
Abstract
Background
Acyl hydrazones are an important class of heterocyclic compounds promising pharmacological characteristics. Malaria is a life-threatening mosquito-borne blood disease caused by a plasmodium parasite. In some places, malaria can be treated and controlled with early diagnosis. However, some countries lack the resources to do this effectively.
Results
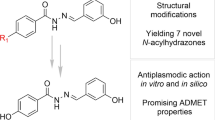
The present work involves the design and synthesis of some novel acyl hydrazone based molecular hybrids of 1,4-dihydropyridine and pyrazole (5a–g). These molecular hybrids were synthesised by condensation of 1,4-dihydropyridin-4-yl-phenoxyacetohydrazides with differently substituted pyrazole carbaldehyde. The final compound (5) showed two conformations (the major, E, s-cis and the minor, E, s-trans) as revealed by NMR spectral data and further supported by the energy calculations (MOPAC2016 using PM7 method). All the synthesised compounds were screened for their in vitro antimalarial activities against chloroquine-sensitive malaria parasite Plasmodium falciparum (3D7) and antimicrobial activity against Gram positive bacteria i.e. Bacillus cereus, Gram negative bacteria i.e. Escherichia coli and antifungal activity against one fungus i.e. Aspergillus niger. All these compounds were found more potent than chloroquine and clotrimazole, the standard drugs.
Conclusions
In vitro antiplasmodial IC50 value of the most potent compound 5d was found to be 4.40 nM which is even less than all the three reference drugs chloroquine (18.7 nM), pyrimethamine (11 nM) and artimisinin (6 nM). In silico binding study of compound 5d with plasmodial cysteine protease falcipain-2 indicated the inhibition of falcipain-2 as the probable reason for the antimalarial potency of compound 5d. All the compounds had shown good to excellent antimicrobial and antifungal activities.

Similar content being viewed by others

Background
Malaria is a public health distress in countries in which this disease is prevalent. 50% of the world population is at risk of contacting the disease. Approximately one million people die annually owing to Plasmodium falciparum malarial infections, the majority of them are young children and pregnant women [1]. Several organised efforts to control the transmission and eradicate the disease have been made through history [2]. Complex life cycle, disease spreading through a mosquito vector, resistance to insecticides and a rapidly growing resistance to malarial parasite to the available drugs are the major reasons behind malaria proliferation [3,4,5]. The parasite is developing resistance against drugs, such as antifoliates and chloroquine, by random mutation [6]. Although five species of Plasmodium family of protozoan parasites can infect humans to cause malaria, P. falciparum and P. vivax are responsible for almost all malaria-related deaths.
Molecular hybridization as a drug discovery strategy involves the rational design of new chemical entities by the fusion (usually via a covalent linker) of two drugs, both active compounds and/or pharmacophoric units recognized and derived from known bioactive molecules [7,8,9,10]. The selection of the two principles in the dual drug is usually based on their observed synergistic pharmacological activities to enable the identification of highly active novel chemical entities.
Pyrazole represents a class of heterocyclic compounds which exhibits significant biological properties such as antimalarial [11,12,13], antispasmodic [14], anti-inflammatory [15], antibacterial [16], analgesic [17], antihyperglycemic [18, 19], antineoplastic [20], antidepressive activities [21]. Similarly, pyridine ring has also been proved to be important scaffold as it has been present in various peptidomimetic and non-peptide falcipain inhibitors [22]. Virtual screening has also witnessed the importance of acyl hydrazones for the synthesis of non-peptide based falcipain inhibitors [23]. Therefore here in this study, we have decided to construct the molecular hybrids based on 1,4-DHP and pyrazole moieties using acyl hydrazone linkage which may possibly circumvent the antiplasmodial drug resistance (Fig. 1).

Drug designing by molecular hybridisation approach for the synthesis of new molecular hybrids
Results and discussion
Synthesis
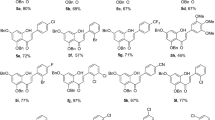
The compound 5(a–g) under investigation was synthesised (Scheme 1) in a 4-step process commencing from a three-component reaction [9] of ethylacetoacetate (2.00 mmol), 4-hydroxybenzaldehyde (1.00 mmol) and ammonium acetate (2.00 mmol) to obtain diethyl 1,4-dihydro-4-(4-hydroxyphenyl)-2,6-dimethylpyridine-3,5-dicarboxylate (1) which was subsequently converted to diethyl 4-(4-((ethoxycarbonyl)methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (2) by alkylation with ethyl bromoacetate. This DHP-based ester 2 was then reacted with hydrazine hydrate (20.00 mmol) to get 2-(4-(3,5-bis(ethoxycarbonyl)-2,6-dimethyl-1,4-dihydropyridin-4-yl)phenoxy)acetic acid hydrazide (3) which was condensed with 3-aryl-1-phenyl-1H-pyrazole-4-carbaldehyde (4) (1.00 mmol) using a catalytic amount of acetic acid in ethanol under reflux condition for 10 h to furnish 4-(4-(((3-aryl-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl)methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate 5(a–g) (92–98%) (Scheme 1; Table 1). The progress of the reaction in all cases was monitored by TLC examination using petroleum ether:ethyl acetate.

Synthesis of diethyl 4-(4-(((3-aryl-1-phenyl-1H-pyrazol-4-yl)methylene aminocarbamoyl)methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5a–g)
Characterisation of compounds and their conformational studies
The structure of these hybrids was ascertained by IR, 1H NMR, 13C NMR, and mass spectral data. The absorption signals corresponding to C=O stretching of amides appeared at 1685–1650 cm−1 and the NH stretching appeared in the region 3315–3244 cm−1 in IR spectra. It is assumed that the compound 5, restricted rotation about imine (C=N) linkage as well as the partial double bond character of hydrazide bond led to the formation of four isomers E, s-cis; E, s-trans; Z, s-cis and Z, s-trans (Fig. 2), where E/Z geometrical isomers with respect to C=N double bond and s-cis/s-trans rotamers with respect to N–C(O) acyl hydrazide [10, 24, 25].

Four possible isomeric form for 5a
Literature survey also reveals that the N-acyl hydrazones synthesised from aromatic carbaldehyde are essentially planar and exist completely in the form of geometric (E)-configuration about the C=N bond due to steric hindrance on the imine bond [10, 24,25,26,27]. The NMR (1H and 13C) spectra of these hydrazones (5a–5g) also gave two sets of resonance signals which confirmed the existence of two conformational isomers in CDCl3 (E, s-cis and E, s-trans) and in agreement with literature, predominant isomer was assigned to the E, s-cis [10, 28,29,30,31]. Therefore, we discarded the formation of Z, s-cis and Z, s-trans isomers.
