
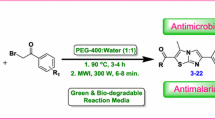
Abstract
Background and objective
A series of thiazolopyrimidine derivatives have been synthesized via multicomponent reaction and tested for biological activities. This research aims to develop a new synthetic method of poly fused pyrimidines under microwave irradiation. 6-Amino-4-aryl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitriles reacted with bromomalono-nitrile to give 3,7-diamino-5-aryl-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitrile more willingly than the isomeric 7H-thiazolo[3,2-a]pyrimidines. Thiazolopyrimidine derivatives reacted with carbon disulphide to produce 11-aryl-11H-1,2,3,4,7,8,9,10-octahydropyrimido[4″,5″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyrimidine-2,4,8,10-tetrathione. The above mentioned reactions were established by using both conventional methods and microwave-assisted irradiation.
Conclusion
This work provides a new method for preparing poly fused pyrimidines. The microwave-assisted technique is preferable due to the yield enhancements attained, time saving, and environmental safety reactions. The newly prepared compounds were verified for their antimicrobial activities. Also, the absorption and emission of some of the prepared compounds were studied.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Background
Pyrimidine derivatives are found to have a wide range of chemotherapeutic effects including angiogenic [1], enzyme inhibitory effects [2, 3] and anti-leshiminal activity [4]. They have also been used as analgesics and anti-parkinsonian agents [5, 6], as modulators of TRPV1 (Transient Receptor Potential Vanilloid Receptor 1) [7], as anticancer agents [8,9,10], as pesticides [11], as phosphate inhibitors [12, 13], for treatment of circulatory system diseases [14]. They are also known to have antimicrobial [15,16,17], anti-inflammatory [18], and anti-insecticidal [19] properties in addition to acetyl cholinesterase inhibitory activity [20]. Thiazolopyrimidine and thiazolo-pyrimidopyrimidine compounds have attracted our interest due to the wide range of biological activities they exhibit. For instance, thiazolopyrimidines are known to exhibit hypoglycemic, hypolipidemic, antidiabetic [21] and antibacterial and anti-tubercular activities [22]. The microwave technique has many benefits over conventional synthetic methods. Reduction of reaction times, minimization energy consumption, management of analytical waste, improving yields and increasing safety for the operator were the main benefits of this technique [23,24,25,26,27,28]. The use of microwave depends on the ability of the reacting molecules to efficiently absorb microwave energy taking advantage of microwave dielectric heating phenomena such as dipolar polarization or ionic conduction mechanisms. This leads to rapid internal heating (in-core volumetric heating) by direct interaction of electromagnetic radiation with the reacting molecules. Even though diverse types of microwave reactors and processing options are available currently, most of the microwave synthetic protocols have been reported in sealed reactors [29]. The rapid heating and high temperatures resulting in microwave chemistry makes it obvious based on the application of the Arrhenius equation, [k = A exp(− Ea/RT)] that transformations that reach completion in hours under conventional heating in a solvent, would be completed in only minutes using superheated solvents under microwave conditions using a autoclave type sealed reactor. In addition the rapid heating generally produced in microwave chemistry may sometimes lead to altered product distributions as compared to reactions conducted under conventional heating if the product distribution is determined by complex temperature dependent kinetics [29, 30]. This may be the reason why in many instances reactions performed under microwave irradiation at an optimized reaction temperature lead to lesser side products in comparison to reactions performed under conventional heating where the reaction temperature is often non-optimal [29,30,31]. Encouraged by the findings of the previously reported work [34,35,36] we herein report the use of microwave-assisted technique for preparing new derivatives of a series of thiazolopyrimidine and thiazolopyrimidothiazolopyrimidine for evaluation of their antimicrobial activity. The absorption and fluorescence emission of some of the prepared compounds were studied in dioxane, revealing that the substituents altered both the absorption and fluorescence emission maxima.
Results and discussion
Chemical characterization
The above discussed medicinal and biological properties of fused pyrimidine derivatives, prompted us to carry out the synthesis of a series of new thiazolopyrimidine and thiazolodipyrimidine derivatives using microwave chemistry in conjunction with conventional chemical synthesis. The reaction of bifunctional reagents with 6-amino-4-aryl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives 1a–d, afforded a simple and efficient approach for the synthesis of the target molecules. The synthesized target molecules were evaluated for their antimicrobial activity. The starting materials 1a–d were obtained by the one pot reaction of aromatic aldehydes, malononitrile and thiourea in an alcoholic sodium ethoxide solution (Scheme 1). Compounds 1a–d were characterized using elemental analysis as well as spectroscopic data. Compounds 1a, b were prepared according to literature procedures [33, 40].

Synthesis of pyrimidine-5-carbonitriles 1a–d
The IR (ʋ, cm−1) spectra of 1a–d showed absorption bands at 3350, 3270 and 3180 (NH, NH2), 3050, 2980 (CH), 2217 (CN). 1H-NMR (DMSO-d6) of 1d, as an example, showed signals δ (ppm) at 5.02 (s, 1H, pyrimidine H-4), 6.65 (s, 2H, NH2, D2O exchangeable), 7.23 (d, 2H, J = 7.8 HZ, aromatic protons), 7.51 (d, 2H, J = 7.8 HZ, aromatic protons), 8.65 (s, 1H, NH, D2O exchangeable) and 9.53 (s, 1H, NH, D2O exchangeable). Its 13C-NMR (DMSO-d6) showed signals δ (ppm) at 54.5 (pyrimidine C-4), 62.3 (pyrimidine C-5), 112.2, 117.1, 127.1, 133.2, 141.2, 160.5 (aromatic carbons + CN and pyrimidine C-6) and 175.3 (C=S). Mass spectrum of 1d, as an example, showed the molecular ion Peak at m/z 247 (8.5%) corresponding to the molecular formula C11H9N4FS (“Experimental”).
Each of 1a–d reacted with equimolar amount of monobromomalononitrile (2), in ethanolic potassium hydroxide solution, yielded in each case a single product which could be formulated to be either 5H-thiazolo[3,2-a]pyrimidine structure 3 or its isomeric structure 7H-thiazolo[3,2-a]pyrimidine 4 (Scheme 2). Preferring structure 3 over 4 was based on the comparison of the 1H-NMR spectral data for compounds 1 and 3. Thus, the 1H-NMR spectrum of 3b as an example revealed, in addition to the methoxy group, aromatic and NH2 proton signals, a singlet (1H) at δ 6.41 assigned to the pyrimidine H-5. The downfield for the pyrimidine H-5 in 3b compared with the pyrimidine H-4 in 1b, which appeared at δ = 5.12 ppm, indicates that the moiety nearby H-5 in 3b differs from that of H-4 in 1b. Therefore, structure 3 could be initially assigned for the reaction products.

