
Abstract
Neem (Azadirachta indica) extract is well-known as a natural pesticide for the control of agricultural pests. Azadirachtin A and its structural analogues are considered as active compounds. However, the amounts of azadirachtins varies in neem extracts, providing a variety of insecticidal activities. In this study, a novel method of automated online solid-phase extraction coupled with liquid chromatography/quadrupole-time-of-flight mass spectrometry (SPE-LC–Q-TOF–MS) was developed and validated for simultaneous quantification of five azadirachtins (azadirachtins A, B, D, H and I) in seed and leaf extracts of A. indica. Different experimental parameters (such as SPE cartridge, injection volume and washing step) were optimized. The optimized SPE-LC–Q-TOF–MS method showed good recovery (82.0–102.8%), linearity (r2 ≥ 0.9991) and precision (0.83–4.83%). The limit of detections (LODs) for the five analytes ranged from 0.34 to 0.76 ng mL−1. The validated method was successfully applied for determination of the analytes in the neem leaves and seeds from different locations and a neem formulation. The online SPE-LC–Q-TOF–MS method was found to be a simple, precise and accurate and can be used as a powerful tool for quality control of neem extracts or its formulations.
Similar content being viewed by others


Introduction
Neem (Azadirachta indica) belongs to the family Meliaceae that is well-known for its insecticidal and biomedical properties [1]. For example, the leaf and seed extracts are applied to treat infestations of lice, a common use in Europe [2]. The neem extract has been found to possess many bioactive properties, such as antioxidant [3], antiviral [4], antitumor [5], antimalarial [6] as well as antifungal [7] activities. The neem extracts are rich in limonoids, which could be responsible for these widespread activities. Among the limonoids, azadirachtin A and its structural analogues are considered as active compounds in natural bio-pesticides, which are also considered to be biodegradable and environmental safety [8].
The amounts of azadirachtins in neem extracts varies in different parts of the plant, providing a variety of pesticidal activities [9]. The neem based formulations may show the wide variability in the content of the active principles, which affects the efficacy, reliability and quality of the products [10]. Therefore, each azadirachtin compound and its exact concentration are important for the quality control of neem extracts or its formulations. The analytical methods in relation to neem metabolites have been developed, such as enzyme-linked immunosorbent assay [11], high performance liquid chromatography (HPLC) [12,13,14] and liquid chromatography–mass spectrometry (LC–MS) [15, 16]. The HPLC methods often applied in the quantification of azadirachtins, but its absorption wavelength is at very short zone where the solvents peaks absorb strongly [9]. Furthermore, the interfering components can not be easily removed by simple purification methods.
Online solid-phase extraction (online-SPE) method could be a good choice for sample purification. Online-SPE technology is a fully automated method for sample preparation that allows direct injection of samples for analysis [17]. This procedure is not only faster than manual samples pre-treatment, but can improve reproducibility [18]. Online-SPE coupled with LC–MS has been successfully applied for qualitative and quantitative analysis of the chemical constituents in plant samples [19].
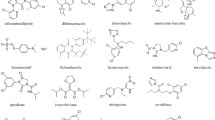
Online SPE coupled with liquid chromatography/quadrupole-time-of-flight tandem mass spectrometry (LC–Q-TOF–MS) is a powerful strategy, that could be used for the analysis of five azadirachtins (Fig. 1), including azadirachtin A (AZ-A), azadirachtin B (AZ-B), azadirachtin D (AZ-D), azadirachtin H (AZ-H) and azadirachtin I (AZ-I). The aim of this study was to develop and validate a fully automated online SPE-LC–Q-TOF–MS method for determination of the five azadirachtins in the leaf and seed extracts of A. indica.

Chemical structures of the five investigated azadirachtins A, B, D, H and I
Materials and methods
Plant materials and chemicals
Different seeds (No. S1, No. S2 and No. S3) of A. indica were collected from Yuanmou County (101°51′E, 25°40′N), Yuanjiang County (102°02′E, 23°61′N), and Jianshui County (102°86′E, 23°22′N), Yunnan Province, China, respectively, in August 2017. Neem leaves (No. L1 and L2) were collected from Yuanjiang County (102°02′E, 23°61′N), Yunnan Province, China. The neem leaves were air dried under shade, ground to powder, and stored at − 20 °C. The neem seeds were manually removed from the fruits and ground in an iced mortar with liquid nitrogen.