In 1H-NMR of acyl hydrazones (5a–5g), splitting of signals were observed for methylene (–O–CH2–), imine (N=CH), amide (CONH) and other protons which envisaged the existence of their two isomers i.e. E, s-cis and E, s-trans. For E, s-cis isomer, singlet for methylene (–O–CH2–) protons were observed at δ 4.54–4.61 ppm (1.65–1.70 H i.e. 82.41–85.23%). Similarly, signals for both imine (N=CH) proton and amide (CONH) proton also appeared as singlet at δ 8.32–8.74 ppm (0.83–0.85 H i.e. 83.5–85%) and δ 9.39–9.91 ppm (0.84–0.85 H i.e. 84.15–85.15%) respectively. In case of E, s-trans isomer singlets for methylene (–O–CH2–), imine (N=CH) and amide (CONH) protons were observed at δ 4.77–4.91 ppm (0.29–0.35 H i.e. 14.7–17.59%), 8.55–8.66 ppm (0.15–0.16 H i.e. 14.94–16.5%), 8.81–10.04 ppm (0.15–0.16 H i.e. 14.85–15.85%) respectively. The percentage of both E, s-cis and E, s-trans isomers at 25 °C were found in the range of 82–86 and 12–18%, respectively (Additional file 1: Table S1) as derived by integration area in NMR spectrum for methylene (–O–CH2–), imine (N=CH) and amide (CONH) protons.
Compound 5a was use as model to study the conformational isomers of hydrazone by means of IR, 1H-NMR, 13C-NMR, mass, 1H-1H COSY, 1H-13C HMBC spectra. In the 1H-NMR (Fig. 3), the protons of –OCH2 of test compound 5a resonated at δ 4.57 with 85.23% abundance for E, s-cis conformation and at δ 4.91 with 14.77% abundance for E, s-trans conformation (Fig. 3) and approximately same ratio is found in the case of N=CH proton at δ 8.32 ppm (16.17%, E, s-trans conformation) and 8.55 ppm (83.83%, E, s-cis conformation) and for the CONH proton signals at δ 9.79 ppm (15.85%, E, s-trans conformation) and δ 9.91 ppm (84.15%, E, s-cis conformation). The difference between the intensities of the two signals indicates the predominant formation of E, s-cis isomer. In 13C spectra (Fig. 3), some carbons also showed two peaks instead of one, such as two peaks for –OCH2 were observed at δ 67.30 and 65.50 ppm (Fig. 3). In ESI–MS mass spectra of compound 5a, m/z value was observed at 666.12 [M+H]+. In order to understand the effect of solvent on isomer distribution, the NMR of compound 5a was taken in DMSO-d 6 . Interestingly the ratio for E, s-trans and E, s-cis isomers were found to be in 2:3 ratio (Fig. 4). This may be due to the solvation and stability of different conformation in different solvent.

Assignment of various characteristic peaks and 2D correlation of 5a

Comparison of δ of two isomers of 5a in CDCl3 and DMSO
The PM7 calculations using MOPAC2016 [32] on DELL LATITUDE E5410 on the stability of E, s-cis and E, s-trans conformation were made to corroborate the experimental results which demonstrated the higher stability of the E, s-cis isomer. Our aim was to compute semi-empirical derived properties that would be useful as starting points for understanding the ratio of conformational isomerism of N-acyl hydrazone in different solvent. Model compound 5a was considered to study the geometric isomerism. As we had information about isomer distribution of 5a compound in DMSO and CDCl3, we modelled E, s-cis and E, s-trans isomers to analyse the structures for conformational analysis of the amide (HNCO) group. As expected for 5a, two minimum-energy conformers were found at about 0° and 180°, corresponding to the syn (E, s-cis) and anti(E, s-trans) arrangements. The difference in the heat of formation ∆Hf, as calculated by the PM7 method, was found to be 13.74719 kcal/mol in CHCl3 and 3.17416 kcal/mol in DMSO, favouring the E, s-cis isomer. The results that we obtained are summarized in Additional file 1: Table S2. There was considerable energy difference between the E, s-cis and E, s-trans conformer in CHCl3 and DMSO (Additional file 1: Table S2). Thus we concluded that theoretical calculations, experimental results and literature proved that E, s-cis conformation was predominant conformation over E, s-trans conformation.
In vitro antimalarial study
All the synthesised molecular hybrids of DHP and pyrazole 5a–5g were screened for their in vitro anti-malarial activity against chloroquine-sensitive strain of P. falciparum (3D7) using chloroquine as reference drug. The number of schizonts alive at different concentrations (mg/ml) of compounds 5a–5g was shown in Table 2. The results of the biological evaluation were expressed as the drug concentration resulting in 50% inhibition (IC50) of parasite. The antiplasmodial IC50 values of synthesised compounds 5a–5f are depicted in Table 3. Compound 5d was found to be most active with an IC50 4.40 nM, followed by compound 5c and 5b with IC50 values of 8.08 and 8.66 nM, respectively. A comparison of % inhibition of all synthesised compounds is shown in Fig. 5. All the newly synthesised compounds were found to be twice potent except compound 5d which is four times potent than the reference drug chloroquine.

Graphical representation of % inhibition of compounds 5a–5g
In vitro antibacterial study
All the synthesized compounds 5a–5g were evaluated in vitro for their antimicrobial activity against one Gram positive bacterium strain i.e. Bacillus cereus, one Gram negative bacterium strain i.e. Escherichia coli and antifungal activity against one yeast i.e. Aspergillus niger by agar well diffusion method [33]. Several compounds displayed more than 90% inhibition. As compared to reference drug Tetracycline, the acyl hydrazones 5b (ZOI = 15 mm), 5c (ZOI = 14 mm) and 5e (ZOI = 17 mm) revealed very good activity against Bacillus cereus. As compared to reference drug clotrimazole, the compounds 5a (ZOI = 15 mm), 5b (ZOI = 13 mm), 5c (ZOI = 16 mm), 5d (ZOI = 17 mm), 5e (ZOI = 14 mm), 5f (ZOI = 17 mm), and 5g (ZOI = 15 mm) revealed excellent activity against Aspergillus niger (Table 4; Fig. 6).