Synthesis of thiazolopyrimidine 3
The IR (ʋ, cm−1) spectra of 3a–d displayed absorption bands characterized for 2NH2 and 2CN groups. 1H-NMR (DMSO-d6) for compound 3b, as an example, showed signals δ (ppm) at 6.53, 6.95 characterized 2NH2 (D2O exchangeable) groups. Its 13C-NMR (DMSO-d6) showed signals δ (ppm) at 52.5 (pyrimidine C-5), 56.7 (OCH3), 60.3 (thiazole C-2), 81.2 (pyrimidine C-6), 113.2, 117.1 (2CN), 125.1, 129.3, 135.2, 145.2, 159.5, 160.2 (aromatic carbons + C-8a and thiazole C-3) and 165.3 (pyrimidine C-7). Its mass spectrum showed the molecular ion Peak at m/z 324 (11.4%) corresponding to the molecular formula C15H12N6OS. Compounds (3a–d) gave compatible elemental and spectral data (“Experimental”). Comparing compounds formed by the traditional method and those prepared by the microwave assisted conditions indicates reduction of the reaction time to 8 min instead of 24 h standing. Also, the reaction yields were increased from 42–55 to 69–88%. Compound 3, as typical dienaminonitriles, allowed for hetero-annelations performing access to fused pyrimidines. They could be used as precursors for the preparation of pyrimidothiazolopyrimidopyrimidines. Thus, a mixture of each of 3a, b were heated under reflux with an excess of carbon disulphide to yield, in each case, the corresponding 11-aryl-11H-1,2,3,4,7,8,9,10-octahydropyrimido[4″,5″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyrimidine-2,4,8,10-tetrathione 5a, b (Scheme 3).

Synthesis of pyrimidothiazolopyrimidopyrimidine 5, 6
Finally, treatment of 3a, b with formic acid by heating several hours yielded 11-aryl-9H-1,3,6,7-tetrahydropyrimido-[5″,4″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyrimidine-4,10-dione 6a, b (Scheme 3). Compounds 5, 6 gave compatible elemental and spectral data (“Experimental”). Formation of 6 is assumed to proceed via condensation reaction followed by partial hydrolysis and finally removal of two molecules of water (Scheme 4).

Mechanism for the formation of 6
Compound 6b could be synthesized in step wise sequence by heating 5-(4-methoxyphenyl) 7-thioxo-5,6,7,8-tetrahydro-3H-pyrimido[4,5-d]pyrimidin-4-one (7) [33] with bromomalononitrile (2) in ethanolic potassium hydroxide solution to produce 7-amino-5-(4-methoxyphenyl)-4-oxo-3,5-dihydro-4H-pyrimido[4,5-d]thiazolo[3,2-a]pyrimidine-8-carbonitrile (8). Compound 8 gave compatible elemental and spectral data (“Experimental”). On boiling under reflux compound 8 with formic acid for several hours yielded the desired compound. The obtained product was identical in all aspects (m.p., mixed m.p., IR spectra) to product 6b (Scheme 5).

Synthesis of 6b in step wise sequence
As an extension of alkylation and cycloalkylation, compound 1a was heated under reflux with a mixture of chloroacetic acid, aromatic aldehyde and anhydrous sodium acetate in acetic acid/acetic anhydride solution to give 2-arylmethylene-7-amino-5-(4-chloropenyl)-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (9a, b), in good yields (Scheme 6). IR (ύ, cm−1) spectra of 9 display absorption bands around 3350 and 3240 (NH2), 2213 (CN) and 1695 (CO). 1H-NMR spectrum (DMSO-d6) of 9b, as an example, shows signals at δ 3.41 ppm (s, 3H, CH3), 5.07(s, 1H, pyrimidine H-5), 7.20–7.67 (m, 8H, aromatic protons + 1H, methine proton) and 8.3 (s, 2H, NH2, D2O exchangeable). Mass spectrum of 9b, as an example, gives the molecular ion peaks at m/z 422 (35.4%), 424 (12.2%) and the base peak at m/z 302. In support of structure 9, compound 9b, as an example, could be synthesized step wisely. Thus, when compound 1a was heated under reflux with chloroacetic acid and sodium acetate in acetic acid, it gave the 2-carboxymethylthio derivative 10. The latter compound could be cyclized by heating with acetic acid/acetic anhydride at 100 °C to give 7-amino-5-(4-chloropenyl)-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (11). Upon heating under reflux 11 with p-methoxybenzaldehyde in acetic acid, in presence of anhydrous sodium acetate, 9b was obtained (Scheme 7). Compounds 10 and 11 gave the expected values in elemental analyses and spectral data (“Experimental”).

Formation of 2-arylmethylene-7-amino-5-(4-chloropenyl)-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile

Supporting of structure 9
We have recently been attentive in carrying out synthesis of some heterocyclic compounds, with expected biological activity, under environmentally friendly, time saving microwave-assisted conditions [34,35,36,37,38,39]. Accordingly, we resynthesized the previously described compounds 1a–d, 3a–d, 5a, b and 6a, b under microwave conditions, aiming to increase reaction yields and reduce the reaction times, the difference in the outcome of the MW-assisted and thermal reactions are shown in Table 1. The outcomes of these preparations indicated that reaction yields were improved by 17–23% compared to the conventional methods. Also reaction times were considerably reduced. Figure 1 summarizes the outcome of using microwave technique for the preparation of the abovementioned compounds.

UV–Vis absorption spectra of the prepared compounds: a = 1a, 3a, 6a; b = 5a, 6a, 6b, 9b; c = 3b, 3d, 10; d = 1b, 1d; e = 1c, 3c, 5b, 9a
Biological evaluation
Antimicrobial evaluation
The newly prepared compounds were verified for their antimicrobial action against different microorganisms such as: Escherichia coli, Pseudomonas putida, Bacillus subtilis, Streptococcus lactis, Aspergillus niger, Penicillium sp. and Candida albicans. The initial screening of the investigated compounds was achieved using the filter paper disc-diffusion method. Compounds 1a, b, 3a, b, 5a, 6a, 8, 9a and 10 showed moderate to slight inhibitory action towards the microorganisms. Other compounds showed slight to no sensitivity at all to the mentioned organisms, the results are listed in Table 2.
Fluorescence and absorption spectra
The UV–Vis absorption spectra of all compounds as well as the fluorescence spectra of the compounds exhibiting fluorescence in solution were measured in 1,4-dioxane. It is clear from Fig. 1 that the prepared compounds exhibit UV–Vis absorption spectra in the region of 250–500 nm with a maximum absorption at 326 nm. The difference in the intensity of the prepared compounds depends on the difference of their chemical structures. The probabilities of compounds towards excitation from the ground state to the singlet excited state (absorption cross-section σa) by absorbing photons at wavelength of 326 nm were calculated using Eq. (1) as follows [40]: σa = 0.385 × 10−20 ε where: the molar absorptivity ε was calculated from Beer–Lambert law Eq. (2):
where: A: absorbance, I0 and I: intensities of incident and emerged light from the sample, C: molar concentration of compounds and L is the light path (1 cm).
The absorption and emission spectral maxima are listed in Table 3. The fluorescence properties of the compounds depend on the presence of electron-with donating and electron-withdrawing substituents on the acceptor part. The acceptor part of 2-carboxymethylthio derivative 10 contains carboxyl group when compared with other compounds. Hence, due to the less positive inductive effect of 10, the donating tendency becomes less and compound 10 exhibits high quantum yield φ f of 0.73, much higher than other compounds.
No fluorescence was detected in solution for all studied compounds except 10, 3a and 5a (Table 3; Figs. 1, 2). Compounds 3a and 5a exhibited intense fluorescence while compounds 10 exhibited high quantum yield φ f of 0.68 and 0.63 and 0.73 respectively, which may be due to the presence a polycyclic compounds with tetrathione moiety and electron-withdrawing substituents, enabling extended conjugation (Table 3). Simultaneously, it was observed that only compound 10 showed fluorescence in both solution and solid phase, and the fluorescence maximum in solid phase was shifted bathochromically by about 50 nm compared with the maximum in solution. Conversely, compounds 3a and 5a exhibited fluorescence only in solution.