HPLC-grade methanol (MeOH) and acetonitrile (ACN) were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Sodium acetate was purchased from CNW Technologies GmbH (Dusseldorf, Germany). Standards of azadirachtins A, B, D, H and I were prepared in our laboratory with purity greater than 95% using HPLC method [20]. Neem pesticide formulation (0.6% azadirachtin EC) was purchased from the market.
Sample preparation
Sample extraction was based on the previous study with some modifications [21]. A portion (0.10 g) of well-homogenized powdered leaves or seeds was weighted in a 40 mL glass bottle. After adding 20 mL of 70% (v/v) acetonitrile to the bottle, the mixture was extracted in an ultrasonic cleaning bath (KQ-800E, 800W, Kunshan Ultrasonic Instruments Co., Ltd., Kunshan, China) for 30 min. As to the seed samples, the extraction step was repeated twice. The leaf samples were extracted only once. After centrifugation at 5000 rpm for 5 min, 1 mL of supernatant was transferred into a 10 mL volumetric flask and diluted to volume with water.
The neem pesticide formulation (50 μL) was dissolved in 10 mL of acetonitrile and extracted by ultrasonic assisted method for 5 min. One mL of sample was transferred into a 10 mL volumetric flask and diluted to volume with water. The final sample solution was passed through a syringe filter membrane (0.22 µm) before injection.
Online SPE-LC system conditions
Online SPE-LC separation was performed on a Symbiosis™ Pico system (Spark Holland, Emmen, Netherlands) equipped with an auto-sampler with a 100 µL sample loop, a high pressure dispenser (HPD) module and two binary LC pumps. SPE cartridges were used for sample concentration and cleanup. Three different SPE cartridges, including HySphere™ C18 HD (10 × 2 mm i.d., 7 μm), HySphere™ Resin SH (10 × 2 mm i.d., 15–25 μm) and HySphere™ Resin GP (10 × 2 mm i.d., 10–12 μm) were tested. Sample was injected and loaded onto the cartridge for online sample clean-up and concentration. Different sample volumes (5, 10, 20, 35 and 50 µL) were tested. The flow rate of loading phase was maintained at 700 µL min−1 and kept for 1 min. All the tests were carried out in triplicate. The loading phase selected was 10% MeOH. High pressure dispenser (HPD) mode with peak focusing was selected. The SPE parameters were listed in Table 1.
The washing step was optimized to remove interferences from the SPE column. The optimized washing step was carried out using spiked standard samples, including AZ-A (375 ng mL−1), AZ-B (75 ng mL−1), AZ-D (50 ng mL−1), AZ-H (25 ng mL−1) and AZ-I (12.5 ng mL−1). After the washing step, the target analytes were eluted from the SPE cartridge, followed by remixing with the LC eluent, resulting in a total flow rate of 400 μL min−1 onto an analytical column. The chromatographic separation was performed on a C18 column (150 mm × 2.1 mm i.d., 3.5 µm, Zorbax Eclipse XDB, Agilent USA) at 25 °C. The LC mobile phase consisted of H2O (solvent A) and ACN (solvent B) with 10 μM sodium acetate, respectively. The gradient program was as follows: 0–2 min, 10% B; 2–2.08 min, 10–50% B; 2.08–2.5 min, 50–40% B; 2.5–7 min, 40% B; 7–7.08 min, 40–90% B; 7.08–10 min, 90% B; 10–10.08 min, 90–10% B; 10.08–12 min, 10% B. The flow rate was set at 0.25 mL min−1 in the first 2 min, then the flow rate was set at 0.4 mL min−1.