Bilogical assay for antibacterial activity. Activity against bacteria (B. cereus), activity against fungi (A. niger)
In silico studies
Docking analysis
The complex life cycle associated with Plasmodium falciparum provides a number of targets which can be explored to discover new drugs for treatment of malaria. During life span, a parasite plays an important role in metabolite synthesis, membrane transport and haemoglobin degradation. Targets which are involved in these processes can be used to inhibit parasitic growth by their inhibition. Evidences indicate that the falcipain family proteases, namely FP2 and FP3 are promising targets involved in haemoglobin hydrolysis. Thus, inhibiting these targets could prevent haemoglobin hydrolysis which indeed hindered parasitic growth [34,35,36,37,38,39]. Falcipain inhibitors can be broadly divided into three categories [40]; (i) peptide based, (ii) peptidomimetic inhibitors, and (iii) non-peptidic inhibitors. Most of the falcipain inhibitors identified so far are peptide and peptidomimetic based inhibitors [40], however their utility as therapeutic agents is limited for their susceptibility due to metabolic degradation and their poor absorption through cell membranes. Thus, it would be of great interest to discover non-peptide inhibitors, which are less exposed to degradation by host proteases and thereby, more likely to offer in vivo activity. This strategy yielded several non-peptide inhibitors [41,42,43].
In silico studies of many pyrazole based hydrazone derivatives have been revealed to inhibit malarial cysteine protease [13]. In an effort to investigate the plausible mode of action for antimalarial activity and to predict orientation of the molecules at the active site, docking simulations were performed using Auto Dock Vina program [44]. The plasmodial cysteine protease falcipain-2 is chosen for docking because it is an important target for antimalarial chemotherapy [45]. For the survival of P. falciparum parasite, the free amino acids are produced by hydrolysis of hemoglobin, which is carried out by trophozoites of P. falciparum in an acidic food vacuole [45]. The inhibition of falcipain-2 (FP2) direct to a noticeable cutback in hemoglobin hydrolysis by trophozoites. The crystal structure of falcipain-2 was co-crystallized with inhibitor E64 (N-[N-(l-3-trans-carboxyirane-2-carbonyl)-l-leucyl]-agmatine), which was covalently-bonded to the enzyme as to block substrates from reaching the catalytic triad as defined by Gly83, Cys42 and His174 residues (Fig. 7). The binding energy obtained for E64 is 7.1 kcal/mol. Docking results suggested that compound 5d could bind the active site of falcipain-2 (Fig. 8) with binding energy of 8.5 kcal/mol. Binding energy of E64 was obtained by its re-docking non-covalently due to the limitation of available docking software in performing covalent docking. Owing to these inherent differences in the binding mechanism, thus it cannot be assumed that the DHP-pyrazole-hydrazones could possess higher potency than E64 by judging from their binding energies.

The interaction of ligand E64 into the binding sites of FP-2(PDB ID: 3BPF)

Interactions of the 5d into the binding sites of FP-2(PDB ID: 3BPF)
Compound 5d, which had the highest antimalarial activity, could bind the active site of falcipain-2 via the interaction scheme shown in Fig. 8. The oxygen of ester group of dihydropyridine and acyl hydrazone form conventional hydrogen bonding with Glycine (Gly83) and Glutamine (Gln36) and Cysteine (Cys42) respectively. In addition, it elicited the hydrophobic interactions with other amino acids viz., Trp206 (pi–pi interaction), Val152, Ala157 and Cys42 (pi-alkyl). Methylene group made carbon hydrogen bonds with Asparagine (Asn173). The hydrogen bonding interaction site i.e., Gly83 and Cys42 for ligand E64 and 5d are common (Fig. 7). The docked pose of 5d with highest binding affinity is shown in Fig. 8. It can be noticed from Fig. 8 that mainly hydrogen bonding and hydrophobic interactions (pi–pi interaction and pi-alkyl interaction) are responsible for fixing of the compound 5d. Some important interactions of 5d with different amino acids have been listed in Additional file 1: Table S3. The role of Cys42 residue in the inhibition of FP2 by E64 has been well known in literature [46, 47]. All these facts show that binding of compound 5d to these active site residues might be the cause of antimalarial activity. The docking results suggested that the antimalarial activity of the DHP-pyrazole-hydrazone derivatives might be due to their inhibitions of falcipain-2.
Conclusion
1,4-dihydropyridin-4-yl-phenoxyacetohydrazides with differently substituted pyrazole carbaldehyde were synthesised using molecular hybridisation. The resulting compound (5) exists in two conformations (i.e., E, s-cis and E, s-trans) as revealed from conformational studies. All the synthesised compounds were screened for their in vitro antimalarial activities against chloroquine-sensitive malaria parasite P. falciparum 3D7 and exhibit good inhibition as compared to standard drug chloroquine. In vitro antiplasmodial IC50 value of compound 5d was found to be 4.40 nM which is lower than that of all the three reference drugs chloroquine (18.7 nM), Pyrimethamine (11 nM) and Artimisinin (6 nM). In silico binding study of compound 5d with plasmodial cysteine protease falcipain-2 shows that inhibition of falcipain-2 could be the probable reason for the potency shown by compound 5d. The results obtained from in vitro and in silico studies suggest that these compounds can be used as potent anti-malarial agents after their cytotoxicity evaluation. All the synthesized compounds 5a–5g show moderate to good antimicrobial activity against Gram negative bacterium strain i.e. Escherichia coli and excellent antifungal activity against Aspergillus niger compared to reference drug.
Experimental
All the chemicals used were purchased from Spectrochem, Avra and Sigma Aldrich and were used as received. Silica gel 60 F254 (Precoated aluminium plates) from Merck was used to monitor reaction progress. Melting points were determined on Buchi Melting Point M-560 apparatus and are uncorrected. IR (KBr) spectra were recorded on Perkin Elmer FTIR spectrophotometer and the values are expressed as νmax cm−1. The 1H and 13C spectra were recorded on Bruker top spin and Jeol JNM ECX-400P at 400 MHz and 100 MHz respectively. Mass spectra were recorded at Bruker Micro TOF Q-II. The chemical shift values are recorded on δ scale and the coupling constants (J) are in Hertz. Pyrazole carbaldehydes were prepared according to the procedure described in literature [48].
General procedure for synthesis of acyl hydrazones (5a–5g)
To a clear solution of 3 (1.00 mmol) in ethanol (10 ml), 1.00 mmol of pyrazole carbaldehyde (4) and a catalytic amount of glacial acetic acid were added and the reaction mixture was refluxed for 10 h. The progress of the reaction was monitored by TLC using ethyl acetate-petroleum ether, (70:30, v/v). After completion, the reaction mixture was poured onto crushed ice. The precipitate formed was collected by vacuum filtration and washed with cold ethanol to afford pure products (5a–5g) in 92–98% yield. The products were characterized by IR, 1H NMR, 13C NMR and Mass spectra.