Emission spectra of the prepared compounds: a = 10; b = 3a; c = 5a; d = 1a–d, 2a–d, 3b, 3c, 3d; e = 6–9
Experimental
General
A Gallenkamp melting point apparatus was used to determine melting points and IR spectra (KBr discs) were recorded on a Shimadzu FTIR-8201PC Spectrophotometer. 1H-NMR and 13C-NMR spectra were verified on a Varian Mercury 300 MHz and a Varian Gemini 200 MHz spectrometers using TMS as an internal standard and DMSO-d6 as a solvent and the chemical shifts were expressed as δ (ppm) units. Shimadzu GCMS-QP1000EX instrument were used to record Mass spectra using an inlet type sample injection at 70 eV. The Microanalytical Center of Cairo University performed the microanalyses. Microwave reactions were performed with a Millstone Organic Synthesis Unit (Micro SYNTH with touch control terminal) with a continuous focused microwave power delivery system in a pressure glass vessel (10 mL) sealed with a septum under magnetic stirring. A calibrated infrared temperature control was used to monitor the temperature of the reaction mixture under the reaction vessel with a pressure sensor connected to the septum of the vessel to control the pressure. Ultraviolet–visible absorption spectra were measured on a PerkinElmer Lambda 35 Spectrophotometer at room temperature. Steady-state fluorescence spectra were measured on a PerkinElmer LS 55 spectrophotometer. The prepared compounds were dissolved in precleaned amber glass vials (1-cm cell) containing dioxane as solvent in concentration of 1 × 10−5 M (King Khalid University).
Compounds 1a, b were prepared according to literature procedures [33, 41].
6-Amino-4-aryl-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carbonitriles 1a–d
Method A A solution of thiourea (0.76 g, 0.01 mol), malononitrile (0.66 g, 0.01 mol) and the appropriate aromatic aldehyde in sodium ethoxide (sodium metal 0.23 g, 0.01 mol in absolute ethanol 30 mL) was stirred at room temperature for 24 h. Then the mixture was poured onto ice-cold water and neutralized by dilute HCl. The solid precipitate so-formed was filtered off, washed with water and crystallized from ethanol.
Method B The same reactants of method A in 5 mL sodium ethoxide solution were heated in microwave oven at 500 W and 140 °C for about 10 min. Compounds 1a–d was produced by treating the reaction mixture in a similar manner of method A.
6-Amino-4-(4-chlorophenyl)-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carbonitrile (1a). The aromatic aldehyde used was 4-chlorobenzaldehyde (1.40 g, 0.01 mol), (yield 88%, 2.32 g) according to method B. Compound 1a was obtained as yellow crystals, yield for method A, 52%, m.p. 121–123 °C. 1H-NMR: δ (ppm) 1.52 (s, 1H, –SH), 3.41 (s, 1H, NH, D2O exchangeable), 4.31 (s, 2H, NH2, D2O exchangeable), 4.81 (s, 1H, –CH), 6.87–7.23 (m, 4H, Ar–H). 13C-NMR: δ (ppm) 45.8 (pyrimidine C-4), 68.2 (pyrimidine C-5), 117.2 (CN), 126.5, 127.6, 128.4, 129.0, 133.1, 158.3 (aromatic carbons + pyrimidine C-6) and 170.1 (C=S). IR (KBr) ʋ: 3370, 3252 and 3180 (NH + NH2), 3050, 2950 (CH), 2215 (CN), 1640, 1543 cm−1 (Aromatic C=C). MS (70 eV): (M+2) m/z 266 (5.8%), (M+) 264 (18.6%). Anal. Calcd. For C11H9N4SCl (264.5): C (49.91%), H (3.43%), N (21.16%), S (12.12), Cl (13.44%); Found: C (49.85%), H (3.38%), N (20.87%), S (11.87%), Cl (13.38).
6-Amino-4-(4-methoxyphenyl)-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carbonitrile (1b). The aromatic aldehyde used was 4-methoxybenzaldehyde (1.22 mL, 0.01 mol), (yield 74%, 1.92 g) according to method B. Compound 1b was obtained as fine yellow crystals, yield for method A, 48%, m.p. 120–122 °C. 1H-NMR: δ (ppm) 1.45 (s, 1H, –SH), 3.41 (s, 1H, NH, D2O exchangeable), 3.86 (s, 3H, –OCH3), 4.40 (s, 2H, NH2, D2O exchangeable), 4.67 (s, 1H, –CH), 6.76–7.12 (m, 4H, Ar–H). 13C-NMR: δ (ppm) 46.2 (–OCH3), 52.3 (pyrimidine C-4), 60.0 (pyrimidine C-5), 117.3 (CN), 126.5, 127.6, 128.4, 129.0, 133.1, 158.3 (aromatic carbons + pyrimidine C-6) and 168.4 (C=S). IR (KBr) ʋ: 3377, 3260 and 3180 (NH + NH2), 3050, 2925 (CH), 2213 (CN), 1645, 1543 cm−1 (Aromatic C=C). MS (70 eV): (M+) m/z 260 (13.5%). Anal. Calcd. For C12H12N4OS (260.32): C (55.37%), H (4.65%), N (21.52%), S (12.30); Found: C (55.31%), H (4.59%), N (21.10%), S (11.87%).
6-Amino-4-(4-nitrophenyl)-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carbonitrile (1c). The aromatic aldehyde used was 4-nitrobenzaldehyde 1.51 g (0.01 mol). Compound 1c was obtained as fine yellow crystals, yield for method A, 52%, 1.43 g and for method B, 87%, 2.39 g), m.p. 200–203 °C. 1H-NMR: δ (ppm) 4.95 (s, 1H, pyrimidine H-4), 6.61(s, 2H, NH2, D2O exchangeable), 7.48 (d, 2H, Ar–H, J = 7.4 Hz), 7.82 (d, 2H, Ar–H, J = 7.4 Hz), 8.84 (s, 1H, NH, D2O exchangeable) and 9.53 (s, 1H, NH, D2O exchangeable). 13C-NMR: δ (ppm) 53.5 (pyrimidine C-4), 62.2 (pyrimidine C-5), 117.2 (CN), 124.5, 127.6, 144.4, 150.0, 168.1 (aromatic carbons + pyrimidine C-6) and 173.1 (C=S). IR (KBr) ʋ: 3350, 3270 and 3180 (NH + NH2), 3050, 2980 (CH), 2217 (CN), 1605, 1500 cm−1 (Aromatic C=C). MS (70 eV): (M+) m/z 275 (16.2%). Anal. Calcd. for C11H9N5O2S (275.25): C (47.99%), H (3.29%), N (25.44%), S (11.64); Found: C (47.83%), H (3.14%), N (25.35%), S (11.53%).
6-Amino-4-(4-florophenyl)-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carbonitrile (1d). The aromatic aldehyde used was 4-flourobenzaldehyde, 1.07 mL (0.01 mol). Compound 1d was obtained as yellow crystals, yield, for method A, 45%, 1.11 g and for method B 83%, 2.05 g) m.p. 246–247 °C. 1H-NMR: δ (ppm) 5.02 (s, 1H, pyrimidine H-4), 6.65 (s, 2H, NH2, D2O exchangeable), 7.23 (d, 2H, J = 7.8 HZ, aromatic protons), 7.51 (d, 2H, J = 7.8 HZ, aromatic protons), 8.65 (s, 1H, NH, D2O exchangeable) and 9.53 (s, 1H, NH, D2O exchangeable). 13C-NMR: δ (ppm) 54.5 (pyrimidine C-4), 62.3 (pyrimidine C-5), 112.2, 117.1, 127.1, 133.2, 141.2, 160.5 (aromatic carbons + CN and pyrimidine C-6) and 175.3 (C=S). IR (KBr) ʋ: 3300, 3220 and 3140 (NH + NH2), 3050, 2980 (CH), 2217 (CN), 1605, 1500 cm−1 (Aromatic C=C). MS (70 eV): (M+) m/z 248 (11.2%). Anal. Calcd. For C11H9N4SF (248.24): C (53.21%), H (3.64%), N (22.56%), S (12.91), F (7.64); Found: C (53.24%), H (3.53%), N (22.38%), S (12.51%), F (6.97%).