MS spectrometry
The quantitative analysis of the five analytes was carried out using an Agilent 6540 Q-TOF–MS system (Agilent Technologies, Santa Clara, CA, USA) equipped with a jet stream ESI interface. The MS data were obtained in a MS scan mode. Mass spectra were recorded from m/z 50 to 800 in positive ionization mode. The optimized mass analysis conditions were as follows: drying gas (N2) flow rate, 10 L min−1; drying gas temperature, 350 °C; nebulizer, 310 kPa; sheath gas temperature, 250 °C; capillary voltage, 4000 V; fragmentor voltage, 140 V; nozzle voltage, 500 V; octopole RF voltage, 750 V. All the operations and data analysis were controlled using an integrated software system including Symbiosis Pico in Analyst™ version 1.2.00 (Spark Holland) and MassHunter B.04.00 software (Agilent Technologies, USA).
Calibration curves and limits of detection
Stock solutions of the five analytes (AZ-A AZ-B AZ-D, AZ-H and AZ-I) were prepared in methanol at concentrations of 3000, 1200, 800, 400 and 200 μg mL−1, respectively. Working solutions were prepared by diluting aliquots of stock solutions with 10% methanol. The desired calibration concentrations were obtained using two-fold serial dilutions. The calibration curves for the five analytes were constructed by plotting the peak area (EIC signal of MS) against the concentration at least seven concentrations. According to ICH guideline [22], the limit of detection (LOD) and limit of quantification (LOQ) were calculated as 3.3σ/S and 10σ/S, where S is the slope of the calibration plot and σ is the standard deviation of the response.
Accuracy, precision and repeatability
The accuracy of the method was calculated by spike-recovery experiments, which was evaluated by adding three concentration levels (low, middle and high) of standard solutions into the seed and leaf samples. The samples of each level were spiked in triplicates. Then the mixtures were analyzed according to the developed method.
Intra- and inter-day variations were used to test the precision of the proposed method. For intra-day precision, the solution of seed sample was analyzed for six replicates in 1 day. For inter-day test, the seed sample was analyzed in duplicates for 3 days consecutively. Six independent samples (sample No. S2) were analyzed in parallel for the measurement of repeatability. All of these treatments were judged with relative standard deviation (RSD).
Method application
The final developed method has been applied for the identification and simultaneous quantification of five azadirachtins in the seeds and leaves of neem, and a commercial product of neem pesticide formulation. The identification of the five analytes was performed by comparing accurate mass and their retention times with those of standard compounds.
Statistical analysis
Statistical significance was carried out applying one-way ANOVA followed by Duncan’s test at p = 0.05, using SPSS Statistics version 20.0 (SPSS Inc., Chicago, IL, USA). Origin Pro software (Version: 8.5.0 SR1) was used to fit the data and draw the figures.
Results and discussion
Optimization of LC–Q-TOF–MS conditions
Different mobile phase compositions such as acetonitrile–water and methanol–water solvents were tested. To obtain stable product ions and high responses, 10 μM sodium acetate was added into the mobile phase. The gradient mode of acetonitrile–water solvents as the mobile phase, were better than methanol–water for a satisfactory MS response and chromatographic resolution. The positive ionization mode was selected for the quantification and identification of the five analytes for its most intense response. A good separation of all the five analysts were obtained in a short runtime (8 min). Furthermore, MS parameters including fragmentor voltage and drying gas temperature were optimized. The extraction ion current (EIC) chromatograms of the five analytes are shown in Fig. 2.

Liquid chromatography/quadrupole-time-of-flight mass spectrometry (LC–Q-TOF–MS) extraction ion current (EIC) of five standards. Peaks a, b, c, d and e correspond to azadirachtins I, H, D, A, and B
Optimization of online-SPE conditions
Recovery of online SPE cartridges
The choice of SPE adsorbent material is an important factor for obtaining high recovery [23]. The sample purification step was necessary to remove the possible interference for the determination of azadirachtins using LC or LC–MS [24, 25]. The azadirachtins possess the characteristics of medium polarity, and therefore medium-polar SPE cartridges were considered. Three different SPE cartridges were evaluated. The results showed that HySphere™ C18 HD cartridge provided a good recovery and reproducibility (Fig. 3). Thus, the HySphere C18 HD cartridge was selected in this study. In our laboratory, HySphere C18 HD cartridges could be used repeatedly at least ten times by washing with 1 mL of methanol followed aqueous solvents each time. This means a decrease in the cost and low consumption of organic solvents.

