
Abstract
Background
Ligustrazine has potent effects of thrombolysis, neuroprotection and vascular protection, which were important for effectively protecting the nervous system. Previous study in our laboratory reported that ligustrazine-benzoic acid derivatives have been shown to exhibit beneficial effect against CoCl2-induced neurotoxicity in differentiated PC12 cells. To further improve ligustrazine’s neuroprotection, we integrated the ligustrazine and phenolic acid fragments into one molecule via an amide bond based on structural combination.
Results
In this study, 12 novel ligustrazine-phenolic acid derivatives were synthesized and nine others were prepared by improved methods. Furthermore, these compounds were evaluated for their protective effects against CoCl2-induced neurotoxicity in differentiated PC12 cells. The amides conjunctional derivatives exhibited promising neuroprotective activities in comparison with ligustrazine. In addition, the most active congener (E)-3-(2,3,4-trimethoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L10, EC50 = 25 μM), which is 2 times higher than that of ligustrazine, may be a potential candidate for intervention in neurological diseases. Structure-activity relationship was discussed briefly.
Conclusions
Results of series of ligustrazinyl amides enrich the study of ligustrazine derivatives with neuroprotective effects. Our completed work supports that the attempt to apply structure combination to discover more efficient neuroprotection lead compounds is viable.

Ligustrazinyl Amides L1-L21 with Neuroprotective Effects.
Similar content being viewed by others
Background
Neurological disorders, such as Stroke, Alzheimer’s disease (AD) and Parkinson’s disease (PD), threaten millions of patients with ever growing numbers in ageing societies [1-3]. To discover drugs with nerve functional recovery as a treatment or prevention of neurological disorder is of great significance [4]. To date, despite the remarkable progress achieved in theory, effective approaches to recover damaged nerve are not yet to be found [1,5]. Therefore, to discover more effective drugs for the treatment in injured nerve cell remains an important area of drug discovery [6,7].
Nowadays, many new drugs have been generated from natural products [8-11]. Ligustrazine (2,3,5,6-tetramethylpyrazine, TMP), derived from the traditional Chinese medicine Rhizoma Chuanxiong (Ligusticum chuanxiong Hort.) which was widely used to treat Stroke and cerebrovascular disease (CVD) in China [12,13]. Recent studies have indicated that ligustrazine has potent effects of thrombolysis, neuroprotection and vascular protection, which were important for effectively protecting the nervous system [14-21]. In addition, many phenolic acid ingredients, such as caffeic acid, protocatechuic acid, salicylic acid, ferulic acid, vanillic acid, etc., also showed interesting neuroprotective activities [21-24].
To further improve ligustrazine’s neuroprotective effect, inspired by the potent neuroprotective effects of ligustrazine-benzoic acid derivatives, we integrated the ligustrazine and phenolic acid fragments into one molecule based on structural combination [17,21,25]; a series of novel ligustrazine-benzoic analogues was constructed via an amide bond rather than an ester bond in our previous research on ligustrazine-benzoic acid derivatives. A recent study has reported that part of ligustrazinyl amides congener structures exhibited good proliferative activities on human umbilical vascular endothelial cells (HUVECs) [25]. Their protective effects against neurotoxicity were evaluated in differentiated PC12 cells. Structure-activity relationship was discussed briefly.
Results and discussion
Chemistry
All the target compounds were synthesized via the routes outlined in Scheme 1, Scheme 2 and Scheme 3. The key intermediate (3,5,6-trimethylpyrazin-2-yl)methanamine (L) was prepared according to our previous study with minor improvements. Compound B (TMP-Br) was synthesized from anhydrous ligustrazine and N-bromosuccinimide (NBS) in carbon tetrachloride via free radical reaction, the crude product was used directly in the next reaction without further purification. The mixture of TMP-Br and phthalimide potassium in CH3CN that was refluxing for 2 h gave compound C. Intermediate L was obtained by reaction of C and 80% hydrazine hydrate in absolute ethanol refluxing for 5 h.

Synthetic routes to ligustrazine intermediate L. Reagents and Conditions: (i) CCl4, NBS, hv, reflux, 2 h, 65%; (ii) CH3CN, phthalimide potassium, reflux, 2 h, 64%; (iii) CH3CH2OH, N2H4 · H2O, reflux, 5 h, 88%.

Synthetic routes to ligustrazine derivatives L1–L15, L17–L21. Reagents and Conditions: (i) anhydrous CH2Cl2, EDCI/(CH3CH2)3 N, r.t., 12 h; (ii) anhydrous DMF (L11 anhydrous CH2Cl2), EDCI/HOBt, r.t., 12 h; (iii) anhydrous DMF (L21 DMI), EDCI/HOBt, r.t., 12 h.