3,7-Diamino-5-aryl-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitriles (3a–d)
Method A To a warm ethanolic potassium hydroxide solution [prepared by dissolving KOH (0.56 g, 0.01 mol) in ethanol (50 mL)] of each of 1a–d [(1a, 2.64 g; 1b, 2.60 g; 1c, 2.75 g; 1d, 2.48 g; 0.01 mol)],bromomalononitrile (2) (1.45 g, 0.01 mol) was added portion-wise and stirred at room temperature for 24 h. Whereby the solid product that separated upon dilution with water was filtered off and crystallized from the proper solvent.
Method B The same reactants of method A in 5 mL ethanolic potassium hydroxide solution were heated in microwave oven at 500 W and 140 °C for 5–8 min. compounds 3a–d was produced by treating the reaction mixture in a similar manner of method A.
3,7-Diamino-5-(4-chlorophenyl)-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitriles (3a) was crystallized from dil. dioxane as brown crystals, yield for method A, 60%, 1.96 g and for method B, 82%, 2.68 g) m.p. 220–222 °C. 1H-NMR: δ (ppm) 6.10 (s, 1H, pyrimidine H-5), 6.83 (s, 2H, NH2, D2O exchangeable), 7.24 (s, 2H, NH2, D2O exchangeable), 7.73 (d, 2H, J = 7.4 HZ, aromatic protons) and 7.95 (d, 2H, J = 7.4 HZ, aromatic protons). 13C-NMR: δ (ppm) 55.6 (pyrimidine C-5), 59.3 (thiazole C-2), 81.1 (pyrimidine C-6), 113.9, 117.3 (2CN), 127.1, 129.4, 133.2, 141.5, 158.8, 159.3 (aromatic carbons + C-8a and thiazole C-3) and 167.2 (C-7). IR (KBr) ʋ: 3310, 3240 (NH2), 3030, 2984 (CH) and 2217, 2219 (2CN). MS (70 eV): (M+2) m/z 330 (2.8%), (M+) 328 (9.4%). Anal. Calcd. For C14H9N6SCl (328.75): C (51.14%), H (2.75%), N (25.56%), S (9.75), Cl (10.78); Found: C (51.10%), H (2.56%), N (25.14%), S (9.51%), Cl (10.21%).
3,7-Diamino-5-(4-methoxyphenyl)-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitriles (3b) was crystallized from ethanol as beige crystals, yield for method A 50%, 1.62 g and for method B 76%, 2.46 g) m.p. 224–226 °C. 1H-NMR: δ (ppm) 3.85 (s, 3H, OCH3), 6.41 (s, 1H, pyrimidine H-5), 6.63 (s, 2H, NH2, D2O exchangeable), 7.12 (s, 2H, NH2, D2O exchangeable), 7.75 (d, 2H, J = 7.3 HZ, aromatic protons) and 7.95 (d, 2H, J = 7.3 HZ, aromatic protons). 13C-NMR: δ (ppm) 52.5 (pyrimidine C-5), 56.7 (OCH3), 60.3 (thiazole C-2), 81.2 (pyrimidine C-6), 113.2, 117.1 (2CN), 125.1, 129.3, 135.2, 145.2, 159.5, 160.2 (aromatic carbons + C-8a and thiazole C-3) and 165.3 (pyrimidine C-7). IR (KBr) ʋ: 3310, 3240 (NH2), 3030, 2984 (CH) and 2217, 2220 (2CN). MS (70 eV): (M+) m/z 324 (10.4%). Anal. Calcd. For C15H12N6OS (324.31): C (55.54%), H (3.70%), N (25.91%), S (9.88); Found: C (55.31%), H (3.63%), N (25.14%), S (9.53%).
3,7-Diamino-5-(4-nitrophenyl)-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitriles (3c) was crystallized from ethanol as brown crystals, yield for method A, 43%, 1.45 g and for method B, 74%, 2.50 g) m.p. 243–245 °C. 1H-NMR: δ (ppm) 5.84 (s, 1H, pyrimidine H-5), 6.56 (s, 2H, NH2, D2O exchangeable), 6.81 (s, 2H, NH2, D2O exchangeable), 7.75 (d, 2H, J = 7.4 HZ, aromatic protons) and 8.35 (d, 2H, J = 7.3 HZ, aromatic protons). 13C-NMR: δ (ppm) 52.5 (pyrimidine C-5), 59.3 (thiazole C-2), 82.3 (pyrimidine C-6), 115.2, 118.3 (2CN), 125.3, 129.4, 135.2, 144.2, 159.5, 160.5 (aromatic carbons + C-8a and C-3) and 164.7 (pyrimidine C-7). IR (KBr) ʋ: 3310, 3240 (NH2), 3035, 2985 (CH) and 2218, 2223 (2CN). MS (70 eV): (M+) m/z 339 (7.8%). Anal. Calcd. For C14H9N7O2S (339.29): C (49.55%), H (2.67%), N (28.89%), S (9.45); Found: C (49.35%), H (2.60%), N (28.16%), S (9.11%).
3,7-Diamino-5-(4-florophenyl)-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitriles (3d) was crystallized from dioxane as brown crystals, yield for method A, 43%, 1.34 g and for method B, 74%, 2.30 g) m.p. 251–253 °C. 1H-NMR: δ (ppm) 5.91 (s,1H, pyrimidine H-5), 6.63 (s, 2H, NH2, D2O exchangeable), 6.22 (s, 2H, NH2, D2O exchangeable), 7.55 (d, 2H, J = 7.4 HZ, aromatic protons) and 7.84 (d, 2H, J = 7.4 HZ, aromatic protons). 13C-NMR: δ (ppm) 53.6 (pyrimidine C-5), 58.5 (thiazole C-2), 80.2 (pyrimidine C-6), 114.0, 117.3 (2CN), 127.1, 129.4, 133.2, 141.5, 158.8, 159.3 (aromatic carbons + C-8a and C-3) and 165.3 (C-7). IR (KBr) ʋ: 3310, 3240 (NH2), 3030, 2984 (CH) and 2217, 2219 (2CN). MS (70 eV): (M+) m/z 312 (4.6%). Anal. Calcd. For C14H9N6SF (312.29): C (53.84%), H (5.38%), N (26.90%), S (10.26), F (6.08); Found: C (53.75%), H (5.26%), N (26.27%), S (9.87%), F (5.77%).
11-Aryl-11H-1,2,3,4,7,8,9,10-octahydropyrimido[4″,5″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyrimidine-2,4,8,10-tetrathione 5a, b
Method A Each of compounds 3a, b (3a, 1.09 g, 3b, 1.08 g; 0.03 mol) was heated under reflux with an excess of carbon disulphide (20 mL) for 8 h. The reaction mixture was left to cool, the solid that precipitated was filtered off and crystallized from the proper solvent.
Method B Each of compounds 3a, b (3a, 1.09 g, 3b, 1.08 g; 0.03 mol) in 6 mL carbon disulphide were heated in microwave oven at 500 W and 140 °C for 8 min. The reaction mixture was treated in a similar manner to method A to yield compounds 5a, b.