Comparison of recoveries for the five analytes, including azadirachtin A, B, D, H and I, based on three type of SPE cartridges. Standard deviation represented by error bars (n = 3)
Injection volume
The amount of sample loaded on SPE cartridge affects the sensitivity of the analytical method [26]. The effect of sample injection volume on peak area of the analytes was investigated. Peak areas were plotted versus injection volumes to produce five linear curves (Fig. 4). All the curves showed a good linear relationship (r2 > 0.997). No sample breakthrough was observed within the tested range. The peak areas of the five azadirachtins increased with the increasing of sample volumes, thus the increasing of method sensitivity. To establish a more sensitive method for determination of the five azadirachtins, a relatively larger volume (50 µL) was selected as injection volume using the auto-sampler.

Linear curves of injection volumes and peak areas of the five azadirachtins
Optimization of methanol percentage for loading phase
After injection, the sample was withdrawn into a sample loop and then carried over by the loading phase from a high pressure dispenser (HPD) pump. The composition of methanol in the loading phase effects the recovery of the analytes [27]. The loading phase composition of methanol and water were evaluated in the range of 0–30% with the increment of 10% each time. The satisfactory recoveries were acquired using pure water or 10% MeOH as the loading phase (Fig. 5). Additionally, a significant inverse relation was observed between the methanol percentage of the loading phase and the absolute recoveries of the analytes. The reason for this is the fact that the loading phase with high percentage of methanol could lead to premature column breakthrough.

Comparison of the recoveries of five analytes, including azadirachtin A, B, D, H and I, with four different percentages of methanol during loading phase (n = 3)
Optimization of methanol percentage for washing phase
After sample loading, the composition of washing phase was a significant factor for cleanup step [28]. Five different percentages of methanol were investigated ranging from 0 to 40% with an increment of 10% each time. The recoveries of the analytes were tested for the influence of methanol percentage during the washing phase. The recoveries of all the analytes decreased obviously while the 40% methanol was used (Fig. 6). Therefore, 30% methanol was selected as washing phase as it allowed the best recoveries in the case of remove interferences.

Comparison of the recoveries of five analytes, including azadirachtin A, B, D, H and I, with five different percentages of methanol during washing phase (n = 3)
Method validation
The calibration curves, linear ranges, limits of detection (LOD) and limits of quantification (LOQ) values of five azadirachtins were carried out using an online-SPE-LC–Q-TOF–MS method (Table 2).
The correlation coefficient values (r2 ≥ 0.9991) demonstrated good correlation with given concentration ranges. The external calibration curves were constructed by using polynomial regression. The sensitivity expressed as LOD and LOQ were less than 0.76 and 2.30 ng mL−1, respectively.
The RSD values of the peak areas of the five analytes were with the range of 2.12–4.55%. The results for intra-day (0.83–4.62%) and the inter-day (1.67–4.83%) showed good precision. Meanwhile, the retention time variations (RSD) were less than 0.11 and 0.26%, respectively (Table 3).
Good recoveries of 82.0–102.8% with RSD of 0.04–8.11% were obtained in this study (Table 4).
Analysis of neem samples
The proposed method was successfully applied to analyze the five azadirachtins in A. indica from different locations. The contents of the seed and leaf extracts (n = 3) of five azadirachtins and also the neem formulation (n = 3) are shown in Table 5.