Synthetic routes to ligustrazine derivatives L16. Reagents and Conditions: (i) DMF, benzyl bromide, K2CO3, 85°C, 12 h; (ii) H2O/CH3CH2OH, 10% KOH, 70°C, 2 h; (iii) TMP-NH2, DMF, EDCI/HOBt, r.t., 12 h; (iv) CH3OH, Pd/C, H2, r.t., 12 h.
The single-step coupling reaction between L and the cinnamic acids were performed using EDCI and (CH3CH2)3 N in anhydrous CH2Cl2, to afford ligustrazine derivatives (L1–L10, as shown in Method 1 of Scheme 2). In Method 2 and 3, the carboxylic acids and HOBt were firstly transformed to active ester in the presence of EDCI, and then reacted with compound L, obtaining the target compounds (L11–15, L17–21).
In Scheme 3, the starting compound 16 was first perbenzylated and then transformed to free carboxylic acid. The coupling reaction between L and the hydroxyl-perbenzylated benzoic acid 16b was performed using EDCI and HOBt in anhydrous CH2Cl2. A final deprotection step afforded the targeted compound L16. The chemical structures of all target compounds (Table 1) were confirmed by 1H-NMR, 13C-NMR and high resolution mass (HRMS).
Biological activities
Protective effect on injured neuronal-like PC12 cells
Setting ligustrazine as the positive control drug, all the synthesized compounds were tested for their protective effects on neuronal-like PC12 cells damaged by CoCl2. The proliferation rates (P%) of injured PC12 cells were assessed by methyl thiazolyl tetrazolium (MTT) assay. The proliferation rates (%) at different concentration and 50% effective concentrations (EC50) for protecting damaged PC12 cells of the ligustrazine derivatives were outlined in Table 2.
From the obtained results, it was observed that ligustrazine and most of its derivatives presented protective effects on injured differentiated PC12 cells, and several ligustrazine derivatives exhibited competitive positive activities (with lower EC50 values) than TMP (EC50 = 65 μM). Among them, L10 and L11 displayed promising neuroprotective activities (EC50 = 25, 27 μM, respectively), in which compound L10 presented 2 times higher potency than TMP.
Among L1–L10, compounds that introduced methyl and methoxy group on the phenyl ring performed better neuroprotective activities than L1 without any group substituted. It can be concluded that with the increase of the number of methoxy group at the phenyl moiety, the activities increase considerably (L10 > L3, L6, L8 > L4, L5, L7). However, this rule did not work in the case of L9 (EC50 = 102 μM) on the injured PC12 cells model. The structure-activity relationship analysis was consistent with the protective effects of ligustrazine-based stilbene derivatives on damaged ECV-304 cells [26]. These findings may provide a new framework for the design of new ligustrazine derivatives as neuroprotective drugs.
It should be noticed that ligustrazinyl amides L12–L16 with phenolic hydroxyl substituted approximately followed a tendency in activity 4-OH, 3-OCH3 > 4-OH > 2-OH > 3-OH > 3, 4-OH. This was similar to our previous research that the ligustrazine-benzoic acid derivatives were synthesized via an ester bond [21]. Moreover, compounds L15 and L11 were derived from salicylic acid and acetylsalicylic acid, respectively. But L11 (EC50 = 27 μM) displayed observable protective action, and it is much better than L15 (EC50 = 92 μM). This implied that acetyl group may be an effective group, led to improvement of the neuroprotective activities. In addition, the introduction of trans olefinic bond may contribute to enhance the neuroprotective activity, such as L20 > L12, L17 > L13, L19 > L15 and L21 > L16.
Furthermore, previous studies proved that compounds L2, L4, L19 and L20 could stimulate the proliferation of cultured human umbilical vascular endothelial cells [25]; current study also exhibited that L2, L4, L19 and L20 exhibited neuroprotective activities. Based on the above evidences, we reason that the new synthetic ligustrazinylated derivatives possess multiple pharmacological activities such as neuroprotection and protection against vascular endothelial cell injury, suggesting that they may be more efficacious than either a neuroprotective agent or vascular protective drug alone.
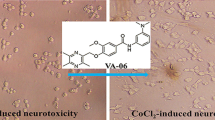
Effects of L10 on PC12 cells in morphology
Observed under optical microscopy (OLMPUS, Japan), as shown in Figure 1-A, we found that undifferentiated PC12 cells that maintained under normal conditions were small and proliferated to form clone-like cell clusters without neural characteristics. By exposure to NGF, normal differentiated PC12 cells showed fine dendritic networks similar to those nerve cells (Figure 1-B). In contrast, CoCl2-insulted differentiated PC12 cells developed mild cell body swelling and some cells shrunk the dendritic networks even lost neurites demonstrating round shape (Figure 1-C). Pretreatment with L10 alleviated morphological manifestations of cells damage compared to model cells (Figure 1-D).

Effects of L10 on differentiated PC12 cell injury in morphology (×200). (A) Control PC12 cells maintained under normal conditions. (B) PC12 cells exposed to NGF. (C) Differentiated PC12 cells exposed to CoCl2 insult. (D) Differentiated PC12 cells pre-incubated with L11 then exposed to CoCl2 insult.