11-(4-Chlorophenyl)-11H-1,2,3,4,7,8,9,10-octahydropyrimido[4″,5″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyrimidine-2,4,8, 10-tetrathione (5a) was crystallized from dioxane as grey crystals, yield for method A, 55%, 0.88 g and for method B, 82%, 1.31 g, m.p. 248–250 °C. 1H-NMR: δ (ppm) 5.88 (s, 1H, pyrimidine H-10), 7.42–7.73 (m, 5H, Ar–H + NH, D2O exchangeable), 9.31 (s, 1H, NH, D2O exchangeable) and 12.85 (br, 2H, 2NH, D2O exchangeable). 13C-NMR: δ (ppm) 58.5 (pyrimidine C-10), 81.2 (C-4a), 110.2 (C-9a), 127.1, 132.2, 144.2, 156.3, 158.2, 166.7 (aromatic carbons + C-12a + C-5a + C-6a) and 171.3, 174.2, 188.4, 190.3 (4C=S). IR (KBr) ʋ: 3305, 3200 (NH), 3030, 2984 (CH), 1605, 1500 cm−1 (aromatic C=C). MS (70 eV): (M+2) m/z 483 (0.9%), (M+) 481 (3.2%). Anal. Calcd. For C16H9N6S5Cl (481.02): C (39.94%), H (1.88%), N (17.46%), S (33.32%), Cl (7.36%); Found: C (39.76%), H (1.78%), N (16.80%), S (32.86%), Cl (7.11%).
11-(4-Methoxyphenyl)-11H-1,2,3,4,7,8,9,10-octahydropyrimido[4″,5″:4′,5′] thiazolo-[3′,2′-a]pyrimido[4,5-d]pyrimidine-2,4,8,10-tetrathione (5b) was crystallized from dioxane as brown crystals, yield for method A, 43%, 0.68 g and for method B, 76%, 1.2 g, yield 43%, 2.04 g, m.p. 254–257 °C. 1H-NMR: δ (ppm) 3.42 (s, 3H, OCH3), 5.86 (s, 1H, pyrimidine H-10), 7.25–7.57 (m, 5H, Ar–H + NH, D2O exchangeable), 9.15 (s, 1H, NH, D2O exchangeable) and 12.24 (br, 2H, 2NH, D2O exchangeable). 13C-NMR: δ (ppm) 55.5 (pyrimidine C-10), 61.2 (OCH3), 79.4 (C-4a), 111.3 (C-9a), 125.1, 129.5, 143.5, 155.3, 158.2, 166.7 (aromatic carbons + C-12a + C-5a + C-6a) and 171.3, 173.4, 186.4, 188.6 (4C=S). IR (KBr) ʋ: 3305, 3200 (NH), 3030, 2984 (CH), 1605, 1500 cm−1 (aromatic C=C). MS (70 eV): (M+) m/z 476 (6.1%). Anal. Calcd. For C17H12N6OS5 (476.59): C (42.83%), H (2.53%), N (17.63%), S (33.64); Found: C (42.65%), H (2.50%), N (17.23%), S (33.22%).
11-Aryl-9H-1,3,6,7-tetrahydropyrimido[5″,4″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyri-midine-4,10-dione 6a, b
Method A Each of compounds 3a, b (3a, 1.09 g; 3b, 1.08 g; 0.03 mol) was heated under reflux with an formic acid (80%, 20 mL) for 10 h. The reaction mixture was left to cool and the solid that precipitated was filtered and crystallized from the proper solvent.
Method B Each of compounds 3a, b (3a, 1.09 g; 3b, 1.08 g; 0.03 mol) in 5 mL formic acid (80%) were heated in microwave oven at 500 W and 140 °C for 8 min. compounds 6a, b was obtained by treating the reaction mixture in a similar manner to method A.
11-(4-Chlorophenyl)-9H-1,3,6,7-tetrahydropyrimido[5″,4″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyri-midine-4,10-dione (6a) was crystallized from dil. Dimethyl formamide as grey crystals, yield for method A, 39%, 0.49 g and for method B, 72%, 0.92 g, m.p. 272–275 °C. 1H-NMR: δ (ppm) 3.44 (s, 3H, OCH3), 4.89 (s, 1H, pyrimidine H-6), 7.31–7.25 (m, 4H, Ar–H), 8.13 (s, 1H, H-2), 8.34 (s, 1H, H-8) and 10.31 (s, 2H, 2NH, D2O exchangeable). 13C-NMR: δ (ppm) 58.5 (pyrimidine C-6), 119.4 (C-4a), 127.1, 129.2, 132.2, 138.3, 143.2, 147.3, 152.4 (aromatic carbons + C-6a + C-8 and C-12a), 154.4, 156.2 (C-5a + C-10a) and 162.4, 165.5 (2C=O). IR (KBr) ʋ: 3305, 3200 (NH), 3030, 2984 (CH), 1675 cm−1 (C=O). MS (70 eV): (M+2) m/z 386 (3.2%), (M+) m/z 384 (11.4%). Anal. Calcd. For C16H9N6O2SCl (384.75): C (49.94%), H (2.35%), N (21.84%), S (8.33%), Cl (9.21%); Found: C (49.82%), H (2.31%), N (21.53%), S (7.87%), Cl (8.75%).
11-(4-Methoxyphenyl)-9H-1,3,6,7-tetrahydropyrimido[5″,4″:4′,5′]thiazolo[3′,2′-a]pyrimido[4,5-d]pyri-midine-4,10-dione (6b) was crystallized from dil. Dimethyl formamide as grey crystals, yield for method A, 39%, 0.49 g and for method B, 70%, 0.88 g, m.p. 263–264 °C. 1H-NMR: δ (ppm) 3.8 (s, 3H, OCH3), 5.12 (s, 1H, pyrimidine H-6), 7.32–7.44 (m, 4H, Ar–H), 8.35 (s, 2H, H-2 + H-8) and 10.57 (s, 2H, 2NH, D2O exchangeable). 13C-NMR: δ (ppm) 56.6 (pyrimidine C-6), 62.3 (OCH3), 111.6 (C-4a), 120.4, 126.1, 133.3, 143.1, 147.5, 152.3 (aromatic carbons + C-6a + C-8 and C-12a), 154.4, 156.2 (C-5a + C-10a) and 162.4 165.5 (2C=O). IR (KBr) ʋ: 3305, 3200 (NH), 3030, 2984 (CH), 1675 cm−1 (C=O). MS (70 eV): (M+) m/z 380 (7.6%). Anal. Calcd. For C17H12N6O3S (380.31): C (53.68%), H (3.17%), N (22.09%), S (8.43%); Found: C (53.32%), H (2.89%), N (21.43%), S (7.87%).