Because seeds contain the highest concentrations of azadirachtins, most commercial preparations of neem are derived from seed extracts [29]. The commercial products of the neem extracts are usually evaluated by measuring the content of azadirachtin A [30]. Azadirachtins A was the most frequently detected compound in all the neem samples, and the five analytes were also found in the neem formulation (Table 5). According to the previous reports, the neem seeds are considered to be the most abundant source, of which the content of azadirachtin A can reach up to 5419.08 μg g−1, whereas the content of azadirachtin A in the neem leaves was 182.42 μg g−1 [31]. In this study, the contents of azadirachtin A ranged from 3862.9 to 4852.1 μg g−1 in neem seeds. The content of azadirachtin A in the neem leaf extract (sample No. L2) was 969.9 μg g−1. The main mass data of the five azadirachtins from neem samples are shown in Additional file 1: Table S1. The contents of azadirachtins in neem seeds were higher than those in neem leaves. Generally, the environmental factors such as climatic and soil conditions can affect chemical composition of the plants. In the previous studies [32, 33], wide variations have been found in azadirachtin contents of neem seeds from different provenances and also between individual trees of a particular location. It has been proved that the variations in azadirachtins are attributed to individual genetic differences among neem trees other than climatic factors [33]. Additionally, azadirachtin is very labile when exposed to air, moisture and sunlight. Its instability to UV radiation may also affect the percentage of azadirachtin present in neem seeds or leaves [25].
Neem extracts and pure azadirachtin are one of the most significant insecticides authorized for organic farming crop protection in many countries, which are used to control agricultural pests [34]. An analysis of A. indica is very important as quality control, since the primary interest is its insecticide activity [35]. Therefore, the selected five azadirachtins found in all the neem seeds were suitable as marker compounds for quality control of the neem extracts. Furthermore, these results indicate the proposed method is a useful tool for determination of the five markers in A. indica from different locations. Further studies on the qualitative and quantitative analysis of the other limonoids found in traces and existed synergy among constituents in the extracts of A. indica are needed.
Conclusions
A fully automated online SPE-LC–Q-TOF–MS method was developed for the simultaneous determination of five azadirachtins in the seed and leaf extracts of A. indica. The online SPE-LC system was able to provide high throughput sample preparation, good reproducibility and large volume sample injection. The Q-TOF–MS system enabled the identification of the five azadirachtins with high selectivity. The method was validated and found to be precise, accurate and sensitive. The proposed method was successful applied to quantify the five azadirachtins in different neem samples and a neem formulation. The online SPE-LC–Q-TOF–MS method can be used as a tool for quality control of neem plant or its formulations.
References
Farah MA, Ateeq B, Ahmad W (2006) Antimutagenic effect of neem leaves extract in freshwater fish, Channa punctatus evaluated by cytogenetic tests. Sci Total Environ 364:200–214
Edwards SE, Rocha IDC, Williamson EW, Heinrich M (2015) Phytopharmacy: an evidence-based guide to herbal medical products, 1st edn. Wiley-blackwell, Chichester
Manikandan P, Letchoumy PV, Gopalakrishnan M, Nagini S (2008) Evaluation of Azadirachta indica leaf fractions for in vitro antioxidant potential and in vivo modulation of biomarkers of chemoprevention in the hamster buccal pouch carcinogenesis model. Food Chem Toxicol 46:2332–2343
Faccin-Galhardi LC, Yamamoto KA, Ray S, Ray B, Linhares REC, Nozawa C (2012) The in vitro antiviral property of Azadirachta indica polysaccharides for poliovirus. J Ethnopharmacol 142:86–90
Wu Q, Kohli M, Bergen HR III, Cheville JC, Karnes RJ, Cao H, Young CYF, Tindall DJ, McNiven MA, Donkena KV (2014) Preclinical evaluation of the supercritical extract of Azadirachta indica (neem) leaves in vitro and in vivo on inhibition of prostate cancer tumor growth. Mol Cancer Ther 13:1067–1077
