE activity of synthesized compounds, 10a-m were performed based on the modified Ellman’s method [36] comparing with donepezil as the reference drug. Compounds were initially screened in vitro against AChE, and none of them exhibited inhibitory properties against the AChE enzyme. Interestingly, half of the 1,2,3-triazole- chromenone derivatives showed remarkable and selective inhibitory potency towards BuChE, which exerted a more prominent role at later stages of the disease [37].
As can be seen in Table 1, the BuChEI activity directly depended on the electronic property of substituents and their positions on the benzyl moiety connected to 1,2,3-triazole ring. Results showed that compound 10h possessing 3,4-diF on the aryl ring induced the best BuChE inhibitory activity (IC50 = 21.71 μM); however, the elimination of 3-F completely changed the activity in such a manner that compound 10g showed no inhibitory activity. meta-Fluorinated derivative 10f was found to be a moderate inhibitor as the calculated IC50 value was 59.58 μM and para-fluorinated derivative 10g showed no inhibitory activity towards BuChE (IC50 > 100 μM).

Considering the inhibitory activity of other halogenated derivatives 10i-m depicted that chlorinated compounds 10i and 10j showed no inhibitory activity (IC50 > 100 μM). In the case of brominated derivatives 10k-m, compounds 10l possessing Br at 3-position of aryl ring showed moderate activity with IC50 value of 65.96 μM.
In the case of the electron-donating substituent (Me and OMe), relatively good results were obtained. The presence of methyl group at 2-position of aryl moiety (compound 10b) led to relatively good anti-BuChE activity (IC50 = 35.73 μM); however, the presence of the same group at 4-position of compound 10c completely diminished inhibitory activity (IC50 > 100 μM). Another point comes back to the derivative 10d containing OMe group at 3-position on the benzyl ring which led to higher activity (IC50 = 23.44 μM). Finally, the absence of substituent on the aryl ring (compound 10a) also depicted good BuChE inhibitory activity (IC50 = 34.41 μM).
The in vitro anti-ChE assay showed that the un-substituted benzyl derivative (10a) along with the meta-substituted analogs (10d, 10f, and 10l) had significant anti-BuChE activity. Introduction of extra small-size halogen groups such as F (compound 10h) resulted in the most potent activity with an IC50 value of 21.71 µM.
Kinetic study of BuChE inhibition
The kinetic study was performed to investigate the mechanism of inhibition by compound 10h against BuChE. Graphical analysis of the reciprocal Lineweaver–Burk plot of compound 10h described a mixed-type inhibition pattern (Fig. 2a) in which compound 10h may bound to the BuChE or it already bound to the substrate. Also, the Ki value was calculated using the secondary plot as 38.3 µM (Fig. 2b)

a Kinetic study of BuChE inhibition by compound 10h. b Inhibition constant (Ki) of compound 10h
Inhibition of AChE-induced and self-induced Aβ aggregation
Aβ peptide is the major constituent of senile plaques in the brains of patients with AD. In this respect, the effect of the most potent compound 10h was assessed for the inhibition against Aβ1-42 aggregation and AChE-induced Aβ1-40 peptide aggregation using the Thioflavin T (ThT) assay. Comparing with donepezil and tacrine as the reference compounds, it can be understood that 10h was more potent than both controls in inhibiting Aβ1-42 self-aggregation, as depicted 32.6% inhibition at 10 µM (Table 2). Furthermore, compound 10h inhibited AChE-induced Aβ aggregation by 29.4% at 100 μM.
Neuroprotective studies on PC12 cell line
Compound 10h was selected to study the neuroprotective ability using PC12 cell injury induced by Aβ25–35 by MTT assay. This compound depicted no neuroprotective effect on Aβ-induced on PC12 cells up to 50 μM. It can be understood that bioisosteric replacement of iminochromene moiety (compound F, Fig. 1) with chromenone ring did not induce the desired neuroprotectivity.
Metal chelating
Compound 10h was tested for its metal chelating ability towards Fe2+, Cu2+, and Zn2+ ions (Fig. 3) The UV spectrum of methanolic solution (20 µM) of that compound showed two characteristic absorption peaks at 309.9 and 386.7 nm. After the interaction of compound 10h with the above mentioned ions for 30 min, small shifts as well as absorption intensity changes were observed in the spectra confirming biometal-ligand interactions.
Interaction of compound 10h with Zn2+ ions demonstrated two absorption peaks at 299.2 and 388.7 nm. Similar changes were observed in the case of Fe2+ ions and those absorptions were observed at 303.5 and 390.9 nm. When compound 10h was treated with Cu2+ ions, two peaks were observed at 303.5 and 382.4 nm. The stoichiometry of complex 10h-Cu2+ was also studied (Fig. 4) due to important role of copper ions in AD [38]. The concentration of the test compound 10h was 20 μM and the final concentration of Cu2+ ranged from 0–44 μM with 4 μM intervals at 303.5 nm.

The absorbance change of compound 10h alone and in the presence of Zn2+, Fe2+, and Cu2+ ions in the wavelength range from 200 to 600 nm. The experiments were performed in triplicate

Determination of the stoichiometry of complex 10h-Cu+2 using molar ratio method. The experiments were performed in triplicate
The plot was obtained by the corresponding absorption against the mole fraction of Cu2+ to ligand 10h. According to the plot, the ratio 1:1 complexation ration of 10h-Cu2+ can be seen at the fracture point of the plot with the mole fraction of 0.6.
Docking study of BuChE
As discussed in the introduction and designing sections, the volume of the BuChE active site is considerably bigger than the one found in the AChE, so BuChE can accommodate bulkier inhibitors, and this may constitute the basis for the selectivity of these derivatives. An overlay of the best pose for 10h with BuChE was depicted in Fig. 5. Chromenenone core is mostly surrounded by residues of CAS pocket, while 3,4-difluoro benzyl moiety oriented towards PAS pocket. More specifically, the carbonyl group of the chromenenone ring form a hydrogen bond with the oxygen of Ser198 of the CAS while the another hydrogen bond interaction was seen between the oxygen of chromenenone pendant group and Ser198. Also, the 1,2,3-triazole ring formed a third hydrogen bond with Trp82 of the anionic subsite.