Diethyl4-(4-(((3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl) methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5a)
White solid; M.p.: 154 °C; Yield 98%; IR (KBr, cm−1) νmax: 3488, 3244, 3087, 1669, 1483, 1225, 1010; 1H NMR (400 MHz, CDCl3) δ 9.91 (s, 1H, CONH, 84.15%), 9.79 (s, 1H, CONH, 15.85%), 8.55 (s, 1H, NH=CH, 83.83%), 8.32 (s, 1H, NH=CH, 16.17%), 8.25 (s, 1H, Pyr–H, 84.69%), 7.95 (s, 1H, Pyr–H, 15.31%), 7.80–7.75 (m, 2H, ArH(n), 16.66%), 7.74–7.69 (m, 2H, ArH(n), 83.34%), 7.67–7.61 (m, 2H, ArH(m), 16.44%), 7.61–7.53 (m, 2H, ArH(m), 83.56%), 7.51–7.41 (m, 2H, ArH(o)), 7.32 (m, 1H, ArH(p)), 7.21 (m, 2H, ArH(f)), 7.10 (m, 2H, ArH(L)), 6.77 (m, 2H, ArH(g), 18.13%), 6.75 (m, 2H, ArH(g), 81.87%), 6.53 (s, 1H, NH, 83.16%), 6.50 (s, 1H, NH, 16.84%), 4.97 (s, 1H, C4–H, 15.74%), 4.95 (s, 1H, C4–H, 84.26%), 4.91 (s, 2H, –OCH2, 14.77%), 4.57 (s, 2H, –OCH2, 85.23%), 4.12–3.99 (m, 4H, CH2), 2.27 (s, 6H, CH3, 83.20%), 2.26 (s, 6H, CH3, 16.80%), 1.20 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3) δ 167.84, 164.93, 164.31, 161.84, 156.40, 155.51, 152.21, 144.49, 142.50, 142.36, 139.25, 130.59, 130.51, 130.45, 130.37, 129.55, 129.25, 128.93, 128.21, 128.18, 127.37, 127.28, 126.91, 119.36, 119.22, 116.16, 115.99, 115.85, 115.64, 115.47, 114.23, 114.14, 103.74, 77.40, 77.09, 76.77, 67.30, 59.79, 38.89, 38.55, 19.25, 19.21, 14.27. MS (m/z) 666.12; Anal. Calcd. For C37H36FN5O6: C, 66.76; H, 5.45; F, 2.85; N, 10.52; O, 14.42. Found: C, 66.81; H, 5.39; F, 2.91; N, 10.47; O, 14.37.
Diethyl4-(4-(((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl) methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5b)
White solid; M.p.: 144 °C; Yield 96%; IR (KBr, cm−1) νmax: 3488, 3244, 2987, 1655, 1483, 1255; 1H NMR (400 MHz, CDCl3) δ 9.42 (s, 1H, CONH), 8.74 (s, 1H, NH=CH, 14.71%), 8.66 (s, 1H, NH=CH, 85.29%), 8.33 (s, 1H, Pyr–H, 16.4%), 8.16 (s, 1H, Pyr–H, 83.6%), 7.77 (m, 2H, ArH), 7.59 (m, 2H, ArH), 7.51–7.47 (m, 2H, ArH), 7.46 (m, 2H, ArH), 7.34 (m, 1H, ArH), 7.23 (m, 2H, ArH), 6.79 (m, 2H, ArH), 5.66 (s, 1H, NH, 83.33%), 5.64 (s, 1H, NH, 16.67%), 4.95 (s, 1H, C4–H, 16.82%), 4.93 (s, 1H, C4–H, 83.18%), 4.61 (s, 2H, –OCH2, 84.5%), 4.54 (s, 2H, –OCH2, 15.5%), 4.11–4.03 (m, 4H, CH2), 2.31 (s, 6H, CH3), 1.21 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3) δ 167.77, 164.74, 155.44, 144.09, 142.44, 142.14, 139.33, 134.91, 130.01, 129.71, 129.47, 129.13, 127.49, 127.23, 119.37, 116.03, 114.17, 104.10, 77.45, 77.13, 76.81, 67.33, 59.91, 38.97, 19.65, 14.38; Anal. Calcd. for C37H36ClN5O6: C, 65.14; H, 5.32; Cl, 5.20; N, 10.27; O, 14.07. Found: C, 65.09; H, 5.27; Cl, 5.16; N, 10.31; O, 14.11.
Diethyl4-(4-(((3-(4-methylphenyl)-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl) methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5c)
White solid; M.p.: 152 °C; Yield 94%; IR (KBr, cm−1) νmax: 3317, 1669, 1512, 1211, 1096, 752; 1H NMR (400 MHz, CDCl3) δ 9.37 (s, 1H, CONH), 8.69 (s, 1H, NH=CH, 15.46%), 8.65 (s, 1H, NH=CH, 84.54%), 8.33 (s, 1H, Pyr–H, 15%), 8.15 (s, 1H, Pyr–H, 85%), 7.81–7.74 (m, 2H, ArH), 7.53 (m, 2H, ArH), 7.46 (m, 2H, ArH), 7.33 (m, 1H, ArH), 7.29 (m, 2H, ArH), 7.23 (m, 2H, ArH), 6.79 (m, 2H, ArH), 5.71 (s, 1H, NH, 84.11%), 5.66 (s, 1H, NH, 15.89%), 4.97 (s, 1H, C4–H, 17.27%), 4.93 (s, 1H, C4–H, 82.73%), 4.77 (s, 2H, –OCH2, 17.59%), 4.60 (s, 2H, –OCH2, 82.41%), 4.12–4.01 (m, 4H, CH2), 2.41 (s, 3H, CH3, 84.90%), 2.39 (s, 3H, CH3, 15.10%), 2.31 (s, 6H, CH3, 84.85%), 2.30 (s, 6H, CH3, 15.15%), 1.20 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3) δ 167.77, 164.64, 155.52, 152.83, 144.19, 142.72, 142.43, 139.73, 139.02, 129.60, 129.45, 128.67, 127.25, 126.89, 119.34, 115.88, 114.17, 103.98, 77.45, 77.14, 76.82, 59.86, 38.97, 21.42, 19.56, 14.38; Anal. Calcd. For C38H39N5O6: C, 68.97; H, 5.94; N, 10.58; O, 14.51. Found: C, 69.01; H, 5.89; N, 10.62; O, 14.47.