7-Amino-5-(4-methoxyphenyl)-4-oxo-3,5-dihydro-4H-pyrimido[4,5-d]thiazolo[3,2-a]pyrimidine-8-carbonitrile (8)
To a warm ethanolic potassium hydroxide solution of each of 7 (2.88 g, 0.01 mol), bromomalononitrile (2) (1.45 g, 0.01 mol) was added portion-wise with stirring. The reaction mixture was then left overnight at room temperature, whereby the solid product that separated upon dilution with water was filtered off and crystallized from ethanol as orange crystals, yield 22%, 0.77 g, m.p. 224–225 °C. 1H-NMR: δ (ppm) 3.6 (s, 3H, OCH3), 5.12 (s, 1H, pyrimidine H-5), 6.23 (s, 2H, NH2, D2O exchangeable), 7.15 (d, 2H, J = 7.1 HZ, aromatic protons) and 7.64 (d, 2H, J = 7.2 HZ, aromatic protons), 8.34 (s, 1H, H-2) and 10.55 (s, 1H, NH, D2O exchangeable). 13C-NMR: δ (ppm) 56.2 (OCH3), 59.0 (C-8), 60.1 (C-5), 113.5 (CN), 116.3, 123.3, 126.2, 132.6, 148.5 (aromatic carbons + C-4a + C-2 and C-10a), 156.7, 158.2 (C-9a + C-7) and 162.3 (C=O). IR (KBr) ʋ: 3280, 3220 and 3160 (NH + NH2), 3050, 2980 (CH), 2213 (CN), 1672 cm−1 (C=O). MS (70 eV): (M+) m/z 352 (9.3%). Anal. Calcd. For C16H12N6O2S (352.31): C (54.54%), H (3.42%), N (23.85%), S (9.10%); Found: C (54.11%), H (3.31%), N (23.53%), S (8.82%).
2-Arylmethylene-7-amino-5-(4-chloropenyl)-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (9a, b)
Method A A solution of 1a (2.64 g; 0.01 mol) and chloroacetic acid (1.04 g, 0.01 mol), in a mixture of glacial acetic acid (20 mL) and acetic anhydride (20 mL) containing 1 g fused sodium acetate was heated under refluxed for 3 h with (0.01 mol) of each of benzaldehyde and p-methoxybenzaldehyde. The reaction mixture was poured onto water; the precipitated solid was filtered off, washed with water, dried and recrystallized from the proper solvent.
Method B A solution of 11 (2.73 g, 0.01 mol) in a mixture of glacial acetic acid (20 mL)/acetic anhydride (8 mL) containing 1 g fused sodium acetate was heated with p-methoxybenzaldehyde (1.49 g, 0.01 mol) under reflux for 1 h. The obtained solid was found to be identical in all aspects (IR, m.p., mixed m.p.) with compound 9b.
7-Amino-5-(4-chloropenyl)-2-phenylylmethylene-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (9a) was crystallized from acetic acid as yellow fine crystals, yield, 63%, 2.46 g, m.p. 233–235 °C. 1H-NMR: δ (ppm) 5.12 (s, 1H, pyrimidine H-5), 6.81 (s, 2H, NH2, D2O exchangeable), 7.23–7.89 (m, 10H, 9 Ar–H + methine–H). 13C-NMR: δ (ppm) 46.6 (pyrimidine C-5), 71.8 (pyrimidine C-6), 113.6 (C-2), 116.3 (CN), 126.4, 127.1, 129.2, 133.3, 136.1, 142.0, 144.5 (aromatic carbons + methine C), 157.4, 159.2 (C-8a + C-7) and 165.5 (C=O). IR (KBr) ʋ: 3345, 3260 (NH2), 3026, 2984 (CH), 2213 (CN), 1695 cm−1 (C=O). MS (70 eV): (M+2) m/z 394 (6.8%), (M+) m/z 392 (21.6%). Anal. Calcd. For C20H13N4OSCl (392.81): C (61.14%), H (3.33%), N (14.26%), S (8.16%), Cl (9.02%); Found: C (61.03%), H (3.12%), N (13.92%), S (7.87%), Cl (8.63%).
7-Amino-5-(4-chloropenyl)-2-(4-methoxyphenylyl)methylene-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (9b) was crystallized from dioxane as deep yellow crystals, yield, 70%, 2.87 g, m.p. 245–246 °C. 1H-NMR: δ (ppm) 3.41 (s, 3H, OCH3), 5.07 (s, 1H, pyrimidine H-5), 7.20–7.67 (m, 9H, 8 Ar–H + methine-H), 8.3 (s, 2H, NH2, D2O exchangeable). 13C-NMR: δ (ppm) 48.4 (pyrimidine C-5), 61.1 (OCH3), 69.8 (pyrimidine C-6), 113.3 (C-2), 116.4 (CN), 126.7, 126.9, 128.2, 131.5, 136.1, 141.2, 145.3 (aromatic carbons + methine C), 157.1, 158.8 (C-8a + C-7) and 165.3 (C=O). IR (KBr) ʋ: 3350, 3240 (NH2), 3024, 2985 (CH), 2213 (CN), 1688 cm−1 (C=O). MS (70 eV): (M+2) m/z 424 (12.2%), (M+) m/z 422 (35.4%). Anal. Calcd. For C21H15N4O2SCl (422.82): C (59.64%), H (3.57%), N (13.24%), S (7.58%), Cl (8.38%); Found: C (59.12%), H (3.11%), N (12.93%), S (7.13%), Cl (7.76%).
Reaction of 1a with chloroacetic acid: formation of 10
A solution of 1a (2.64 g; 0.01 mol) in glacial acetic acid (40 mL) containing 1.0 g fused sodium acetate was heated under reflux for 3 h with 0.95 g (0.01 mol) of chloroacetic acid. The reaction mixture was then poured onto water and the precipitated solid was filtered off, dried and recrystallized from dioxane as yellow crystals, yield, 43%, 1.35 g, m.p. 257–258 °C. 1H-NMR: δ (ppm) 3.6 (s, 2H, CH2), 5.0 (s, 1H, pyrimidine H-4), 7.21–7.70 (m, 5H, 4 Ar–H +NH, D2O exchangeable), 8.21 (s, 2H, NH2, D2O exchangeable), 13.10(s, 1H, OH, D2O exchangeable). 13C-NMR: δ (ppm) 41.2 (CH2), 48.4 (pyrimidine C-4), 69.8 (pyrimidine C-5), 116.8 (CN), 126.6, 127.9, 131.8, 141.2 (Ar–C), 159.8 (C-6) and 165.7 (C=O). IR (KBr) ʋ: 3385–3220 (br, OH, NH), 2216 (CN), 1706 cm−1 (C=O). MS (70 eV): (M+2) m/z 324 (4.8%), (M+) m/z 322 (16.4%). Anal. Calcd. For C13H11N4O2SCl (322.71): C (48.38%), H (3.43%), N (17.35%), S (9.93%), Cl (10.98%); Found: C (48.11%), H (3.12%), N (17.03%), S (9.13%), Cl (10.17%).
7-Amino-5-(4-chloropenyl)-3-oxo-2,3-dihydro-5-H-thiazolo[3,2-a]pyrimidine-6-carbonitrile (11)
Method A A solution of 1a (2.64 g; 0.01 mol) in glacial acetic acid (20 mL)/acetic anhydride (8 mL) containing 1.0 g fused sodium acetate was treated with 0.95 g (0.01 mol) chloroacetic acid and refluxed in water bath for 3 h. The reaction mixture was then cooled and the precipitated solid was filtered off, dried and recrystallized from acetic acid as yellow crystals.