Conclusions
In this work, 21 novel ligustrazine-phenolic acid derivatives were designed, synthesized and biologically evaluated for their protective effects against CoCl2-induced neurotoxicity in differentiated PC12 cells. The biological results have demonstrated that most of ligustrazine derivatives exhibited better neuroprotective activities in comparison with ligustrazine. In addition, the most active congener L10 (EC50 = 25 μM), which is 2 times higher than that of ligustrazine, may be a potential candidate for intervention in neurological diseases. Furthermore, structure-activity relationship was discussed briefly.
Altogether, results of series of ligustrazinyl amides enrich the study of ligustrazine derivatives with neuroprotective effects. Our completed work supports that the attempt to apply structure combination to discover more efficient neuroprotection lead compounds is viable. Furthermore, to pursue the optimized neuroprotective agents, ligustrazine-cinnamic acid ether and ligustrazine-cinnamic acid ester derivatives’ neuroprotective effects are ongoing in our lab.
Experimental section
Chemistry
Materials and methods
Reactions were monitored by TLC which was performed on silica gel GF254 (Qingdao Haiyang Chemical Co., China) and spots were visualized by modified bismuth potassium iodide or by irradiation with UV light (254 nm). Nuclear magnetic resonance spectra were recorded using a Bruker AVANCE 500 NMR spectrometer (Fällanden, Switzerland) in the indicated solvents. Chemical shifts are expressed in δ (ppm) relative to tetramethylsilane (TMS). Coupling constants are reported in Hertz (Hz). HRMS spectra were recorded on a Thermo Scientific™ LTQ Orbitrap XL hybrid FTMS instrument (Thermo Technologies, USA). Melting points (uncorrected) were metered on an X-5 micro melting point apparatus (Beijing, China). Flash column chromatography was performed on 200–300 mesh silica gel. All chemicals used were analytical. Solvents were reagent grade or high-performance liquid chromatography grade, and when necessary, were dried by standard methods. Concentration of the reaction solutions involved the use of rotary evaporator at reduced pressure. The yields were calculated by the last step reaction. Among all target compounds, L1, L2, L4, L8, L11, L13, L15, L17, L20 were reported in Liu’s research [25]. Therefore, HRMS were supplemented to confirm the chemical structures.
Preparation of (3,5,6-trimethylpyrazin-2-yl)methanamine (L)
The target compound L was synthesized by three steps. Compound B was prepared according to our previously reported method [27]. The crude product, which caused a strong mucous membrane irritation, could be used in the next step without further purification. To a solution of B (5.750 g, 27.00 mmol) in acetonitrile (100 mL), phthalimide potassium (5.000 g, 27.00 mmol) was added. The mixture was refluxing for 2 h, then concentrated under reduced pressure to afford compound C (4.878 g, 64%), white solid. m.p.:155.3-156.8, HRMS (ESI) m/z: 282.12381 [M + H]+, calcd. for C16H15N3O2 282.12425. The compound C was purified by flash column chromatography and recrystallization from acetone [28]. The important intermediate L was obtained by the reaction of C (4.000 g, 14.23 mmol) and 80% hydrazine hydrate (N2H4 · H2O, 0.85 mL) in absolute ethyl alcohol (100 mL) refluxing for 5 h. The solution was filtered, concentrated under reduced pressure. The residue was dissolved in dichloromethane, filtered and recovered methylene chloride to give a pale yellow semi-solid substance L (3.521 g, 88%), HRMS (ESI) m/z: 152.11829 [M + H]+, calcd. for C8H13N3 152.12827.
Preparation of L1–L10
To a solution of L (1.324 mmol) and the corresponding cinnamic acids (1.322 mmol) in anhydrous CH2Cl2 (20 mL), EDCI (253 mg, 1.324 mmol) and triethylamine ((CH3CH2)3 N, 3.97 mmol) were added. The mixture was stirred at room temperature for 12 h. Then washed with water (2 × 20 mL) and brine (20 mL), successively, dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was purified by flash chromatography and recrystallization from acetone.
N-((3,5,6-trimethylpyrazin-2-yl)methyl)cinnamamide (L1): White solid, yield: 90.7%. HRMS (ESI) m/z: 282.15988 [M + H]+, calcd. for C17H19N3O 282.16064.
(E)-3-(p-tolyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L2): White solid, yield: 83.5%. HRMS (ESI) m/z: 296.17554 [M + H]+, calcd. for C18H21N3O 296.17629.