Diethyl 4-(4-(((1,3-diphenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl)methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5d)
White solid; M.p.: 164 °C; Yield 96%; IR (KBr, cm−1) νmax: 3276, 2987, 1672, 1499, 1211, 1095, 750; 1H NMR (400 MHZ, CDCl3) δ 10.04 (s, 1H, CONH, 15%), 9.39 (s, 1H, CONH, 85%), 8.68 (s, 1H, NH=CH, 13.4%), 8.55 (s, 1H, NH=CH, 86.6%), 8.35 (s, 1H, Pyr–H, 15%), 8.16 (s, 1H, Pyr–H, 85%), 7.79 (m, 2H, ArH), 7.64 (m, 2H, ArH), 7.50 (m, 2H, ArH), 7.47 (m, 1H, ArH), 7.46–7.41 (m, 2H, ArH), 7.33 (m, 1H, ArH), 7.23 (m, 2H, ArH), 6.79 (m, 2H, ArH), 5.65 (s, 1H, NH, 85%), 5.63 (s, 1H, NH, 15%), 4.97 (s, 1H, C4–H, 17.39%), 4.93 (s, 1H, C4–H, 82.61%), 4.61 (s, 2H, –OCH2), 4.11–4.02 (m, 4H, CH2), 2.31 (s, 6H, CH3, 83.69%), 2.30 (s, 6H, CH3, 16.31%), 1.20 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3) δ 167.67, 143.87, 142.42, 130.33, 129.68, 129.51, 128.96, 128.82, 119.38, 114.18, 104.24, 92.62, 77.44, 77.12, 76.80, 67.53, 67.34, 61.71, 59.88, 39.15, 19.74, 14.38; Anal. Calcd. for C37H37N5O6: C, 68.61; H, 5.76; N, 10.81; O, 14.82. Found: C, 68.57; H, 5.81; N, 10.93; O, 14.74.
Diethyl 4-(4-(((3-(4-bromophenyl)-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl) methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5e)
White solid; M.p.: 130 °C; Yield 95%; IR (KBr, cm−1) νmax: 3517, 3317, 3087, 1684, 1483, 1211, 1096, 767; 1H NMR (400 MHz, CDCl3) δ 9.44 (s, 1H, CONH), 8.81 (s, 1H, NH=CH, 16.33%), 8.65 (s, 1H, NH=CH, 83.67%), 8.33 (s, 1H, Pyr–H, 16.5%), 8.16 (s, 1H, Pyr–H, 83.5%), 7.77 (m, 2H, ArH), 7.63–7.59 (m, 2H, ArH), 7.52 (m, 2H, ArH), 7.50–7.45 (m, 2H, ArH), 7.34 (m, 1H, ArH), 7.24 (m, 2H, ArH), 6.79 (m, 2H, ArH), 5.69 (s, 1H, NH, 82.35%), 5.67 (s, 1H, NH, 17.65%), 4.95 (s, 1H, C4–H, 17.65%), 4.93 (s, 1H, C4–H, 82.35%), 4.61 (s, 2H, –OCH2, 84.65%), 4.54 (s, 2H, –OCH2, 15.35%), 4.11–4.03 (m, 4H, CH2), 2.31 (s, 6H, CH3, 80.61%), 2.30 (s, 6H, CH3, 19.39%), 1.21 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3) δ 184.71, 167.74, 164.71, 155.45, 151.38, 144.06, 142.44, 142.12, 139.33, 132.15, 132.07, 131.98, 131.89, 131.09, 130.53, 130.28, 129.84, 129.70, 129.46, 128.24, 127.49, 127.21, 119.84, 119.36, 116.04, 114.19, 104.12, 77.44, 77.12, 76.81, 67.35, 59.89, 38.99, 19.64, 14.39; Anal. Calcd. For C37H36BrN5O6: C, 61.16; H, 4.99; Br, 11.00; N, 9.64; O, 13.21. Found: C, 61.13; H, 5.01; Br, 10.96; N, 9.67; O, 13.17.
Diethyl4-(4-(((3-(4-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl) methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5f)
White solid; M.p.: 134 °C; Yield 92%; IR (KBr, cm−1) νmax: 3317, 1684, 1497, 1197, 1096; 1H NMR (400 MHz, CDCl3) δ 9.44 (s, 1H, CONH, 84.16%), 8.81 (s, 1H, CONH, 15.84%), 8.64 (s, 1H, NH=CH, 83.96%), 8.51 (s, 1H, NH=CH, 16.04%), 8.32 (s, 1H, Pyr–H, 15.15%), 8.15 (s, 1H, Pyr–H, 84.85%), 7.78–7.75 (m, 2H, ArH), 7.59–7.53 (m, 2H, ArH), 7.45 (m, 2H, ArH), 7.31 (m, 1H, ArH), 7.21 (m, 2H, ArH), 7.02–6.98 (m, 2H, ArH), 6.80–6.74 (m, 2H, ArH), 5.80 (s, 1H, NH), 4.97 (s, 1H, C4–H, 16.95%), 4.93 (s, 1H, C4–H, 83.05%), 4.60 (s, 2H, –OCH2, 85.4%), 4.58 (s, 2H, –OCH2, 14.6%), 4.11-4.01 (m, 4H, CH2), 3.87 (s, 3H, –OCH3, 17.67%), 3.85 (s, 3H, –OCH3, 82.33%), 2.30 (s, 6H, CH3, 82.21%), 2.29 (s, 6H, CH3, 17.79%), 1.20 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (101 MHz, CDCl3) δ 167.73, 164.60, 160.18, 155.46, 153.25, 144.09, 142.73, 142.43, 139.47, 130.35, 130.04, 129.76, 129.64, 129.46, 127.22, 126.90, 124.58, 119.81, 119.30, 115.76, 114.35, 114.28, 114.19, 104.10, 77.45, 77.13, 76.81, 67.36, 59.87, 55.47, 38.97, 19.60, 14.38; Anal. Calcd. For C38H39N5O7: C, 67.34; H, 5.80; N, 10.33; O, 16.52. Found: C, 67.31; H, 5.85; N, 10.29; O, 16.48.