The nitrogen of the dimethylamino propenone interacted with Pro285 via Van der Waals interaction. The Van der Waals interaction was also constructed between 1,2,3-triazole moiety and Ala326 of the PAS residue. The 3,4-difluoro benzyl ring also showed π–π and van der Waals interactions with Tyr332. These results strongly supported the high potency of compound 10h against BuChE.

Schematic representation showing interactions of compound 10h with the surrounding residues of BuChE (PDB code: 4BDS) [39]
It is concluded that not only dimethylamino propenone attached to the chromenone ring is an essential factor for designing an active and selective BuChE inhibitor, but also the nature of substituents on the aryl moieity connected to 1,2,3-triazole is the necessary element to afford a higher BuChE inhibitory effect. Further computer-aided lead optimization to improve anti-BuChE can be performed via replacing of the dimethylamino propenone pendant group with a cyclic amine to evaluate the size and bulkiness for the selective BuChE inhibition. Besides, the 1,2,3-triazole moiety can be substituted by different aliphatic spacer containing CO, NHCO, NH to improve the capability of H-bonding interaction with the active site.
In silico ADME evaluation
In silico ADME/T studies of the synthesized compounds was performed using https://lmmd.ecust.edu.cn/admetsar2/ and https://preadmet.bmdrc.kr, to evaluate pharmacokinetic properties of possible and potential candidates/drug molecules which can be helpful for future anti-AD drug developments [40]. As shown in Table 3, most of the compounds showed drug-like characteristics based on Lipinski's rule of five (MW < 500, cLogP < 5, HB donor ≤ 5, HB acceptor ≤ 10). Our results indicated that lipophilicity and solubility of the derivatives were drug-like. Furthermore, molecular weight, cLogP, and blood–brain barrier were well within the standard ranges.
Materials and methods
Instrumental methods
Melting points of synthesized compounds were determined on a Kofler hot stage apparatus. 1H and 13C NMR spectra were determined on a Varian FT-500, using TMS as an internal standard. IR spectra were recorded using KBr disks on a Bruker Tensor 27 FTIR spectrophotometer. Elemental analysis was carried out with an Elemental Analyzer system GmbH VarioEL CHN mode.
Synthesis of 2-hydroxy-4-(prop-2-yn-1-yloxy)benzaldehyde (3)
Compound 3 was prepared from the reaction of 2,4-dihydroxybenzaldehyde 1 and propargyl bromide 2 in the presence of potassium carbonate (K2CO3) and potassium iodide (KI) in acetone at 50 ºC, according to the literature [34].
Synthesis of 3-acetyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (5)
A few drops of piperidine were added to the mixture of compound 3 (1 mmol) and ethyl acetoacetate (2.5 mmol) 4 in ethanol (10 mL) and it was stirred overnight at room temperature to obtain yellow precipitates. After completion of the reaction (checked by TLC), they were filtered off and used for the next step with no further purification. It was also completely characterized and compared with reported in the literature [41].
Synthesis of (E)-3-(3-(dimethylamino)acryloyl)-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (7)
The mixture of compound 5 (1 mmo l) and DMA-DMF 6 (2 mmol) in 1,4-dioxane (10 mL) was heated at reflux for 6 h. Then, the solvent was evaporated under vacuum and the residue was purified using plate chromatography with ethyl acetate as eluent.
Yield: 55%; M.p. 98–100 °C. IR (KBr): 2925, 2850, 2150, 1715, 1642, 1598 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.58 (s, 1H, H4), 7.93 (d, J = 12.4 Hz, 1H, CH), 7.54 (d, J = 8.3 Hz, 1H, H5), 6.93–6.91 (m, 2H, H6, H8), 6.34 (d, J = 12.4 Hz, 1H, CH), 4.77 (s, 2H, CH2), 3.17 (s, 3H, CH3), 2.97 (s, 3H, CH3), 2.60 (1H, CH) ppm. 13C NMR (CDCl3, 125 MHz): 182.3, 162.1, 160.1, 156.4, 155.0, 145.9, 131.1, 123.3, 113.5, 113.4, 101.3, 95.2, 77.2, 77.0, 56.3, 45.2, 37.6 ppm.
Synthesis of 1,2,3-triazole-dimethylaminoacryloyl-chromenone hybrids 10a-m
The final step was performed by the click reaction of compound 7 and in situ prepared azides 9. For this purpose, a solution of benzyl chloride/bromide derivative 8 (1.1 mmol), sodium azide (0.06 g, 0.9 mmol), and triethylamine (0.13 g, 1.3 mmol) in water (4 mL) and tert-butyl alcohol (4 mL) was stirred at room temperature for 30 min. Then, compound 7 (0.5 mmol) and CuSO4.5H2O (7 mol%) were added to the mixture and it was continued for 24 h. Upon completion of the reaction checked TLC), the mixture was diluted with water, extracted with chloroform, and dried over anhydrous Na2SO4. After evaporation of the solvent, the residue was recrystallized from ethyl acetate and petroleum ether to give pure product 10. In the case of some compounds, they were purified using plate chromatography with ethyl acetate as eluent.
(E)-7-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10a)
Yield: 54%; M.p. 186–188 °C. IR (KBr): 2920, 2853, 1715, 1640, 1597, 1558 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.55 (s, 1H, triazole), 7.91 (d, J = 12.5 Hz, 1H, CH), 7.60 (s, 1H, H4), 7.50 (d, 1H, J = 8.5 Hz, H5), 7.38–7.29 (m, 5H, H2′, H3′, H4′, H5′, H6′), 6.92–6.90 (m, 2H, H6, H8), 6.33 (d, J = 12.5 Hz, 1H, CH), 5.55 (s, 2H, CH2), 5.24 (s, 2H, CH2), 3.17 (s, 3H, CH3), 2.96 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.4, 171.7, 162.9, 156.2, 151.4, 145.6, 144.0, 134.7, 133.3, 131.1, 129.8, 128.9, 128.2, 122.6, 121.5, 113.3, 101.2, 92.2, 63.4, 57.2, 47.6, 38.1 ppm. Anal. calcd. for C24H22N4O4: C, 66.97; H, 5.15; N, 13.02. Found: C, 66.71; H, 5.38; N, 12.86.