(E)-3-(3,4-dimethoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L3): White solid, yield: 86.3%, m.p.: 150.8–151.4°C. 1H NMR (500 MHz, CDCl3): δ 7.62 (d, J = 15.6 Hz, 1H), 7.24 (s, 1H), 7.12 (d, J = 8.3 Hz, 1H), 7.07 (s, 1H), 6.86 (d, J = 8.3 Hz, 1H), 6.45 (d, J = 15.6 Hz, 1H), 4.61 (d, J = 3.7 Hz, 2H), 3.92 (s, 3H), 3.91 (s, 3H), 2.53 (s, 3H), 2.50 (br, 6H); 13C NMR (125 MHz, CDCl3): δ 166.2, 150.7, 149.8, 149.2, 147.9, 145.1, 141.0, 128.0, 122.1, 118.6, 111.2, 109.9, 56.1, 56.0, 41.4, 21.5, 21.5, 20.2. HRMS (ESI) m/z: 342.18167 [M + H]+, calcd. for C19H23N3O3 342.18177.
(E)-3-(4-methoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L4): White solid, yield: 85.8%. HRMS (ESI) m/z: 312.17093 [M + H]+, calcd. for C18H21N3O2 312.17120.
(E)-3-(2-methoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L5): White solid, yield: 90.1%, m.p.: 153.3–153.8°C. 1H NMR (500 MHz, CDCl3): δ 7.95 (d, J = 15.8 Hz, 1H), 7.52 (d, J = 7.5 Hz, 1H), 7.33 – 7.30 (m,1H), 7.25 (s, 1H), 6.96 – 6.93 (m, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.67 (d, J = 15.8 Hz, 1H), 4.61 (d, J = 3.9 Hz, 2H), 3.88 (s, 3H), 2.52 (s, 3H),2.51 (s, 3H), 2.50 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 166.6, 158.3, 149.7, 148.0, 145.2, 136.5, 130.9, 128.9, 124.0, 121.5, 120.7, 111.2, 55.5, 41.4, 21.5, 20.2. HRMS (ESI) m/z: 312.17102 [M + H]+, calcd. for C18H21N3O2 312.17120.
(E)-3-(2,3-dimethoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L6): White solid, yield: 83.6%, m.p.: 182.7–183.4°C. 1H NMR (500 MHz, CDCl3): δ 7.94 (d, J = 15.9 Hz, 1H), 7.31 (s, 1H), 7.16 (d, J = 7.8 Hz, 1H), 7.06 – 7.03 (m, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.64 (d, J = 15.9 Hz, 1H), 4.61 (d, J = 2.7 Hz, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 2.51 (s, 3H), 2.50 (br, 6H); 13C NMR (125 MHz, CDCl3): δ 166.3, 153.3, 149.9, 148.4, 148.1, 148.0, 145.1, 135.8, 129.3, 124.3, 122.5, 119.5, 113.5, 61.4, 56.0, 41.5, 21.6, 21.6, 20.3. HRMS (ESI) m/z: 342.18155 [M + H]+, calcd. for C19H23N3O3 342.18177.
(E)-3-(3-methoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L7): White solid, yield: 91.3%, m.p.: 117.2–117.9°C. 1H NMR (500 MHz, CDCl3): δ 7.64 (d, J = 15.6 Hz, 1H), 7.32 (s, 1H), 7.30 – 7.27 (m, 1H), 7.14 (d, J = 7.3 Hz, 1H), 7.06 (s, 1H), 6.90 (d, J = 7.9 Hz, 1H), 6.56 (d, J = 15.6 Hz, 1H), 4.60 (s, 2H), 3.83 (s, 3H), 2.53 (s, 3H), 2.50 (br, 6H); 13C NMR (125 MHz, CDCl3): δ 165.9, 159.9, 149.8, 148.0, 147.9, 144.9, 141.1, 136.3, 129.9, 121.0, 120.6, 115.5, 113.1, 55.4, 41.4, 21.5, 21.5, 20.2. HRMS (ESI) m/z: 312.17117 [M + H]+, calcd. for C18H21N3O2 312.17120.
(E)-3-(2,5-dimethoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L8): White solid, yield: 88.5%. HRMS (ESI) m/z: 342.18118 [M + H]+, calcd. for C19H23N3O3 342.18177.
(E)-3-(3,4,5-trimethoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L9): White solid, yield: 81.6%, m.p.: 154.7–155.4°C. 1H NMR (500 MHz, CDCl3): δ 7.60 (d, J = 15.5 Hz, 1H), 7.29 (s, 1H), 6.77 (s, 2H), 6.48 (d, J = 15.5 Hz, 1H), 4.61 (s, 2H), 3.89 (s, 6H), 3.87 (s, 3H), 2.53 (s, 3H), 2.50 (br, 6H); 13C NMR (125 MHz, CDCl3): δ 165.9, 153.5, 149.9, 148.0, 147.9, 144.9, 141.2, 139.5, 130.5, 120.0, 105.1, 61.1, 56.3, 41.4, 21.6, 21.5, 20.2. HRMS (ESI) m/z: 372.19232 [M + H]+, calcd. for C20H25N3O4 372.19233.