Diethyl4-(4-(((3-(4-nitrophenyl)-1-phenyl-1H-pyrazol-4-yl)methyleneaminocarbamoyl) methoxy)phenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (5g)
Yellow solid; M.p.: 136 °C; Yield 94%; IR (KBr, cm−1) νmax: 3317, 1655, 1497, 1325, 1211, 1082, 853, 752; 1H NMR (400 MHz, CDCl3) δ 9.59 (s, 1H, CONH, 85.15%), 9.21 (s, 1H, CONH, 14.85%), 8.66 (s, 1H, NH=CH, 85.11%), 8.36 (s, 1H, NH=CH, 14.89%), 8.33 (s, 1H, Pyr–H, 14.94%), 8.32 (s, 1H, Pyr–H, 85.06%), 8.30 (m, 2H, ArH), 7.90–7.84 (m, 2H, ArH), 7.78 (m, 2H, ArH), 7.49 (m, 2H, ArH), 7.37 (m, 1H, ArH), 7.22 (m, 2H, ArH), 6.78 (m, 2H, ArH), 5.82 (s, 1H, NH, 15.53%), 5.78 (s, 1H, NH, 84.47%), 4.93 (s, 1H, C4–H, 84.62%), 4.92 (s, 1H, C4–H, 15.38%), 4.89 (s, 2H, –OCH2, 15.42%), 4.61 (s, 2H, –OCH2, 84.58%), 4.11–4.01 (m, 4H, CH2), 2.31 (s, 6H, CH3, 14.85%), 2.30 (s, 6H, CH3, 85.15%), 1.20 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (101 MHz, CDCl3) δ 167.79, 164.96, 155.48, 147.77, 144.13, 142.44, 141.60, 139.15, 138.70, 129.78, 129.42, 129.35, 127.83, 127.74, 124.09, 119.43, 116.79, 114.20, 104.05, 77.44, 77.12, 76.81, 67.41, 59.92, 38.99, 19.62, 14.38; Anal. Calcd. For C37H36N6O8: C, 64.15; H, 5.24; N, 12.13; O, 18.48. Found: C, 64.19; H, 5.28; N, 12.17; O, 18.44.
Anti-malarial activity
Parasite cultivation
The anti-malarial activity of synthesised acyl hydrazones (5a–5g) was assessed against chloroquine-sensitive P. falciparum (3D7) isolate. P. falciparum was cultivated in human A Rh+ red blood cells using RPMI 1640 medium (Sigma, India) supplemented with AB Rh+ serum (10%), 5% sodium bicarbonate (Sigma, India) and 40 μg/mL of gentamycin sulphate 17 (Sigma, India).
In vitro test for anti-malarial activity
The in vitro activity of P. falciparum intra erythrocytic stage on synthesised compounds was evaluated by Schizonts maturation Inhibition (SMI) method [49]. Accordingly, the compounds were dissolved in DMSO and serially diluted with RPMI 1640 medium to reach 1 mg/ml before use. The cultures, before testing, were synchronized by treatment with 5% d-sorbitol with a parasitemia of 0.6–0.8%. Each well received 10 μl of parasite-infected erythrocytes, 5% hematocrit and 90 μl of different compound dilutions. Chloroquine and solvent controls contained similar volumes of the solvent, as that of test wells. The plates were incubated at 37 °C for 24 h. After confirmation of the presence of 10% mature schizonts in control wells, the blood from each well was harvested and a thick film was prepared on a glass slide. The blood films were stained for 40 min with Giemsa stain at a dilution of 10% in double distilled water. Three independent optical-microscopy readings of the number of schizonts with three or more nuclei were carried out in 200 parasitized red blood cells for each dilution and duplicate. Growth inhibition was expressed as the percentage of schizonts in each concentration, compared with controls.
Calculation and analysis
The number of schizonts counted per well was directly entered into the nonlinear regression software, HN NonLin V 1.1 [50], which was particular for the analysis of in vitro drug sensitivity assay for malaria. Individual dose response curves were generated and their IC50 values were determined.
Antimicrobial activity
Test microorganisms
3 microbial strains were selected on the basis of their clinical importance in causing diseases in humans. One Gram-positive bacteria (Bacillus cereus); one Gram-negative bacteria (Escherichia coli) and one yeast, (Aspergillus niger) were screened for evaluation of antibacterial and antifungal activities of the synthesized pyrazoles. All the microbial cultures were procured from Microbial Type Culture Collection (MTCC), IMTECH, Chandigarh. The bacteria were sub cultured on nutrient agar whereas yeast on malt yeast agar.
In-vitro antibacterial activity
The antimicrobial activity of synthesised acyl hydrazones (5a–5g) was evaluated by the agar-well diffusion method. All the microbial cultures were adjusted to 0.5 McFarland standard, which is visually comparable to a microbial suspension of approximately 1.5 × 108 cfu/ml. Agar medium (20 ml) was poured into each Petri plate and plates were swabbed with 100 µl inocula of the test microorganisms and kept for 15 min for adsorption. Using sterile cork borer of 8 mm diameter, wells were created into the seeded agar plates which were loaded with a 100 µl volume with concentration of 8.0 mg/ml of each compound reconstituted in the dimethylsulphoxide (DMSO). All the plates were incubated at 37 °C for 24 h. Antimicrobial activity of each compound was evaluated by measuring the zone of growth inhibition against the test organisms with zone reader (Hi Antibiotic zone scale). DMSO was used as a negative control whereas Tetracycline was used as positive control for bacteria and clotrimazole for fungi. This procedure was performed in triplicates for each organism.
Computational details
Energy calculation with PM7 Hamiltonian method
All calculation was carried out with PM7 Hamiltonian method using MOPAC2016 program [32] on DELL LATITUDE E5410. All chemical structures were drawn in Marvin Sketch 15.12.7.0 [51]. The structures under study were optimized using default value of GNORM and properties were calculated. The calculations were performed in solution phase using chloroform (dielectric constant = 4.8) and dimethylsulfoxide (dielectric constant = 46.70) solvents in Andreas Klamt’s COSMO implicit solvation model.
Docking studies
The crystal structure of plasmodial cysteine protease falcipain-2 was obtained from the Brookhaven Protein Data Bank http://www.rcsb.org/pdb (PDB entry: 3BPF). To carry outdocking studies, the 2D-structures of 5d were drawn in Marvin Sketch 15.12.7.0 [51]. Then explicit hydrogens were added and this was converted to 3D and its energy was minimized. Co-crystallized ligand was removed from pdb file 3BPF and protein molecule was prepared by deleting solvent molecules using UCSF Chimera 1.10 [52]. Incomplete side chains were replaced using Dun Brack Rotamer library [53]. Hydrogens were added and gasteiger charges were calculated using AMBERff14SB and antechamber [54]. The prepared file was saved as pdb format and used for further studies. These structures of ligand 5d and proteins were transformed into pdbqt format with Auto Dock Tools [55]. Docking studies were carried out by using Auto Dock Vina 1.1.2 [44]. Grid center was placed on the active site. The centers and sizes of grid box were as follows: center_x = − 58.5196350008, center_y = − 1.19310953271 and center_z = − 17.0068559885, size_x = 25.0, and size_y = 25.0, size_z = 25.0. Exhaustiveness of the global search algorithm was set to be 100. Then, finally docking results were viewed in Discovery Studio Visualizer 16.1.0.15350 [56].