(E)-3-(3-(Dimethylamino)acryloyl)-7-((1-(2-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (10b)
Yield: 59%; M.p. 203–205 °C. IR (KBr): 2924, 2855, 1712, 1640, 1596, 1558 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.58 (s, 1H, triazole), 7.92 (d, J = 12.3 Hz, 1H, CH), 7.52 (d, 1H, J = 8.3 Hz, H5), 7.46 (s, 1H, H4), 7.32–7.18 (m, 4H, H3′, H4′, H5′, H6′), 6.93–6.91 (m, 2H, H6, H8), 6.35 (d, J = 12.3 Hz, 1H, CH), 5.57 (s, 2H, CH2), 5.24 (s, 2H, CH2), 3.18 (s, 3H, CH3), 2.98 (s, 3H, CH3), 2.29 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.4, 162.4, 160.2, 156.5, 154.9, 145.9, 143.0, 137.0, 132.0, 131.1, 130.7, 129.5, 129.3, 126.7, 123.5, 122.7, 113.3, 113.0, 101.3, 95.2, 62.4, 52.5, 45.3, 37.6, 19.0 ppm. Anal. calcd. for C25H24N4O4: C, 67.55; H, 5.44; N, 12.60. Found: C, 67.31; H, 5.70; N, 12.44.
(E)-3-(3-(Dimethylamino)acryloyl)-7-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (10c)
Yield: 55%; M.p. 199–201 °C. IR (KBr): 2923, 2855, 1715, 1640, 1596, 1558 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.92 (d, J = 12.3 Hz, 1H, CH), 7.55 (s, 1H, H4), 7.51 (d, 1H, J = 8.3 Hz, H5), 7.19–7.17 (m, 4H, H2′, H3′, H5′, H6′), 6.94–6.91 (m, 2H, H6, H8), 6.34 (d, J = 12.3 Hz, 1H, CH), 5.51 (s, 2H, CH2), 5.24 (s, 2H, CH2), 3.17 (s, 3H, CH3), 2.97 (s, 3H, CH3), 2.36 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.5, 162.4, 160.2, 156.6, 1549, 145.9, 143.1, 138.9, 131.2, 130.7, 129.8, 128.2, 123.3, 122.8, 113.3, 133.2, 101.2, 95.2, 62.4, 54.6, 45.6, 37.6, 21.2 ppm. Anal. calcd. for C25H24N4O4: C, 67.55; H, 5.44; N, 12.60. Found: C, 67.40; H, 5.26; N, 12.71.
(E)-3-(3-(Dimethylamino)acryloyl)-7-((1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (10d)
Yield: 52%; M.p. 209–211 °C. IR (KBr): 2923, 2855, 1712, 1640, 1595, 1557 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.92 (d, J = 12.4 Hz, 1H, CH), 7.59 (s, 1H, H4), 7.52 (d, 1H, J = 8.4 Hz, H5), 7.30 (t, J = 7.9 Hz, 1H, H5′), 6.94–6.81 (m, 5H, H6, H8, H2′, H4′, H6′), 6.35 (d, J = 12.4 Hz, 1H, CH), 5.52 (s, 2H, CH2), 5.25 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 3.17 (s, 3H, CH3), 2.97 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.4, 162.4, 160.2, 156.6, 154.9, 145.9, 144.0, 143.2, 135.7, 130.7, 130.3, 123.4, 122.9, 119.3, 114.3, 113.8, 133.3, 112.5, 101.2, 95.2, 62.4, 55.3, 54.3, 45.2, 37.6 ppm. Anal. calcd. for C25H24N4O5: C, 65.21; H, 5.25; N, 12.17. Found: C, 65.41; H, 5.10; N, 12.32.
(E)-3-(3-(Dimethylamino)acryloyl)-7-((1-(2-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (10e)
Yield: 55%; M.p. 180–183 °C. IR (KBr): 2924, 2852, 1712, 1640, 1597, 1558 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.91 (d, J = 12.4 Hz, 1H, CH), 7.67 (s, 1H, H4), 7.51 (d, 1H, J = 8.0 Hz, H5), 7.39–7.38 (m, 1H, H4′), 7.30 (td, J = 7.6, 1.5 Hz, 1H, H3′), 7.17–7.11 (m, 2H, H5′, H6′), 6.94–6.92 (m, 2H, H6, H8), 6.34 (d, J = 12.4 Hz, 1H, CH), 5.61 (s, 2H, CH2), 5.25 (s, 2H, CH2), 3.17 (s, 3H, CH3), 2.97 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.3, 162.4, 160.6 (d, JC-F = 251.2 Hz), 160.1, 158.7, 156.6, 154.8, 145.9, 143.2, 131.1 (d, JC-F = 8.1 Hz), 130.7 (d, JC-F = 3.3 Hz), 124.9 (d, JC-F = 3.4 Hz), 123.3, 121.3, 121.6 (d, JC-F = 14.5 Hz), 115.9 (d, JC-F = 21.03 Hz), 113.3, 113.2, 101.2, 95.2, 62.4, 47.9, 45.2, 37.5 ppm. Anal. calcd. for C24H21FN4O4: C, 64.28; H, 4.72; N, 12.49. Found: C, 64.36; H, 4.57; N, 12.55.