(E)-3-(2,3,4-trimethoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L10): White solid, yield: 85.2%, m.p.: 139.8–140.5°C. 1H NMR (500 MHz, CDCl3): δ 7.81 (d, J = 15.8 Hz, 1H), 7.26 (d, J = 8.6 Hz, 1H),7.24 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.59 (d, J = 15.8 Hz, 1H), 4.61 (d, J = 1.8 Hz, 2H), 3.91 (s, 3H), 3.88 (s, 3H), 3.88 (s, 3H), 2.52 (s, 3H), 2.51 (br, 6H); 13C NMR (125 MHz, CDCl3): δ 166.6, 155.0, 153.2, 149.8, 148.0, 147.9, 145.2, 142.5, 136.1, 123.4, 122.0, 120.1, 107.6, 61.4, 61.0, 56.1, 41.4, 21.6, 21.5, 20.2. HRMS (ESI) m/z: 372.17950 [M + H]+, calcd. for C20H25N3O4 372.19233.
Preparation of L11–L15, L17–L21
Intermediate L (1.324 mmol) and the corresponding phenolic acids (1.322 mmol) were dissolved in anhydrous DMF (L11 CH2Cl2, L21 DMI) (20 mL), EDCI (253 mg, 1.324 mmol) and HOBt (59 mg, 0.44 mmol) were added. The mixture was stirred at room temperature under nitrogen atmosphere for 12 h. The reaction mixture was washed with water (2 × 20 mL) and brine (20 mL), dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was purified by colume chromatography and recrystallization from methanol to give L11–L15 and L17–L21.
2-(((3,5,6-trimethylpyrazin-2-yl)methyl)carbamoyl)phenyl acetate (L11): White solid, yield: 68.9%. HRMS (ESI) m/z: 336.13163 [M + Na]+, calcd. for C17H19N3O3 336.13241.
4-hydroxy-3-methoxy-N-((3,5,6-trimethylpyrazin-2-yl)methyl)benzamide (L12): White solid, yield: 81.8%, m.p.: 171.3–172.0°C. 1H NMR (500 MHz, CDCl3): δ 7.84 (s, 1H), 7.52 (d, J = 1.5 Hz, 1H), 7.35 (dd, J = 8.2, 1.5 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.40 (s, 1H), 3.93 (s, 3H), 2.52 (br, 6H), 2.51 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 167.0, 149.9, 149.0, 148.0, 148.0, 146.8, 145.2, 126.6, 120.0, 114.2, 110.6, 56.1, 41.5, 21.6, 21.5, 20.2. HRMS (ESI) m/z: 302.15002 [M + H]+, calcd. for C16H19N3O3 302.15047.
4-hydroxy-N-((3,5,6-trimethylpyrazin-2-yl)methyl)benzamide (L13): White solid, yield: 76.9%. HRMS (ESI) m/z: 272.13925 [M + H]+, calcd. for C15H17N3O2 272.13990.
3-hydroxy-N-((3,5,6-trimethylpyrazin-2-yl)methyl)benzamide (L14): White solid, yield: 78.4%, m.p.: 210.4–211.3°C. 1H NMR (500 MHz, DMSO-d 6): δ 9.64 (s, 1H), 8.76 (t, J = 5.0 Hz, 1H), 7.31 – 7.19 (m, 3H), 6.90 (dd, J = 7.8, 1.3 Hz, 1H), 4.51 (d, J = 5.2 Hz, 2H), 2.46 (s, 3H), 2.41 (br, 6H); 13C NMR (125 MHz, DMSO-d 6): δ 166.2, 157.3, 149.1, 147.9, 147.8, 147.4, 135.7, 129.3, 118.1, 117.8, 114.3, 42.1, 21.1, 21.1, 20.3. HRMS (ESI) m/z: 272.13913 [M + H]+, calcd. for C15H17N3O2 272.13990.
2-hydroxy-N-((3,5,6-trimethylpyrazin-2-yl)methyl)benzamide (L15): White solid, yield: 84.3%. HRMS (ESI) m/z: 272.13943 [M + H]+, calcd. for C15H17N3O2 272.13990.
(E)-3-(4-hydroxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L17): Little yellow solid, yield: 71.3%. HRMS (ESI) m/z: 296.14035 [M-H]−, calcd. for C17H19N3O2 296.13990.
(E)-3-(3-hydroxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L18): Little yellow solid, yield: 74.8%, m.p.:196.3–197.0°C. 1H NMR (500 MHz, DMSO-d 6): δ 9.60 (s, 1H), 8.52 (t, J = 5.0 Hz, 1H), 7.35 (d, J = 15.8 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.93 (s, 1H), 6.77 (d, J = 8.0 Hz, 1H), 6.65 (d, J = 15.8 Hz, 1H), 4.47 (d, J = 5.2 Hz, 2H), 2.46 (s, 3H), 2.43 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, DMSO-d 6): δ 164.8, 157.7, 149.4, 148.0, 147.8, 147.1, 139.2, 136.1, 129.9, 121.7, 118.8, 116.7, 113.7, 41.7, 21.1, 21.0, 20.2. HRMS (ESI) m/z: 296.14037 [M-H]−, calcd. for C17H19N3O2 296.13990.