Change history
06 February 2018
After publication of the original article [1], the following error was reported in the Results section of the Abstract: “antifungal activity against one yeast i.e. Aspergillus niger” should read: “antifungal activity against one fungus i.e. Aspergillus niger”. The authors would like to confirm all antifungal activity has been screened against fungi not yeast.
References
World Malaria Report 2014 (2014) World Health Organization
Alílio MS, Bygbjerg IC, Breman JG (2004) Are multilateral malarial research and control programs the most successful? Lessons from the past 100 years in Africa. Am J Trop Med Hyg 71:268–278
Towie N (2006) Malaria breakthrough raises spectre of drug resistance. Nature 440:852–853
Tumwebaze P, Conrad MD, Walakira A, LeClair N, Byaruhanga O, Nakazibwe C, Kozak B, Bloome J, Okiring J, Kakuru A, Bigira V, Kapisi J, Legac J, Gut J, Cooper RA, Kamya MR, Havlir DV, Dorsey G, Greenhouse B, Nsobya SL, Rosenthal PJ (2015) Impact of antimalarial treatment and chemoprevention on the drug sensitivity of malaria parasites isolated from Ugandan children. Antimicrob Agents Chemother 59:3018–3030
Fancony C, Brito M, Gil JP (2016) Plasmodium falciparum drug resistance in Angola. Malar J 15:74–85
Sirawaraporn W, Sathitkul T, Sirawaraporn R, Yuthavong Y, Santi DV (1997) Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA 94:1124–1129
Jnr CV, Danuello A, Bolzani VS, Barreiro EJ, Fraga CAM (2007) Molecular hybridization: a useful tool in the design of new drug prototypes. Curr Med Chem 14:1829–1852
Walsh JJ, Bell A (2009) Hybrid drugs for malaria. Curr Pharm Des 15:2970–2985
Ullooraa S, Shabarayab R, Ranganathanc R, Adhikari AV (2013) Synthesis, anticonvulsant and anti-inflammatory studies of new 1,4-dihydropyridin-4-yl phenoxy acetohydrazones. Eur J Med Chem 70:341–349
Syakaev V, Podyachev S, Buzykin B, Latypov S, Habicher W, Konovalov A (2006) NMR study of conformation and isomerization of aryl- and hetero arylaldehyde 4-tert-butylphenoxyacetylhydrazones. J Mol Struct 788:55–62
Cunico W, Cechinel CA, Bonacorso HG, Martins AP (2006) Antimalarial activity of 4-(5-trifluoromethyl-1H-pyrazol-1-yl)-chloroquine analogues. Bioorg Med Chem Lett 16:649–653
Bekhit AA, Hymete A, Asfaw H, Bekhit AD (2012) Synthesis and biological evaluation of some pyrazole derivatives as anti-malarial agents. Arch Pharm 345:147–154
Santanna CMR, Alencastro RB, Rodrigues CR, Barreiro G, Barreiro EJ, Neto JDM, Freitas ACC (1996) A semi empirical study of pyrazole acylhydrazones as potential antimalarial agents. Int J Quantum Chem 60:1835–1843
Sugimoto N, Watanabe H, Ide A (1960) The synthesis of l-α-amino-β-(pyrazolyl-N)-propionic acid in Citrullus vulgaris. Tetrahedron 11:231–233
Hannah J, Kelly K, Patchett AA, Steelman SL, Morgan ER (1975) Substituted pyrazolo corticoids as topical antiinflam-matory agents. J Med Chem 18:168–172
Stauffer SR, Huang YR, Aron ZD, Coletta CJ, Sun J, Katzenellenbogen BS, Katzenellenbogen JA (2001) Triarylpyrazoles with basic side chains: development of pyrazole-based estrogen receptor antagonists. Bioorg Med Chem 9:151–161
Fink BE, Mortensen DS, Stauffer SR, Aron ZD, Katzenellenbogen JA (1999) 1,3,5-triaryl-4-alkyl-pyrazoles bind to the estrogen receptor (ER) with high affinity. Chem Biol 6:205–219
Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA (2000) Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J Med Chem 43:4934–4947
Ashton WT, Hutchins SM, Greenlee WJ, Doss GA, Chang RS, Lotti VJ, Faust KA, Chen TB, Zingaro GJ (1993) Nonpeptide angiotensin II antagonists derived from 1H-pyrazole-5-carboxylates and 4-aryl-1H-imidazole-5-carboxylates. J Med Chem 36:3595–3605
Abdel-Aziz M, Abuo-Rahma GEDA, Hassan AA (2009) Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur J Med Chem 44:3480–3487
Manikannan R, Venkatesan R, Muthusubramanian S, Yogeeswari P, Sriram D (2010) Pyrazole derivatives from azines of substituted phenacyl aryl/cyclohexylsulfides and their antimycobacterial activity. Bioorg Med Chem Lett 20:6920–6924
Verissimo E, Berry N, Gibbons P, Cristiano ML, Rosenthal PJ, Gut J, Ward SA, Neill PM (2008) Design and synthesis of novel 2-pyridone peptidomimetic falcipain 2/3 inhibitors. Bioorg Med Chem Lett 18:4210–4214
Desai PV, Patny A, Sabnis Y, Tekwani B, Gut J, Rosenthal PJ, Srivastava A, Avery M (2004) Identification of novel parasitic cysteine protease inhibitors using virtual screening. The ChemBridge database. J Med Chem 47:6609–6615
Patorski P, Wyrzykiewicz E, Bartkowiak G (2013) Synthesis and conformational assignment of N-(E)-stilbenyloxymethylenecarbonyl-substituted hydrazones of acetone and o-(m- and p-) Chloro-(nitro-)benzaldehydes by means of and NMR spectroscopy. J Spectroscopy 2013:1–12
Ershov AY, Lagoda IV, Yakimovich SI, Pakalnis VV, Zerova IV, Dobrodumov AV, Shamanin VV (2009) Tautomerism and conformational isomerism of mercaptoacetylhydrazones of aliphatic and aromatic aldehydes. Russ J Org Chem 45:660–661
Gonzaga DTG, Silva FCD, Ferreira VF, Wardell JL, Wardell SMSV (2016) Crystal structures of 1-Aryl-1H- and 2-Aryl-2H-1,2,3-triazolyl hydrazones, conformational consequences of different classical hydrogen bonds. J Braz Chem Soc 27:2322–2333
Glidewell C, Low JN, Skakle JMS, Wardell JL (2004) Hydrogen bonding in nitroaniline analogues: 4-nitrobenzaldehyde hydrazone forms hydrogen-bonded sheets of R 4 4(26) rings. Acta Crystallogr Sect C: Cryst Struct Commun 60:33–34
Hamzi I, Barhoumi-Slimi TM, Abidi R (2016) Synthesis, characterization, and conformational study of acylhydrazones of α, β-unsaturated aldehydes. Heteroat Chem 27:139–148