Method B A solution of 10 (3.22 g, 0.01 mol) in glacial acetic acid (20 mL)/acetic anhydride (8 mL) was refluxed in water bath for 3 h. The reaction mixture was then cooled and the precipitated solid was filtered off and dried. The obtained solid was found to be identical in all aspects (IR, m.p., mixed m.p.) with compound 11. Yield, 61%, 1.83 g, m.p. 223–225 °C. 1H-NMR: δ (ppm) 3.8 (s, 2H, CH2), 5.3 (s, 1H, pyrimidine H-4), 7.23 (d, 2H, Ar–H, J = 7.5 Hz), 7.41 (d, 2H, Ar–H, J = 7.5 Hz), 7.88 (s, 2H, NH2, D2O exchangeable). 13C-NMR: δ (ppm) 34.2 (CH2), 48.1 (C-5), 71.8 (C-6), 116.8 (CN), 126.6, 128.6, 131.8, 141.2 (Ar–C), 159.8 (C-8a), 162.3 (C-7) and 166.5 (C=O). IR (KBr) ʋ: 3345, 3230 (NH2), 3020, 2895 (CH), 2215 (CN), 1695 cm−1 (C=O). MS (70 eV): (M+2) m/z 306 (10.2%), (M+) m/z 304 (33.4%). Anal. Calcd. For C13H9N4OSCl (304.71): C (51.23%), H (2.97%), N (18.38%), S (10.52%), Cl (11.63%); Found: C (50.87%), H (2.77%), N (17.93%), S (10.10%), Cl (11.07%).
Antimicrobial screening
The newly prepared compounds were verified for their antimicrobial activity against: (a) Gram-negative: Escherichia coli and Pseudomonas putida; (b) Gram-positive: Bacillus subtilis and Streptococcus lactis; (c) fungi: Aspergillus niger and Penicillium sp.; (d) yeast: Candida albicans.
Media Three media types were used:
-
Media (1) For bacteria (Nutrient Medium), consisting of (g/L distilled water): peptone, 5 and meat extract, 3. at pH 7.0.
-
Media (2) For fungi (Potato Dextrose Medium), consisting of (g/L distilled water): Infusion from potatoes, 4 and D (+) glucose, 20. at pH 5.5.
-
Media (3) For yeast (Universal Medium), consisting of (g/L distilled water): yeast extract, 3; malt extract, 3; peptone, 5 and glucose, 10 at pH 5.5.
For solid media, 2% agar was added. All media were sterilized at 121 °C for 20 min. Procedure (Filter Paper Diffusion Method) [42]. Suitable concentrations of microbial suspensions were prepared from (1 for bacteria to 3 for yeast and fungi)-day-old liquid stock cultures in cubated on a rotary shaker (100 rpm). In the case of fungi, 5 sterile glass beads were added to each culture flask. The mycelia were then subdivided by mechanical stirring at speed No. 1 for 30 min. Turbidity of microorganisms was adjusted with a spectrophotometer at 350 nm to give an optical density of 1.0. Appropriate agar plates were aseptically surface inoculated uniformly by a standard volume (ca. 1 mL) of the microbial broth culture of the tested microorganism, namely E. coli, P. putida, B. subtilis, S. lactis, A. niger, Penicillium sp. and C. albicans. Whatman No. 3 filter paper discs of 10 mm diameter were sterilized by autoclaving for 15 min at 121 °C. Test compounds were dissolved in 80% ethyl alcohol (final concentrations are ~ 70% ethanol, ~ 5% methanol and ~ 5% isopropanol. Contains ~ 20% water) to give final concentration of 5 mg/mL. The sterile discs were impregnated with the test compounds (50 μg/disc). After the impregnated discs have been air dried, they were placed on the agar surface previously seeded with the organism to be tested. Discs were gently pressed with forceps to insure thorough contact with the media. Three discs were arranged per dish, suitably spaced apart, i.e., the discs should be separated by a distance that is equal to or slightly greater than the sum of the diameters of inhibition produced by each disc alone. Each test compound was conducted in triplicate. Plates were kept in the refrigerator at 5 °C for 1 h to permit good diffusion before transferring them to an incubator at 37 °C for 24 h for bacteria and at 30 °C for 72 h for yeast and fungi [32].
Conclusions
New polycyclic fused pyrimidines have been synthesized using both conventional methods and microwave assisted conditions. The latter methods proved very efficient in reducing reaction times, minimization of energy consumption, management of analytical waste and increased safety for the operator as well as better reaction yields. All prepared compounds were verified for their antimicrobial activities. Some compounds showed moderate or weak antimicrobial activity. The absorption and fluorescence emission of some of the prepared compounds were studied in dioxane.
References
Donnini S, Monti M, Castagnini C, Solito R, Botta M, Schenone S, Giachetti A, Ziche M (2007) Pyrazolo-pyrimidine-derived c-Src inhibitor reduces angiogenesis and survival of squamous carcinoma cells by suppressing vascular endothelial growth factor production and signaling. Int J Cancer 120:995–1004
Bazgir A, Khanaposhtani MM, Soorki AA (2008) One-pot synthesis and antibacterial activities of pyrazolo[4′,3′:5,6]pyrido[2,3-d]pyrimidine-dione derivatives. Bioorg Med Chem Lett 18:5800–5803
Shaban NZ, Masoud MS, Mawlawi MA, Awad D, Sadek OM (2012) Effect of some pyrimidine compounds on rat brain monoamine oxidase-B in vitro. J Physiol Biochem 68:475–484
Pandey S, Suryawanshi SN, Gupta S, Srivastava VML (2004) Synthesis and antileishmanial profile of some novel terpenyl pyrimidines. Eur J Med Chem 39:969–973
Amr AEG, Maigali SS, Abdulla MM (2008) ChemInform abstract: synthesis, and analgesic and antiparkinsonian activities of thiopyrimidine, pyrane, pyrazoline, and thiazolopyrimidine derivatives from 2-chloro-6-ethoxy-4-acetylpyridine. Monatsh Chem 139(11):1409
Amr AE, Mohamed AM, Mohamed SF, Abdel-Hafez NA, Hammam AG (2006) Anticancer activities of some newly synthesized pyridine, pyrane, and pyrimidine derivatives. Bioorg Med Chem 14:5481–5488
Branstetter BJ, Breitenbucher JG, Lebsack AD, Xiao W (2008) Thiaolopyrimidine modulators of TRPV1. US patent, WO 005,303
Flefel EE, Salama MA, El-Shahat M, El-Hashash MA, El-Farargy AF (2007) A novel synthesis of some new pyrimidine and thiazolopyrimidine derivatives for anticancer evaluation. Phosphorus Sulphur 182(8):1739
Hammam AEG, Sharaf MA, EI-Hafez NAA (2001) Synthesis and anticancer activity of pyridine and thiazolopyrimidine derivatives using 1-ethylpiperidone as a synthon. Ind J Chem 40:213–221
Said M, Abouzid K, Mouneer A, Ahmedy A, Osman AM (2004) Synthesis and biological evaluation of new thiazolo pyrimidines. Arch Pharm Res 27:471–477
Linder W, Brandes W (1991) Pesticidalthiazolopyrimidine derivatives. U.S. Pat 367820
Duval R, Kolb S, Braud E, Genest D, Garbay C (2009) Rapid discovery of triazolobenzylidene-thiazolopyrimidines (TBTP) as CDC25 phosphatase inhibitors by parallel click chemistry and in situ screening. J Comb Chem 11(6):947–950
Kolb S, Mondésert O, Goddard ML, Jullien D, Villoutreix BO, Ducommun B, Garbay C, Braud E (2009) Development of novel thiazolopyrimidines as CDC25B phosphatase inhibitors. ChemMedChem 4:633–648