(E)-3-(3-(Dimethylamino)acryloyl)-7-((1-(3-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (10f)
Yield: 60%; M.p. 183–185 °C. IR (KBr): 2922, 2850, 1713, 1640, 1597 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.91 (d, J = 12.3 Hz, 1H, CH), 7.61 (s, 1H, H4), 7.51 (d, 1H, J = 8.3 Hz, H5), 7.36 (td, 1H, J = 7.5, 2.2 Hz, H5′), 7.08–7.04 (m, 2H, H4′, H6′), 6.97 (d, 1H, J = 9.2 Hz, H2′), 6.94–6.91 (m, 2H, H6, H8), 6.34 (d, J = 12.3 Hz, 1H, CH), 5.55 (s, 2H, CH2), 5.26 (s, 2H, CH2), 3.17 (s, 3H, CH3), 2.96 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 180.0, 162.3, 160.7 (d, JC-F = 262.2 Hz), 160.1, 156.7, 154.9, 145.8, 143.5, 136.6 (d, JC-F = 19.4 Hz), 130.8 (d, JC-F = 8.1 Hz), 130.7, 123.6, 123.3, 122.9, 115.9 (d, JC-F = 20.9 Hz), 115.1 (d, JC-F = 22.0 Hz), 113.3, 113.2, 101.2, 95.2, 62.4, 53.7, 45.0, 37.6 ppm. Anal. calcd. for C24H21FN4O4: C, 64.28; H, 4.72; N, 12.49. Found: C, 64.47; H, 4.84; N, 12.60.
(E)-3-(3-(Dimethylamino)acryloyl)-7-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (10g)
Yield: 62%; M.p. 183–185 °C. IR (KBr): 2923, 2855, 1714, 1640, 1598 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.56 (s, 1H, triazole), 7.92 (d, J = 12.4 Hz, 1H, CH), 7.58 (s, 1H, H4), 7.51 (d, 1H, J = 8.4 Hz, H5), 7.29 (dd, J = 8.7, 5.1 Hz, 2H, H2′, H6′), 7.07 (t, J = 8.7 Hz, 2H, H3′, H5′), 6.93–6.91 (m, 2H, H6, H8), 6.33 (d, J = 12.4 Hz, 1H, CH), 5.52 (s, 2H, CH2), 5.24 (s, 2H, CH2), 3.16 (s, 3H, CH3), 2.96 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 180.1, 162.4, 160.5 (d, JC-F = 260.5 Hz), 160.1, 156.5, 154.8, 145.1, 143.4, 134.5 (d, JC-F = 19.5 Hz), 130.7, 130.1, 123.3, 122.8, 116.2 (d, JC-F = 21.0 Hz), 113.3, 113.2, 101.2, 95.1, 62.1, 53.5, 45.0, 37.5 ppm. Anal. calcd. for C24H21FN4O4: C, 64.28; H, 4.72; N, 12.49. Found: C, 64.44; H, 4.60; N, 12.27.
(E)-7-((1-(3,4-Difluorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10h)
Yield: 59%; M.p. 188–190 °C. IR (KBr): 2922, 2850, 1718, 1640, 1594 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.91 (d, J = 12.3 Hz, 1H, CH), 7.63 (s, 1H, H4), 7.52 (d, 1H, J = 8.4 Hz, H5), 7.21–7.11 (m, 2H, H5′, H6′), 7.06–7.04 (m, 1H, H2′), 6.93–6.91 (m, 2H, H6, H8), 6.33 (d, J = 12.3 Hz, 1H, CH), 5.52 (s, 2H, CH2), 5.27 (s, 2H, CH2), 3.18 (s, 3H, CH3), 2.97 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.3, 160.1, 156.6, 154.9, 150.5 (d, JC-F = 243.1 Hz), 149.6, (d, JC-F = 240.0 Hz), 145.8, 144.0, 143.6, 132.8, 131.2, 130.7, 124.3, 123.4, 122.9, 118.1 (d, JC-F = 17.2), 117.3 (d, JC-F = 17.6), 113.3, 101.2, 95.2, 62.4, 53.2, 45.2, 37.6 ppm. Anal. calcd. for C24H20F2N4O4: C, 61.80; H, 4.32; N, 12.01. Found: C, 61.63; H, 4.17; N, 11.84.
(E)-7-((1-(2-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10i)
Yield: 58%; M.p. 178–180 °C. IR (KBr): 2923, 2852, 1713, 1640, 1597 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.60 (s, 1H, triazole), 7.91 (d, J = 11.0 Hz, 1H, CH), 7.68 (s, 1H, H4), 7.51 (d, 1H, J = 8.1 Hz, H5), 7.43 (d, 1H, J = 7.9 Hz, H3′), 7.32–7.24 (m, 3H, H4′, H5′, H6′), 6.93–6.87 (m, 2H, H6, H8), 6.33 (d, J = 11.0 Hz, 1H, CH), 5.69 (s, 2H, CH2), 5.29 (s, 2H, CH2), 3.16 (s, 3H, CH3), 2.96 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.7, 171.5, 163.8, 158.5, 156.3, 151.7, 144.5, 133.6, 131.7, 130.7, 130.5, 130.4, 130.0, 127.7, 123.4, 120.1, 113.3, 112.8, 100.6, 96.6, 61.1, 52.3, 46.4, 40.4 ppm. Anal. calcd. for C24H21ClN4O4: C, 62.00; H, 4.55; N, 12.05. Found: C, 61.81; H, 4.38; N, 11.90.
(E)-7-((1-(4-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10j)
Yield: 61%; M.p. 178–180 °C. IR (KBr): 2891, 2850, 1715, 1640, 1597, 1558 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.54 (s, 1H, triazole), 7.90 (d, J = 12.4 Hz, 1H, CH), 7.61 (s, 1H, H4), 7.50 (d, J = 8.5 Hz, 1H, H5), 7.34 (d, J = 8.1 Hz, 2H, H3′, H5′), 7.22 (d, J = 8.1 Hz, 2H, H2′, H6′),
6.92–6.89 (m, 2H, H6, H8), 6.34 (d, J = 12.4 Hz, 1H, CH), 5.52 (s, 2H, CH2), 5.19 (s, 2H, CH2), 3.11 (s, 3H, CH3), 2.95 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.9, 162.4, 160.1, 156.5, 154.9, 145.8, 144.2, 143.4, 134.9, 130.7, 129.5, 129.4, 123.3, 122.9, 113.3, 133.2, 101.2, 95.2, 62.4, 53.5, 45.2, 37.6 ppm. Anal. calcd. for C24H21ClN4O4: C, 62.00; H, 4.55; N, 12.05. Found: C, 62.11; H, 4.40; N, 12.28.