(E)-3-(2-hydroxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L19): Little yellow solid, yield: 77.2%, m.p.: > 210°C. 1H NMR (500 MHz, DMSO-d 6): δ 10.03 (s, 1H), 8.47 (t, J = 5.0 Hz, 1H), 7.66 (d, J = 15.9 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.81 (t, J = 7.4 Hz, 1H), 6.74 (d, J = 15.9 Hz, 1H), 4.46 (d, J = 5.2 Hz, 2H), 2.46 (s, 3H), 2.43 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, DMSO-d 6): δ 165.4, 156.3, 149.4, 148.0, 147.8, 147.3, 134.8, 130.5, 128.1, 121.6, 121.3, 119.3, 116.1, 41.7, 21.1, 21.0, 20.3. HRMS (ESI) m/z: 296.14047 [M-H]−, calcd. for C17H19N3O2 296.13990.
(E)-3-(4-hydroxy-3-methoxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L20): Little yellow solid, yield: 80.1%. HRMS (ESI) m/z: 326.15090 [M-H]−, calcd. for C18H21N3O3 326.15047.
(E)-3-(3,4-dihydroxyphenyl)-N-((3,5,6-trimethylpyrazin-2-yl)methyl)acrylamide (L21): Little yellow solid, yield: 64.3%, m.p.: > 210°C. 1H NMR (500 MHz, DMSO-d 6): δ 9.43 (s, 1H), 9.20 (s, 1H), 8.42 (t, J = 4.9 Hz, 1H), 7.26 (d, J = 15.7 Hz, 1H), 6.96 (s, 1H), 6.83 (d, J = 7.9 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 6.43 (d, J = 15.7 Hz, 1H), 4.45 (d, J = 4.9 Hz, 2H), 2.45 (s, 3H), 2.42 (s, 3H), 2.41 (s, 3H); 13C NMR (125 MHz, DMSO-d 6): δ 165.3, 149.3, 147.9, 147.8, 147.3, 147.3, 145.5, 139.5, 126.3, 120.4, 118.2, 115.8, 113.9, 41.7, 21.0, 21.0, 20.2. HRMS (ESI) m/z: 336.13140 [M + Na]+, calcd. for C17H19N3O3 336.13241.
Preparation of L16
The desired compound 16b was gained according to the method described by Kojima and Tranchimand with minor modifications [29,30]. EDCI (0.253 g, 1.324 mmol) and HOBt (0.059 g, 0.44 mmol) were added to a stirred solution of 16b (0.442 g, 1.324 mmol) and the intermediate L (1.324 mmol) in anhydrous CH2Cl2 (20 mL). The mixture was stirred at room temperature for 12 h. The mixture was washed with water (2 × 20 mL) and brine (20 mL), dried over sodium sulfate, filtered, and concentrated under vacuum. Flash chromatography afforded 16c (0.566 g, 91.6%), white solid, m.p.: 127-128°C, HRMS (ESI) m/z: 490.21033 [M + Na]+, calcd. for C29H29N3O3 490.21066. To a solution of 16c (0.48 g, 1.028 mmol) in methanol (20 mL), was added Pd/C 10% (0.01 g). Then the suspension was stirred under hydrogen atmosphere at room temperature for 12 h. The mixture was filtered, washed with methanol (2 × 20 mL), and methanol evaporated under vacuum to afford L16.
3,4-dihydroxy-N-((3,5,6-trimethylpyrazin-2-yl)methyl)benzamide (L16): White solid, yield: 93.7%, m.p.: > 210°C. 1H NMR (500 MHz, DMSO-d 6): δ 9.45 (s, 1H), 9.13 (s, 1H), 8.52 (t, J = 5.0 Hz, 1H), 7.29 (s, 1H), 7.22 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 8.2 Hz, 1H), 4.48 (d, J = 5.0 Hz, 2H), 2.45 (s, 3H), 2.41 (s, 6H); 13C NMR (125 MHz, DMSO-d 6): δ 165.9, 149.0, 148.4, 147.8, 147.5, 144.8, 125.5, 119.0, 115.1, 114.8, 42.1, 21.0, 21.0, 20.2. HRMS (ESI) m/z: 288.12699 [M + H]+, calcd. for C15H17N3O3 288.13482.