Ohnishi H, Kosuzume H, Inaba H, Okura M, Mochizuki H, Suzuki Y, Fujii R (1983) Effects of AC-1370, a new semisynthetic cephalosporin, on phagocyte functions. Antimicrob Agents Chemother 23:874–880
Rashad AE, Shamroukh AH, Abdel-Megeid RE, El-Sayed WA (2010) Synthesis, reactions, and antimicrobial evaluation of some polycondensedthienopyrimidine derivatives. Synth Commun 40(8):1149–1160
El-Emary TI, Abdel-Mohsen SA (2006) Synthesis and antimicrobial activity of some new 1,3-diphenylpyrazoles bearing pyrimidine, pyrimidinethione, thiazolopyrimidine, triazolopyrimidine, thio- and alkylthiotriazolopyrimidinone moieties at the 4-position. Phosphorus, Sulfur Silicon Relat Elem 181(11):2459–2474
Maddila S, Damu GLV, Oseghe EO, Abafe OA, Venakata RC, Lavanya P (2012) Synthesisand biological studies of novel biphenyl-3,5-dihydro-2H-thiazolo-pyrimidines derivatives. J Korean Chem Soc 56:334–340
Nofal Z, Fahmy HH, Zarea ESW (2011) Synthesis of new pyrimidine derivatives with evaluation of their anti-inflammatory and analgesic activities. Acta Pol Pharm 68(4):507–517
Xing-Hai L, Qiao W, Zhao-Hui S, Dvid EW, James JB, Alden SE, Cheng-Xia T, Jian-Quan W (2017) Synthesis and insecticidal activity of novel pyrimidine derivatives containing urea pharmacophore against Aedes aegypti. Pest Manag Sci 73:953–959
Zhi H, Chen L, Zhang L, Liu S, Wan DCC, Lin H, Hu C (2008) Design, synthesis, andbiological evaluation of 5H-thiazolo[3,2-a]pyrimidine derivatives as a new type ofacetylcholinesterase inhibitors. ARKIVOC 11:266–277
Lee HW, Kim BY, Ahn JB, Kang SK, Lee JH, Shin JS, Ahn SK, Lee SJ, Yoon SS (2005) Molecular design, synthesis, and hypoglycemic and hypolipidemic activities of novel pyrimidine derivatives having thiazolidinedione. Eur J Med Chem 40:862–874
Dong C, Zhi-Hua Z, Yu Chen XY, Shi-Ti Z, Liang-Jing Z, Li-Hong M, Fang L, Bing-Jie F (2016) Synthesis of some new thiazolo[3,2-a]pyrimidine derivatives and screening of their in vitro antibacterial and antitubercularactivities. Med Chem Res 25(2):292–302
Kidwai M, Saxena S, Mohan R, Venkataramanan R (2002) A novel one pot synthesis of nitrogen containing heterocycles: an alternate methodology to the Biginelli and Hantzsch reactions. J Chem Soc Perkin Trans 1(16):1845–1846. https://doi.org/10.1039/B205539M
Gałuszka A, Migaszewski Z, Namieśnik J (2013) The 12 principles of green analytical chemistry and the significance mnemonic of green analytical practices. TrAC 50:78–84. https://doi.org/10.1016/j.trac.2013.04.010
Sosnowski M, Skulski L (2002) Microwave-accelerated iodination of some aromatic amines, using urea-hydrogen peroxide addition compound (UHP) as the oxidant. Molecules 7:867–870
Gregg B, Golden K, Quinn J (2007) Indium (III) trifluoromethanesulfonate as an efficient catalyst forthedeprotection of acetals and ketals. J Org Chem 72:5890–5893
Lerebours R, Wolf C (2007) Palladium(II)-catalyzed conjugate addition of arylsiloxanes in water. Org Lett 9:2737–2740
Marion N, Gealageas R, Nolan S (2007) [(NHC)AuI]-catalyzed rearrangement of allylic acetates. Org Lett 9:2653–2656
Jonathan DM, Kappe CO (2011) A critical assessment of the greenness and energy efficiency of microwave-assisted organic synthesis. Green Chem 13:794–806
Keglevich G, Greiner I, Mucsi Z (2015) An Interpretation of the rate enhancing effect of microwaves—modelling the distribution and effect of local overheating—a case study. Curr Org Chem 19(14):1436–1440. https://doi.org/10.2174/1385272819666150528004505
Keglevich G, Kiss NZ, Grün A, Bálint E, Kovács T (2017) Advantages of the microwave tool in organophosphorus syntheses. Synthesis 49(14):3069–3083. https://doi.org/10.1055/s-0036-1589031
Youssef MM, Amin MA (2012) Microwave assisted synthesis of some new thiazolopyrimidine, thiazolodipyrimidine and thiazolopyrimidothiazolopyrimidine derivatives with potential antioxidant and antimicrobial activity. Molecules 17(8):9652–9667. https://doi.org/10.3390/molecules17089652
Fadda A, Abdel-latif E, Bondock S, Samir A (2008) Synthesis of some new pyrimidine and pyrimido[4,5-d]pyrimidine derivatives. Synth Commun 38:4352–4368
Gomha Sobhi M, Mastoura ME, Faty Rasha A M, Zeinab AM, Yahia NM (2017) Microwave-assisted one pot three-component synthesis of some novel pyrazole scaffolds as potent anticancer agents. Chem Central J 11:37. https://doi.org/10.1186/s13065-017-0266-4
Rasha MF, Mohamed MY, Ayman MSY (2011) Microwave assisted synthesis and unusual coupling of some novel pyrido[3,2-f][1, 4]thiazepines. Molecules 16:4549–4559
Rasha MF, Alaa MG, Ayman MSY (2015) Selective cyclization of S-substituted imidazolethione: synthesis of polysubstituted imidazothiazoles. J Iran Chem Soc 12:1509–1519. https://doi.org/10.1007/s13738-015-0621-0
Youssef AMS, Azab ME, Youssef MM (2012) Bromination and diazo-coupling of pyridinethiones; microwave assisted synthesis of isothiazolopyridine, pyridothiazine and pyridothiazepines. Molecules 17:6930–6943
El-Agrody AM, Al-Dies AM, Fouda AM (2014) Microwave assisted synthesis of 2-amino-6-methoxy-4H-benzo[h] chromene derivatives. Eur J Chem 5(1):133–137. https://doi.org/10.5155/eurjchem.5.1.133-137.923
Rasha AMF, Ayman MSY (2009) Oxothioxopyridinecarbonitriles as precursors for thiazolopyridines, pyrazolopyridotriazines and pyridothiazolopyrimidines. Curr Org Chem 13:1577–1584
Dalal MA, Abdlkader HI, Maram THA (2015) Optical, photo-physical properties and photostability of pyrromethene (PM-597) in ionic liquids as benign green-solvents. J Luminescence 161:221–228
Atapour H, Soukhtanloo M, Massoudi A, Shiri A, Bakavoli M (2016) Synthesis and evaluation of cytotoxity of 6-amino-4-aryl-2-thioxo-1,2,3,4-tetrahydropyimidine-5-carbonitriles. Russ J Bioorg Chem 42:316–322
Coffen DL, Korzan DG (1971) Synthetic quinine analogs. III. Frangomeric and anchimeric processes in the preparation and reactions of α,β-epoxy ketones. J Org Chem 36:390–395
Authors’ contributions
AMY designed research; AMY, AMF and RAMF performed research and analyzed the data. All authors read and approved the final manuscript.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through General Research Project under Grant Number (G.R.P-109-38).
Also, the authors would like to thank Chemistry Department, Faculty of Science, Cairo University for their valuable support of this work assistance.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Sample availability
Samples of the compounds are available from the authors.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Youssef, A.M.S., Fouda, A.M. & Faty, R.M. Microwave assisted synthesis of some new thiazolopyrimidine and pyrimidothiazolopyrimidopyrimidine derivatives with potential antimicrobial activity. Chemistry Central Journal 12, 50 (2018). https://doi.org/10.1186/s13065-018-0419-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0419-0