Udeinya IJ, Mbah AU, Chijioke CP, Shu EN (2004) An antimalarial extract from neem leaves is antiretroviral. Trans Roy Soc Trop Med Hyg 98:435–437
Ali EOM, Shakil NA, Rana VS, Sarkar DJ, Majumder S, Kaushik P, Singh BB, Kumar J (2017) Antifungal activity of nano emulsions of neem and citronella oils against phytopathogenic fungi, Rhizoctonia solani and Sclerotium rolfsii. Ind Crop Prod 108:379–387
Tomé HVV, Martins JC, Corrêa AS, Galdino TVS, Picanço MC, Guedes RNC (2013) Azadirachtin avoidance by larvae and adult females of the tomato leafminer Tuta absoluta. Crop Prot 46:63–69
Morgan ED (2009) Azadirachtin, a scientific gold mine. Bioorg Med Chem 17:4096–4105
Gunasekaran S, Anita B (2010) Analysis of phytochemical variability in neem formulations. Indian J Nat Prod Resour 1:291–295
Hemalatha K, Venugopal NBK, Rao BS (2001) Determination of azadirachtin in agricultural matrixes and commercial formulations by enzyme-linked immunosorbent assay. J AOAC Int 84:1001–1010
Paula JAMD, Brito LF, Caetano KLFN, Rodrigues MCDM, Borges LL, Conceição ECD (2016) Ultrasound-assisted extraction of azadirachtin from dried entire fruits of Azadirachta indica A. Juss. (Meliaceae) and its determination by a validated HPLC-PDA method. Talanta 149:77–84
Forim MR, Silva MFDGFD, Cass QB, Fernandes JB, Vieira PC (2010) Simultaneous quantification of azadirachtin and 3-tigloylazadirachtol in Brazilian seeds and oil of Azadirachta indica: application to quality control and marketing. Anal Methods 2:860–869
Shukla G (2013) Estimation and quantification of secondary metabolites and comparative quantification of azadirachtin in neem (Azadirachta indica) leaf samples collected from different pollution and population levels. Bull Pure Appl Sci 32B:41–48
Schaaf O, Jarvis AP, Esch SAVD, Giagnacovo G, Oldham NJ (2000) Rapid and sensitive analysis of azadirachtin and related triterpenoids from neem (Azadirachta indica) by high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J Chromatogr A 886:89–97
Barrek S, Paisse O, Grenier-Loustalot MF (2004) Analysis of neem oils by LC–MS and degradation kinetics of azadirachtin-A in a controlled environment. Characterization of degradation products by HPLC–MS–MS. Anal Bioanal Chem 378:753–763
Schebb NH, Inceoglu B, Rose T, Wagner K, Hammock BD (2011) Development of an ultra fast online-solid phase extraction (SPE) liquid chromatography electrospray tandem mass spectrometry (LC–ESI-MS/MS) based approach for the determination of drugs in pharmacokinetic studies. Anal Methods 3:420–428
Nováková L, Vlčková H (2009) A review of current trends and advances in modern bio-analytical methods: chromatography and sample preparation. Anal Chim Acta 656:8–35
Chen ML, Fu XM, Liu JQ, Ye TT, Hou SY, Huang YQ, Yuan BF, Wu Y, Feng YQ (2012) Highly sensitive and quantitative profiling of acidic phytohormones using derivatization approach coupled with nano-LC–ESI–Q-TOF-MS analysis. J Chromatogr B 905:67–74
Gao Q, Sun J, Xun H, Yao X, Wang J, Tang F (2017) A new azadirachta from the crude extracts of neem (Azadirachta Indica A. Juss) seeds. Nat Prod Res 31:1739–1746
Yang F, Liu Z, Zhen D, Lin YH, Chen J, Ruan J, Chen G (2011) Determination of botanical insecticides residues in fish by liquid chromatography–electrospray tandem mass spectrometry. Food Anal Methods 4:601–607
ICH guideline Q2 (R1) Validation of analytical procedures: text and methodology. In: International conference on harmonization of technical requirements for registration of pharmaceuticals for human use. 2005. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf. Accessed 1 Jan 2018
Zhao RS, Yuan JP, Jiang T, Shi JB, Cheng CG (2008) Application of bamboo charcoal as solid-phase extraction adsorbent for the determination of atrazine and simazine in environmental water samples by high-performance liquid chromatography-ultraviolet detector. Talanta 76:956–959
Beltrán E, Ibáñez M, Gracialor E, Sancho JV, Hernández F, Thompson DG (2014) Application of liquid chromatography/mass spectrometry in assessment of potential use of azadirachtins (TreeAzinTM) against Asian longhorned beetle. Anal Methods 6:8063–8071
Ramesh A, Balasubramanian M (1999) Rapid preconcentration method for the determination of azadirachtin-A and -B, nimbin and salannin in neem oil samples by using graphitised carbon solid phase extraction. Analyst 124:19–21