Bio-evaluation methods
Protective effect on damaged differentiated PC12 cells
The PC12 cell line (pheochromocytoma) was purchased from Institute of Materia Medica of Chinese Academy of Medical Science. Cells were growed in RPMI 1640 medium supplemented with 5% (v/v) fetal bovine serum (FBS), 10% (v/v) heat inactivated horse serum and 100 U/mL penicillin-streptomycin (Thermo Technologies, USA) and incubated at 37°C in a humidified atmosphere of 5% CO2. When cells achieved the desired density of > 80% confluency original medium was removed and cells were maintained with serum-free media for 14 h. The cells were resuspended in new growth media which consisted of RPMI 1640 supplemented with 10% heat inactivated fetal bovine serum. Cells were plated on 96-well dishes pre-coated with poly-L-lysine at 7 × 103 cells/well, differentiated by treated with 50 ng/mL NGF for 48 h. After these, the differentiated PC12 cells were pretreated with various concentrations (60, 30, 15, 7.5, 3.75 μM) of ligustrazine derivatives for 36 h. Then the cells were induced by CoCl2 (final concentration, 200 mM) for 12 h. Control differentiated cells were injected with new growth media at equal amounts. After MTT solution (20 μL, 5 mg/mL) was added to each well, the plate was incubated for a further 4 h at 37°C. The supernatant was removed carefully without disturbing the attached cells and formazan crystals were solubilized by adding 100 μL DMSO into each well. After shaking for additional 15 min at 37°C, the plates were read for optical density at 490 nm (Thermo Multiskan GO, USA). CoCl2 was dissolved in RPMI 1640 medium. ligustrazine derivatives were dissolved in DMSO [21,31-33].
The proliferation rates of damaged PC12 cells were calculated in the following formula [OD490 (Compd) − OD490 (CoCl2)]/[OD490 (NGF) − OD490 (CoCl2)] × 100%; the EC50 values were using the equation below: −pEC50 = log Cmax − log 2 × (ΣP − 0.75 + 0.25Pmax + 0.25Pmin), Where Cmax = maximum concentration, ΣP = sum of proliferation rates, Pmax = maximum value of proliferation rate and Pmin = minimum value of proliferation rate [18-21,34].
References
Lindvall O, Kokaia Z. Stem cells for the treatment of neurological disorders. Nature. 2006;441:1094–6.
Yu L, Wang N, Zhang Y, Wang Y, Li J, Wu Q, et al. Neuroprotective effect of muscone on glutamate-induced apoptosis in PC12 cells via antioxidant and Ca2+ antagonism. Neurochem Int. 2014;70:10–21.
Feng X, Liang N, Zhu D, Gao Q, Peng L, Dong H, et al. Resveratrol inhibits β-amyloid-induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS One. 2013;8:e59888.
Tereshchenko J, Maddalena A, Bähr M, Kügler S. Pharmacologically controlled, discontinuous GDNF gene therapy restores motor function in a rat model of Parkinson’s disease. Neurobiol Dis. 2014;65:35–42.
Choi SS, Lee SR, Kim SU, Lee HJ. Alzheimer’s disease and stem cell therapy. Exp Neurobiol. 2014;23:45–52.
Ginsberg MD. Current status of neuroprotection for cerebral ischemia: synoptic overview. Stroke. 2009;40:S111–4.
Xian YF, Lin ZX, Mao QQ, Ip SP, Su ZR, Lai XP. Protective effect of isorhynchophylline against β-amyloid-induced neurotoxicity in PC12 cells. Cell Mol Neurobiol. 2012;32:353–60.
Li JW, Vederas JC. Drug discovery and natural products: end of era or an endless frontier? Science. 2009;325:161–5.
Harvey AL. Natural products in drug discovery. Drug Discov Today. 2008;13:894–901.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–35.
Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206–20.
Yu X. Present status and prospect using Ligusticum chuanxiong to prevent and treat stroke. Chin J Integr Med Cardio-/Cerebrovasc Dis. 2006;4:339–41.
Kim H. Neuroprotective herbs for stroke therapy in traditional eastern medicine. Neurol Res. 2005;27:287–301.
Kao TK, Ou YC, Kuo JS, Chen WY, Liao SL, Wu CW, et al. Neuroprotection by tetramethylpyrazine against ischemic brain injury in rats. Neurochem Int. 2006;48:166–76.
Zhang C, Wang SZ, Zuo PP, Cui X, Cai J. Protective effect of tetramethylpyrazine on learning and memory function in D-galactose-lesioned mice. Chin Med Sci J. 2004;19:180–4.
Lu C, Zhang J, Shi X, Miao S, Bi L, Zhang S, et al. Neuroprotective effects of tetramethylpyrazine against dopaminergic neuron injury in a rat model of Parkinson’s disease induced by MPTP. Int J Biol Sci. 2014;10:350–7.
Xu K, Wang PL, Xu X, Chu FH, Lin JX, Zhang YZ, et al. An overview on structural modifications of ligustrazine and biological evaluation of its synthetic derivatives. Res Chem Intermediat. 2013. doi:10.1007/s11164–013–1281–2.
Cheng XC, Liu XY, Xu WF, Guo XL, Zhang N, Song YN. Ligustrazine derivatives. Part 3: design, synthesis and evaluation of novel acylpiperazinyl derivatives as potential cerebrocardiac vascular agents. Bioorg Med Chem. 2009;17:3018–24.