Angelusiu MV, Barbuceanu SF, Draghici C, Almajan GL (2010) New Cu(II), Co(II), Ni(II) complexes with aroyl-hydrazone based ligand. Synthesis, spectroscopic characterization and in vitro antibacterial evaluation. Eur J Med Chem 45:2055–2062
Stadler AM, Harrowfield J (2009) Bis-acyl-/aroyl-hydrazones as multidentate ligands. Inorg Chim Acta 362:4298–4314
Onnis V, Cocco MT, Fadda R, Congiu C (2009) Synthesis and evaluation of anticancer activity of 2-arylamino-6-trifluoromethyl-3-(hydrazonocarbonyl)pyridines. Bioorg Med Chem 17:6158–6165
Stewart JP (2016) Stewart computational chemistry, MOPAC2016, Version: 16.043 W web: http://OpenMOPAC.net
Aneja KR, Sharma C, Joshi R (2011) In vitro efficacy of amaltas (Cassia fistula L.) against the pathogens causing otitis externa. J Microbiol 4:175–184
Drinkwater N, Vinh NB, Mistry SN, Bamert RS, Ruggeri C, Holleran JP, Loganathan S, Paiardini A, Charman SA, Powell AK, Avery AK, McGowan S, Scammells PJ (2016) Potent dual inhibitors of Plasmodium falciparum M1 and M17 aminopeptidases through optimization of S1 pocket interactions. Eur J Med Chem 110:43–64
Singh A, Rosenthal PJ (2001) Comparison of efficacies of cysteine protease inhibitors against five strains of Plasmodium falciparum. Antimicrob Agents Chemother 45:949–951
Sharma M, Chauhan K, Srivastava RK, Singh SV, Srivastava K, Saxena JK, Puri SK, Chauhan PMS (2014) Design and synthesis of a new class of 4-aminoquinolinyl- and 9-anilinoacridinyl schiff base hydrazones as potent antimalarial agents. Chem Biol Drug Des 84:175–181
Sharma RK, Younis Y, Mugumbate G, Njoroge M, Gut J, Rosenthal PJ, Chibale K (2015) Synthesis and structure-activity-relationship studies of thiazolidinediones as antiplasmodial inhibitors of the Plasmodium falciparum cysteine protease falcipain-2. Eur J Med Chem 90:507–518
Rosenthal PJ, Olson JE, Lee GK, Palmer JT, Klaus JL, Rasnick D (1996) Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrob Agents Chemother 40:1600–1603
Shenai BR, Lee BJ, Alvarez-Hernandez A, Chong PY, Emal CD, Neitz RJ, Roush WR, Rosenthal PJ (2003) Structure-activity relationships for inhibition of cysteine protease activity and development of Plasmodium falciparum by peptidyl vinyl sulfones. Antimicrob Agents Chemother 47:154–160
Ettari R, Bova F, Zappala M, Grasso S, Micale N (2010) Falcipain-2 inhibitors. Med Res Rev 30:136–167
Sajid M, McKerrow JH (2002) Cysteine proteases of parasitic organisms. Mol Biochem Parasitol 120:1–21
Schirmeister T, Kaeppler U (2003) Non-peptidic inhibitors of cysteine proteases. Mini Rev Med Chem 3:361–373
Melnyk P, Leroux V, Sergheraert C, Grellier P (2006) Design, synthesis and in vitro antimalarial activity of an acylhydrazone library. Bioorg Med Chem Lett 16:31–35
Trott O, Olson AJ, Vina AD (2010) Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Kerr ID, Lee JH, Pandey KC, Harrison A, Sajid M, Rosenthal PJ, Brinen LS (2009) Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: implications for substrate specificity. J Med Chem 52:852–857
Grazioso G, Legnani L, Toma L, Ettari R, Micale N, Micheli CD (2012) Mechanism of falcipain-2 inhibition by a, b-unsaturated benzo[1,4]diazepin-2-one methyl ester. J Comput Aided Mol Des 26:1035–1043
Arafet K, Ferrer S, Martí S, Moliner V (2014) Quantum mechanics/molecular mechanics studies of the mechanism of falcipain-2 inhibition by the epoxysuccinate E64. Biochemistry 53:3336–3346
Kumar P, Kumar S, Husain K, Kumar A (2011) An efficient synthesis of pyrazolechalcones under solvent free conditions at room temperature. Chin Chem Lett 22:37–40
Lambros C, Vanderberg JP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420
Noedl H (2002) Non linear evaluation of malaria drug sensitivity data (HN-NonLin V1.1) Bangkok, Thailand: Armed Forces Research Institute for Medical Sciences. http://www.meduniwien.ac.at/user/harald.noedl/malaria/download.html
Mavin Sketch 15.12.7.0 ChemAxon Ltd (1998–2015) http://www.chemaxon.com
Pettersen Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Dunbrack RL Jr (2002) Rotamer library in the 21st century. Curr Opin Struct Boil 12:431–440
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graphics Modeling 25:247–260
Forte S, AutoDock Tools (version 1.5.6 rc2) (1999–2010) Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute, 1999–2010. http://mgltools.scripps.edu
Discovery Studio 2016 client (2005–2016) Accelrys Software Inc.
Authors’ contributions
All authors participate in each stage in the preparation of this manuscript like carried the literature study, designing part, designing of synthetic schemes, synthesis and purification of compounds. All authors read and approved the final manuscript.
Acknowledgements
One (KK) of the authors wishes to express his gratitude to the University Grant Commission (UGC) for junior research fellowship.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All samples of the synthesized compounds as well as their data are available from the authors.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised: “one yeast i.e. Aspergillus niger” was changed to “one fungus i.e. Aspergillus niger” in the Results section of the Abstract.
A correction to this article is available online at https://doi.org/10.1186/s13065-018-0374-9.
Additional file
Additional file 1.
Additional tables.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kumar, P., Kadyan, K., Duhan, M. et al. Design, synthesis, conformational and molecular docking study of some novel acyl hydrazone based molecular hybrids as antimalarial and antimicrobial agents. Chemistry Central Journal 11, 115 (2017). https://doi.org/10.1186/s13065-017-0344-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-017-0344-7