Subari SNM, Osman R, Saim N (2017) Direct analysis of six pharmaceuticals using online solid phase extraction liquid chromatography. Am J Appl Sci 14:517–525
Ciofi L, Fibbi D, Chiuminatto U, Coppini E, Checchini L, Bubba MD (2013) Fully-automated on-line solid phase extraction coupled to high-performance liquid chromatography-tandem mass spectrometric analysis at sub-ng/L levels of selected estrogens in surface water and wastewater. J Chromatogr A 1283:53–61
Caboni P, Sarais G, Angioni A, Garcia AJ, Lai F, Dedola F, Cabras P (2006) Residues and persistence of neem formulations on strawberry after field treatment. J Agric Food Chem 54:10026–10032
Liu L, Wen YB, Liu KN, Sun L, Wu M, Han GF, Lu YX, Wang QM, Yin Z (2013) Optimization of on-line solid phase extraction and HPLC conditions using response surface methodology for determination of WM-5 in mouse plasma and its application to pharmacokinetic study. J Chromatogr B 923–924:8–15
Sharma V, Walia S, Kumar J, Nair MG, Parmar BS (2003) An efficient method for the purification and characterization of nematicidal azadirachtins A, B, and H, using MPLC and ESIMS. J Agric Food Chem 51:3966–3972
Rangiah K, Varalaxmi BA, Gowda M (2016) UHPLC-MS/SRM method for quantification of neem metabolites from leaf extracts of Meliaceae family plants. Anal Methods 8:2020–2031
Sidhu OP, Kumar V, Behl HM (2004) Variability in triterpenoids (nimbin and salanin) composition of neem among different provenances of India. Ind Crop Prod 19:69–75
Sidhu OP, Kumar V, Behl HM (2003) Variability in neem (Azadirachta indica) with respect to azadirachtin content. J Agric Food Chem 51:910–915
Drozdzynski D, Kowalska J (2009) Rapid analysis of organic farming insecticides in soil and produce using ultra-performance liquid chromatography/tandem mass spectrometry. Anal Bioanal Chem 394:2241–2247
Forim MR, Cornelio VE, Da SM, Rodrigues-Filho E, Fernandes JB, Vieira PC, Matinez SS, Napolitano MP, Yost RA (2010) Chemical characterization of Azadirachta indica grafted on Melia azedarach and analyses of azadirachtin by HPLC–MS–MS (SRM) and meliatoxins by MALDI-MS. Phytochem Anal 21:363–373
Authors’ contributions
LS performed all experimental work and data analysis; JW participated in the design of the study and writing the manuscript; QG and XM performed samples extraction; YW and YZ contributed to samples collection and pretreatment; HX and XY contributed reagents and chemicals; FT as project leader, participated in the design of the study and participated in sample preparation. All authors read and approved the final manuscript.
Acknowledgements
All authors are thankful to the financial support from the National Key Research and Development Program of China (Grant Number: 2016YFD0600801), China Postdoctoral Science Foundation (Grant Number: 2016M600975), and the Central Public-Interest Scientific Institution Basal Research Fund, China (Grant Number: 1632014009). The authors are thankful to Senior Engineer Xingmin Peng, Research Institute of Resource Insects, Chinese Academy of Forestry, Kunming, China, who authenticated all the plant samples.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data and materials are fully available without restriction.
Consent for publication
The authors declare that the copyright belongs to the journal.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This study was funded by the National Key Research and Development Program of China (Grant Number: 2016YFD0600801), China Postdoctoral Science Foundation (Grant Number: 2016M600975), and the Central Public-Interest Scientific Institution Basal Research Fund, China (Grant Number: 1632014009).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1: Table S1.
Mass data of the five azadirachtins from neem samples by online-SPE-LC-Q-TOF–MS.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Song, L., Wang, J., Gao, Q. et al. Simultaneous determination of five azadirachtins in the seed and leaf extracts of Azadirachta indica by automated online solid-phase extraction coupled with LC–Q-TOF–MS. Chemistry Central Journal 12, 85 (2018). https://doi.org/10.1186/s13065-018-0453-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0453-y