(E)-7-((1-(2-Bromobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10k)
Yield: 58%; M.p. 202–205 °C. IR (KBr): 2924, 2850, 1709, 1641, 1598, 1559 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.92 (d, J = 12.3 Hz, 1H, CH), 7.70 (s, 1H, H4), 7.63 (d, 1H, J = 7.5 Hz, H3′), 7.52 (d, 1H, J = 8.3 Hz, H5), 7.33 (t, 1H, J = 7.5 Hz, H5′), 7.25–7.21 (m, 2H, H4′, H6′), 6.95–6.93 (m, 2H, H6, H8), 6.34 (d, J = 12.3 Hz, 1H, CH), 5.69 (s, 2H, CH2), 5.27 (s, 2H, CH2), 3.21 (s, 3H, CH3), 2.97 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.5, 162.4, 160.1, 156.6, 154.9, 145.9, 143.1, 133.8, 133.4, 133.3, 130.7, 130.6, 130.5, 128.3, 123.6, 123.3, 113.4, 113.3, 100.4, 95.2, 62.0, 54.0, 44.8, 37.2 ppm. Anal. calcd. for C24H21BrN4O4: C, 56.59; H, 4.16; N, 11.00. Found: C, 56.37; H, 4.30; N, 11.21.
(E)-7-((1-(3-Bromobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10l)
Yield: 64%; M.p. 218–220 °C. IR (KBr): 2925, 2856, 1710, 1640, 1595 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.59 (s, 1H, triazole), 7.93 (d, J = 12.4 Hz, 1H, CH), 7.61 (s, 1H, H4), 7.55–7.51 (m, 3H, H5, H2′, H4′), 7.26–7.18 (m, 2H, H5′, H6′), 6.96–6.94 (m, 2H, H6, H8), 6.35 (d, J = 12.4 Hz, 1H, CH), 5.51 (s, 2H, CH2), 5.28 (s, 2H, CH2), 3.19 (s, 3H, CH3), 2.98 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 180.1, 162.3, 160.2, 156.5, 154.9, 145.9, 143.5, 136.4, 133.7, 132.1, 131.1, 130.9, 130.8, 126.6, 122.9, 122.2, 113.4, 113.3, 101.1, 95.0, 62.8, 53.6, 44.8, 38.8 ppm. Anal. calcd. for C24H21BrN4O4: C, 56.59; H, 4.16; N, 11.00. Found: C, 56.31; H, 4.24; N, 11.18.
(E)-7-((1-(4-Bromobenzyl)-1H-1,2,3-triazol-4-yl)methoxy)-3-(3-(dimethylamino)acryloyl)-2H-chromen-2-one (10m)
Yield: 64%; M.p. 173–175 °C. IR (KBr): 2925, 2850, 1715, 1640, 1597 cm−1. 1H NMR (CDCl3, 500 MHz): δ = 8.57 (s, 1H, triazole), 7.91 (d, J = 12.3 Hz, 1H, CH), 7.60 (s, 1H, H4), 7.52–7.51 (m, 3H, H5, H3′, H5′), 7.17 (d, J = 8.4 Hz, 2H, H2′, H6′), 6.93–6.91 (m, 2H, H6, H8), 6.34 (d, J = 12.3 Hz, 1H, CH), 5.51 (s, 2H, CH2), 5.22 (s, 2H, CH2), 3.17 (s, 3H, CH3), 2.96 (s, 3H, CH3) ppm. 13C NMR (CDCl3, 125 MHz): δ = 182.4, 162.9, 160.1, 156.6, 154.9, 146.6, 144.4, 143.5, 133.3, 132.4, 130.7, 129.8, 123.3, 123.1, 122.9, 112.7, 100.8, 95.2, 62.4, 53.6, 45.2, 37.6 ppm. Anal. calcd. for C24H21BrN4O4: C, 56.59; H, 4.16; N, 11.00. Found: C, 56.68; H, 4.38; N, 10.87 (Additional file 1).
Inhibitory activities against AChE and BuChE
All enzymes and reagents required for the assay were obtained from Aldrich. The in vitro anticholinesterase activity of all synthesized compounds 10a-m was assayed using modified Ellman's method using a 96-well plate reader (BioTek ELx808) according to the literature [36, 42]. Initially, the stock solutions of compounds 10 were prepared by dissolving the test compound (1 mg) in DMSO (1 mL) and then, diluted solutions at final concentrations of 1, 10, 20, and 40 μg/mL were prepared using methanol. Each well contained 50 µL potassium phosphate buffer (KH2PO4/ K2HPO4, 0.1 M, pH 8), 25 µL sample solution, and 25 µL enzyme (final concentration 0.22 U/mL in buffer). Control experiments were also performed under the same conditions without enzyme. After incubation at room temperature for 15 min, 125 µL DTNB (3 mM in buffer) was added and the characterization of enzymatic reaction was spectrometrically performed at 405 nm followed by the addition of substrate (ATCI 3 mM in water) after 5–10 min. The IC50 values were determined graphically from inhibition curves (log inhibitor concentration vs. percent of inhibition). Also, the same method was used for the BuChE inhibition assay.
Kinetic characterization of BuChE inhibitory activity
The kinetic study for the inhibition of BuChE by compound 10h was carried out according to Ellman's method used for the inhibition assay using four different concentrations of inhibitor (0, 10.7, 42.9, and 85.8 µM). The Lineweaver–Burk reciprocal plot was provided by plotting 1/V against 1/[S] at variable concentrations of butyrylthiocholine as the substrate (187.5, 750, 1500, 3000 µM). The inhibition constant Ki was achieved by the plot of slopes versus the corresponding concentrations of compound 10h [43, 44].