Li Z, Yu F, Cui L, Zhan P, Wang S, Shen Y, et al. Ligustrazine derivatives. Part 6: design, synthesis and evaluation of novel ligustrazinyl acylguanidine derivatives as potential cardiovascular agents. Med Chem. 2012;8:928–33.
Chen H, Li G, Zhan P, Liu X. Ligustrazine derivatives. Part 5: design, synthesis and biological evaluation of novel ligustrazinyloxy-cinnamic acid derivatives as potent cardiovascular agents. Eur J Med Chem. 2011;46:5609–15.
Wang P, Zhang H, Chu F, Xu X, Lin J, Chen C, et al. Synthesis and protective effect of new ligustrazine-benzoic acid derivatives against CoCl2-induced neurotoxicity in differentiated PC12 cells. Molecules. 2013;18:13027–42.
Picone P, Nuzzo D, Di Carlo M. Ferulic acid: a natural antioxidant against oxidative stress induced by oligomeric A-beta on sea urchin embryo. Biol Bull. 2013;224:18–28.
Huang Y, Jin M, Pi R, Zhang J, Chen M, Ouyang Y, et al. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci Lett. 2013;535:146–51.
More SV, Kumar H, Kang SM, Song SY, Lee K, Choi DK. Advances in neuroprotective ingredients of medicinal herbs by using cellular and animal models of Parkinson’s disease. Evid Based Complement Alternat Med. 2013;2013:957875.
Li Z, Yu F, Cui L, Chen W, Wang S, Zhan P, et al. Ligustrazine derivatives. Part 8: design, synthesis, and preliminary biological evaluation of novel ligustrazinyl amides as cardiovascular agents. Med Chem. 2014;10:81–9.
Deng L, Guo X, Zhai L, Song Y, Chen H, Zhan P, et al. Ligustrazine derivatives. Part 4: design, synthesis, and biological evaluation of novel ligustrazine-based stilbene derivatives as potential cardiovascular agents. Chem Biol Drug Des. 2012;79:731–9.
Wang P, She G, Yang Y, Li Q, Zhang H, Liu J, et al. Synthesis and biological evaluation of new ligustrazine derivatives as anti-tumor agents. Molecules. 2012;17:4972–85.
Al-Qaisi JA, Alhussainy TM, Qinna NA, Matalka KZ, Al-Kaissi EN, Muhi-Eldeen ZA. Synthesis and pharmacological evaluation of a minoacetylenic isoindoline-1,3-dione derivatives as anti-inflammatory agents. Arab J Chem. 2011. doi:10.1016/j.arabjc.2010.12.030.
Tranchimand S, Tron T, Gaudin C, Iacazioa G. First chemical synthesis of three natural depsides involved in flavonol catabolism and related to quercetinase catalysis. Synth Commun. 2006;36:587–97.
Kojima T, Hirasa N, Noguchi D, Ishizuka T, Miyazaki S, Shiota Y, et al. Synthesis and characterization of ruthenium(II)-pyridylamine complexes with catechol pendants as metal binding sites. Inorgan Chem. 2010;49:3737–45.
Liu Z, Tao X, Zhang C, Lu Y, Wei D. Protective effects of hyperoside (quercetin-3-o-galactoside) to PC12 cells against cytotoxicity induced by hydrogen peroxide and tert-butyl hydroperoxide. Biomed Pharmacother. 2005;59:481–90.
Zhou J, Fu Y, Tang XC. Huperzine A and donepezil protect rat pheochromocytoma cells against oxygen-glucose deprivation. Neurosci Lett. 2001;306:53–6.
Hu J, Zhao TZ, Chu WH, Luo CX, Tang WH, Yi L, et al. Protective effects of 20-hydroxyecdysone on CoCl2-induced cell injury in PC12 cells. J Cell Biochem. 2010;111:1512–21.
Liu X, Zhang R, Xu W, Li C, Zhao Q, Wang X. Synthesis of the novel liqustrazine derivatives and their protective effect on injured vascular endothelial cell damaged by hydrogen peroxide. Bioorg Med Chem Lett. 2003;13:2123–6.
Acknowledgements
We gratefully acknowledge support of this work by the National Natural Science Foundation of China (No. 81173519). We also acknowledge the Innovation Team Project Foundation of Beijing University of Chinese Medicine (Lead Compounds Discovering and Developing Innovation Team Project Foundation, No. 2011-CXTD-15).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HL, PW and GL designed the study; GL, XX, KX, FC, JS and SZ performed experiments; GL, BX, YG and HZ collected and analyzed data; GL conducted neuroimaging analyses with the help of YZ; GL and PW wrote the paper. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, G., Xu, X., Xu, K. et al. Ligustrazinyl amides: a novel class of ligustrazine-phenolic acid derivatives with neuroprotective effects. Chemistry Central Journal 9, 9 (2015). https://doi.org/10.1186/s13065-015-0084-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-015-0084-5