Inhibition of Aβ1-42 aggregation and disaggregation of aggregated Aβ1-40 induced by AChE
Inhibition of Aβ1-42 self-aggregation was measured by ThT fluorescence assay. The details of the method were reported in our previous study [45]. To study Aβ42 aggregation inhibition, a reported method, based on the fluorescence emission of ThT was followed. Briefly, the mixtures of Aβ1-40 peptide (Bachem company, Switzerland) and AChE (Sigma, Electrophorus electricus), in the presence or absence of the test inhibitor were incubated for 24 h at room temperature. The final concentrations of Aβ (dissolved in DMSO and diluted 0.215 M sodium phosphate buffer, pH 8), AChE (dissolved in 0.215 M sodium phosphate buffer, pH 8.0), and the test compound are 200 µM, 2 µM and 100 µM, respectively. After co-incubation, 20 µL of the mixed solution was diluted to a final volume of 2 mL with ThT (1.5 µM in 50 mM glycine–NaOH buffer, pH 8.5) and the absorbance was measured with a multi-mode plate reader at the excitation and emission wavelength of λex = 450 nm and λem = 485 nm, respectively [45].
Neuroprotection assay against Aβ-induced damage
MTT reduction assay was used to evaluate the neuroprotective effect of compound 10h on neuronal PC12 cell damage induced by Aβ25-35. The cells were grown in monolayer culture on collagen-coated plates at 37 °C in a humidified atmosphere of 5% CO2. Neuronal PC12 cells were plated at a density of 5 × 105 cells/well on 96-well plates. The cells were pre-incubated with compound 10h for 3 h before human Aβ 25–35 (final concentration of 5 μM) was added. After 24 h, 90 μL the medium was taken out and 20 μL of MTT (0.5 mg/ml dissolved in RPMI containing phenol red) was added and incubated for an additional 2 h at 37 °C. The absorbance (A570 nm) was measured using a Bio-Rad microplate reader (Model 680, Bio-Rad). The details were reported in our previous work [33, 46].
Metal chelation studies
To study the metal chelating ability, the solutions of compound 10h and Fe2+, Cu2+, and Zn2+ ions (from FeSO4, CuCl2.2H2O, and ZnCl2) were prepared in methanol. The mixture of compound 10h (1 mL) and the test ion solutions (1 mL) with the same final concentration of 20 µM in a 1 cm quartz cuvette was incubated at room temperature for 30 min. Then, the absorption spectra were recorded in the range of 200–600 nm.
The stoichiometry of complex 10h-Cu2+ was also studied using the molar ratio method [47, 48]. The concentration of compound 10h was 20 μM and the final concentration of Cu2+ ranged from 0–44 μM with 4 μM intervals at 303.5 nm. The plot was obtained by the corresponding absorption versus mole fraction of Cu2+ to ligand 10h. All experiments were performed in triplicate.
Molecular docking
The molecular docking studies of the most potential ligand was performed on BuChE (PDB code: 4BDS) [39] to observe the binding orientation and consensual binding interactions using AutoDock 4.2. The X-ray crystal structure of the receptor was downloaded from the PDB database. All water and ligand molecules were removed from the structure, and the protein was prepared for docking. The co-crystallized ligand within the PDB structures was defined as a center of the binding site. All ligands were created using Chem3D Ultra software, and energy minimizations were done by the semiempirical MM+ [49]. The compounds were docked into the active site of proteins using default parameters for each ligand with 100 runs and 27,000 as the maximum number of generations. The grid boxes were set with 60, 60, and 60 points in the x, y, and z directions, respectively. All other options were set as default. The calculated geometries were ranked in terms of free energy of binding and the best pose was selected for further analysis. Molecular visualizations were performed by Discovery Studio 4.0 client software [20].
Prediction of ADME descriptors
ADME-Tox properties of the synthesized compounds were performed by using online servers especially https://lmmd.ecust.edu.cn:8000/predict/ and https://preadmet.bmdrc.kr.
Conclusion
In summary, a new series of 1,2,3-triazole-dimethylaminoacryloyl-chromenone derivatives were designed and synthesized as multifunctional anti-Alzheimer’s agents. All the target compounds were synthesized and screened as AChE/BuChE inhibitors. The most active compound was further evaluated by the multiple biological activities including Aβ1-42 aggregation inhibition, metal-chelating properties, and neuroprotective effects against Aβ25-35-induced PC12 cell injury. Our results showed that these compounds had a high inhibitory potency and selectivity towards BuChE with an IC50 value of 21.71 μM for 10h as the most potent BuChE inhibitor. The inhibition kinetic analysis revealed a mixed-type inhibition pattern for this compound. The molecular modeling study of the most potent compound 10h with BuChE indicated that it bound to both CAS and PAS of the BuChE. Moreover, this compound had a significant anti-Aβ aggregation capacity and served as a metal chelator. These results indicated that this hybridization approach could be a successful strategy for the further developments of potential multifunctional candidates against AD.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Alzheimer's disease
- AChE:
-
Acetylcholinesterase
- BuChE:
-
Butylcholinesterase
- CAS:
-
Catalytic active site
- PAS:
-
Peripheral anionic site
- Aβ:
-
Amyloid βeta
- APP:
-
Amyloid precursor protein
- BACE1:
-
β-Site APP cleaving enzyme-1
- NFTs:
-
Neurofibrillary tangles
- IC50 :
-
The half maximal inhibitory concentration
- 1H NMR:
-
Proton nuclear magnetic resonance
- 13C NMR:
-
Carbon-13 nuclear magnetic resonance
References
Maramai S, Benchekroun M, Gabr MT, Yahiaoui S (2020) Multitarget therapeutic strategies for Alzheimer’s disease: Review on emerging target combinations. BioMed Res Int 2020:5120230
Iraji A, Khoshneviszadeh M, Firuzi O, Khoshneviszadeh M, Edraki N (2020) Novel small molecule therapeutic agents for Alzheimer disease: focusing on BACE1 and multi-target directed ligands. Bioorg Chem 97:103649
Najafi Z, Mahdavi M, Saeedi M, Karimpour-Razkenari E, Asatouri R, Vafadarnejad F, Moghadam FH, Khanavi M, Sharifzadeh M, Akbarzadeh T (2017) Novel tacrine-1,2,3-triazole hybrids: in vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur J Med Chem 125:1200–1212
Rahman A (2018) Frontiers in clinical drug research-CNS and neurological disorders. Bentham Science Publishers, Sharjah
Li Q, Yang H, Chen Y, Sun H (2017) Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur J Med Chem 132:294–309
Vafadarnejad F, Karimpour-Razkenari E, Sameem B, Saeedi M, Firuzi O, Edraki N, Mahdavi M, Akbarzadeh T (2019) Novel N-benzylpyridinium moiety linked to arylisoxazole derivatives as selective butyrylcholinesterase inhibitors: synthesis, biological evaluation, and docking study. Bioorg Chem 92:103192
Campanari M-L, Navarrete F, Ginsberg SD, Manzanares J, Sáez-Valero J (2016) García-Ayllón M-S (2016) Increased expression of readthrough acetylcholinesterase variants in the brains of Alzheimer’s disease patients. J Alzheimers Dis 53(3):831–841
Darvesh S, Hopkins DA, Geula C (2003) Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 4(2):131
Maurice T, Strehaiano M, Siméon N, Bertrand C, Chatonnet A (2016) Learning performances and vulnerability to amyloid toxicity in the butyrylcholinesterase knockout mouse. Behav Brain Res 296:351–360
Dolles D, Hoffmann M, Gunesch S, Marinelli O, Möller J, Santoni G, Chatonnet A, Lohse MJ, Wittmann H-J, Strasser A (2018) Structure–activity relationships and computational investigations into the development of potent and balanced dual-acting butyrylcholinesterase inhibitors and human cannabinoid receptor 2 ligands with pro-cognitive in vivo profiles. J Med Chem 61(4):1646–1663
Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, Yu Q-S, Mamczarz J, Holloway HW, Giordano T (2005) Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc Natl Acad Sci 102(47):17213–17218
Soreq H: Human cholinesterases and anticholinesterases: Academic Press; 2012.
Lane RM, Potkin SG, Enz A (2006) Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int J Neuropsychopharmacol 9(1):101–124
Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F (2003) Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem 278(42):41141–41147
Tripathi MK, Sharma P, Tripathi A, Tripathi PN, Srivastava P, Seth A, Shrivastava SK (2020) Computational exploration and experimental validation to identify a dual inhibitor of cholinesterase and amyloid-beta for the treatment of Alzheimer’s disease. J Comput Aided Mol Des 34(9):983–1002
Sharma P, Srivastava P, Seth A, Tripathi PN, Banerjee AG, Shrivastava SK (2019) Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog Neurobiol 174:53–89
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y-S (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30(3):665–676
Zatta P, Drago D, Bolognin S, Sensi SL (2009) Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol Sci 30(7):346–355
Andrisano V, Naldi M, De Simone A, Bartolini M (2018) A patent review of butyrylcholinesterase inhibitors and reactivators 2010–2017. Expert Opin Ther Pat 28(6):455–465
Vafadarnejad F, Mahdavi M, Karimpour-Razkenari E, Edraki N, Sameem B, Khanavi M, Saeedi M, Akbarzadeh T (2018) Design and synthesis of novel coumarin-pyridinium hybrids: In vitro cholinesterase inhibitory activity. Bioorg Chem 77:311–319
Saeedi M, Safavi M, Karimpour-Razkenari E, Mahdavi M, Edraki N, Moghadam FH, Khanavi M, Akbarzadeh T (2017) Synthesis of novel chromenones linked to 1,2,3-triazole ring system: investigation of biological activities against Alzheimer’s disease. Bioorg Chem 70:86–93
Yazdani M, Edraki N, Badri R, Khoshneviszadeh M, Iraji A, Firuzi O (2020) 5,6-Diphenyl triazine-thio methyl triazole hybrid as a new Alzheimer’s disease modifying agents. Mol Divers 24:641–654
Moradi A, Faraji L, Nadri H, Hasanpour Z, Moghadam FH, Pakseresht B, Golshani M, Moghimi S, Ramazani A, Firoozpour L et al (2018) Synthesis, docking study, and biological evaluation of novel umbellipherone/hymecromone derivatives as acetylcholinesterase/butyrylcholinesterase inhibitors. Med Chem Res 27(7):1741–1747
Fang J, Li Y, Liu R, Pang X, Li C, Yang R, He Y, Lian W, Liu A-L, Du G-H (2015) Discovery of multitarget-directed ligands against Alzheimer’s disease through systematic prediction of chemical–protein interactions. J Chem Inf Model 55(1):149–164
Jiang Z, You Q, Zhang X (2019) Medicinal chemistry of metal chelating fragments in metalloenzyme active sites: a perspective. Eur J Med Chem 165:172–197
Kostova I, Bhatia S, Grigorov P, Balkansky S, Parmar VS, Prasad AK, Saso L (2011) Coumarins as antioxidants. Curr Med Chem 18(25):3929–3951
Tao D, Wang Y, Bao X-Q, Yang B-B, Gao F, Wang L, Zhang D, Li L (2019) Discovery of coumarin Mannich base derivatives as multifunctional agents against monoamine oxidase B and neuroinflammation for the treatment of Parkinson’s disease. Eur J Med Chem 173:203–212
Najafi Z, Mahdavi M, Saeedi M, Karimpour-Razkenari E, Edraki N, Sharifzadeh M, Khanavi M, Akbarzadeh T (2019) Novel tacrine-coumarin hybrids linked to 1,2,3-triazole as anti-Alzheimer’s compounds: In vitro and in vivo biological evaluation and docking study. Bioorg Chem 83:303–316
Jalili-Baleh L, Nadri H, Forootanfar H, Samzadeh-Kermani A, Küçükkılınç TT, Ayazgok B, Rahimifard M, Baeeri M, Doostmohammadi M, Firoozpour L et al (2018) Novel 3-phenylcoumarin-lipoic acid conjugates as multi-functional agents for potential treatment of Alzheimer’s disease. Bioorg Chem 79:223–234
Jiang N, Huang Q, Liu J, Liang N, Li Q, Li Q, Xie S-S (2018) Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem 146:287–298
Jalili-Baleh L, Forootanfar H, Küçükkılınç TT, Nadri H, Abdolahi Z, Ameri A, Jafari M, Ayazgok B, Baeeri M, Rahimifard M et al (2018) Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer’s disease based on coumarin and lipoic acid scaffolds. Eur J Med Chem 152:600–614
Sang Z, Wang K, Shi J, Liu W, Cheng X, Zhu G, Wang Y, Zhao Y, Qiao Z, Wu A et al (2020) The development of advanced structural framework as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur J Med Chem 192:112180
Iraji A, Firuzi O, Khoshneviszadeh M, Tavakkoli M, Mahdavi M, Nadri H, Edraki N, Miri R (2017) Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur J Med Chem 141:690–702
Jiang Y, Liu G, Wang X, Hu J, Zhang G, Liu S (2015) Cytosol-specific fluorogenic reactions for visualizing intracellular disintegration of responsive polymeric nanocarriers and triggered drug release. Macromolecules 48(3):764–774
Lewis WG, Green LG, Grynszpan F, Radić Z, Carlier PR, Taylor P, Finn M, Sharpless KB (2002) Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew Chem Int Ed 41(6):1053–1057
Ellman GL, Courtney KD, Andres V Jr, Featherstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7(2):88–95
Roldán-Peña JM, Romero-Real V, Hicke J, Maya I, Franconetti A, Lagunes I, Padrón JM, Petralla S, Poeta E, Naldi M et al (2019) Tacrine-O-protected phenolics heterodimers as multitarget-directed ligands against Alzheimer’s disease: Selective subnanomolar BuChE inhibitors. Eur J Med chem 181:111550
Drew SC (2017) The case for abandoning therapeutic chelation of copper ions in Alzheimer’s Disease. Front Neurosci 11:317
Nachon F, Carletti E, Ronco C, Trovaslet M, Nicolet Y, Jean L, Renard P-Y (2013) Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl-and butyryl-cholinesterase. Biochem J 453(3):393–399
Chandrasekaran B, Abed SN, Al-Attraqchi O, Kuche K, Tekade RK. Chapter 21—Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. In: Dosage Form Design Parameters. edn. Edited by Tekade RK: Academic Press; 2018. p. 731–755.
Liu G, Shi G, Sheng H, Jiang Y, Liang H, Liu S (2017) Doubly caged linker for and-type fluorogenic construction of protein/antibody bioconjugates and in situ quantification. Angew Chem 129(30):8812–8817
Rastegari A, Nadri H, Mahdavi M, Moradi A, Mirfazli SS, Edraki N, Moghadam FH, Larijani B, Akbarzadeh T, Saeedi M (2019) Design, synthesis and anti-Alzheimer’s activity of novel 1, 2, 3-triazole-chromenone carboxamide derivatives. Bioorg Chem 83:391–401
Saeedi M, Mohtadi-Haghighi D, Mirfazli SS, Mahdavi M, Hariri R, Lotfian H, Edraki N, Iraji A, Firuzi O, Akbarzadeh T (2019) Design and synthesis of selective acetylcholinesterase inhibitors: arylisoxazole-phenylpiperazine derivatives. Chem Biodivers 16(2):e1800433
Mahdavi M, Hariri R, Mirfazli SS, Lotfian H, Rastergari A, Firuzi O, Edraki N, Larijani B, Akbarzadeh T, Saeedi M (2019) Synthesis and biological activity of some benzochromenoquinolinones: tacrine analogs as potent anti-Alzheimer’s agents. Chem Biodivers 16(4):e1800488
Jalili-Baleh L, Nadri H, Moradi A, Bukhari SNA, Shakibaie M, Jafari M, Golshani M, Homayouni Moghadam F, Firoozpour L, Asadipour A et al (2017) New racemic annulated pyrazolo[1,2-b]phthalazines as tacrine-like AChE inhibitors with potential use in Alzheimer’s disease. Eur J Med Chem 139:280–289
Yazdani M, Edraki N, Badri R, Khoshneviszadeh M, Iraji A, Firuzi O (2019) Multi-target inhibitors against Alzheimer disease derived from 3-hydrazinyl 1,2,4-triazine scaffold containing pendant phenoxy methyl-1,2,3-triazole: design, synthesis and biological evaluation. Bioorg Chem 84:363–371
Iraji A, Firuzi O, Khoshneviszadeh M, Nadri H, Edraki N, Miri R (2018) Synthesis and structure-activity relationship study of multi-target triazine derivatives as innovative candidates for treatment of Alzheimer’s disease. Bioorg Chem 77:223–235
Karamać M (2009) Chelation of Cu (II), Zn (II), and Fe (II) by tannin constituents of selected edible nuts. Int J Mol Sci 10(12):5485–5497
Iraji A, Adelpour T, Edraki N, Khoshneviszadeh M, Miri R, Khoshneviszadeh M (2020) Synthesis, biological evaluation and molecular docking analysis of vaniline-benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem 14(1):28
Acknowledgments
The authrs acknowledged the support from Tehran University of Medical Sciences and National Institute for Medical Research Development.
This paper is dedicated to the memory of our unique teacher in Chemistry and Medicinal Chemistry, Professor Abbas Shafiee (1937–2016)
Funding
This work was supported by grants from National Institute for Medical Research Development with project No. 958756.
Author information
Authors and Affiliations
Contributions
MS contributed to the design and characterization of compounds as well as preparation of the manuscript. HKA synthesized compounds. AI performed docking study and contributed to the preparation of the manuscript. AR performed the biological assay. SNAB and OF supervised the biological tests. TA supervised all phases of the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The manuscript does not contain studies with animal subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
The supplementary file include copies of NMR spectra and elemental analysis report.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Karimi Askarani, H., Iraji, A., Rastegari, A. et al. Design and synthesis of multi-target directed 1,2,3-triazole-dimethylaminoacryloyl-chromenone derivatives with potential use in Alzheimer's disease. BMC Chemistry 14, 64 (2020). https://doi.org/10.1186/s13065-020-00715-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-020-00715-0