
Abstract
Background
Intraoperative hypotension is common in patients having non-cardiac surgery and is associated with serious complications and death. However, optimal intraoperative blood pressures for individual patients remain unknown. We therefore aim to test the hypothesis that personalized perioperative blood pressure management—based on preoperative automated blood pressure monitoring—reduces the incidence of a composite outcome of acute kidney injury, acute myocardial injury, non-fatal cardiac arrest, and death within 7 days after surgery compared to routine blood pressure management in high-risk patients having major abdominal surgery.
Methods
IMPROVE-multi is a multicenter randomized trial in 1272 high-risk patients having elective major abdominal surgery that we plan to conduct at 16 German university medical centers. Preoperative automated blood pressure monitoring using upper arm cuff oscillometry will be performed in all patients for one night to obtain the mean of the nighttime mean arterial pressures. Patients will then be randomized either to personalized blood pressure management or to routine blood pressure management. In patients assigned to personalized management, intraoperative mean arterial pressure will be maintained at least at the mean of the nighttime mean arterial pressures. In patients assigned to routine management, intraoperative blood pressure will be managed per routine. The primary outcome will be a composite of acute kidney injury, acute myocardial injury, non-fatal cardiac arrest, and death within 7 days after surgery.
Discussion
Our trial will determine whether personalized perioperative blood pressure management reduces the incidence of major postoperative complications and death within 7 days after surgery compared to routine blood pressure management in high-risk patients having major abdominal surgery.
Trial registration
ClinicalTrials.gov NCT05416944. Registered on June 14, 2022.
Similar content being viewed by others


Background
About 2% of patients having inpatient non-cardiac surgery die within the first month after surgery in Europe [1] and the USA [2]. If the first month after surgery would be considered a disease, it would be the third leading cause of death worldwide [3]. Postoperative deaths are a consequence of postoperative complications—including acute kidney injury and acute myocardial injury [2]. To improve postoperative outcomes, modifiable risk factors for complications should be identified and addressed. One of these modifiable risk factors may be intraoperative hypotension.
Intraoperative hypotension is common in patients having non-cardiac surgery and is associated with serious complications—such as postoperative acute kidney and myocardial injury [4,5,6]—and even death [6, 7]. The association between intraoperative hypotension and serious complications is supported by many observational database studies [8]—but the extent to which the relation is causal remains unclear.
Optimal intraoperative blood pressures for individual patients also remain unknown. On a population basis, intraoperative mean arterial pressures less than 60–70 mmHg are associated with organ injury [4, 5, 9], leading to the recommendation that intraoperative mean arterial pressures should generally be kept above 65 mmHg [10, 11].
Universally targeting higher intraoperative mean arterial pressures (e.g., 75 mmHg vs. 60 mmHg) does not reduce postoperative complications in patients having major non-cardiac surgery [12]. In contrast, the randomized INPRESS trial suggests that individualized blood pressure management targeting preoperative resting blood pressures reduces postoperative complications compared to routine blood pressure management [13]. However, in the INPRESS trial, individual blood pressure targets were defined based on single preoperative blood pressure measurements which do not adequately reflect individual blood pressure profiles [13,14,15,16]. Preoperative automated blood pressure monitoring is thought to be the optimal method to establish baseline values before surgery [17]. We thus propose preoperative automated blood pressure monitoring to define individual intraoperative blood pressure targets.
We aim to test the hypothesis that personalized perioperative blood pressure management—based on preoperative automated blood pressure monitoring—reduces the incidence of a composite outcome of acute kidney injury, acute myocardial injury, non-fatal cardiac arrest, and death within 7 days after surgery compared to routine blood pressure management in high-risk patients having major abdominal surgery.
Methods/design
Trial design
The proposed trial will be conducted in accordance with the ethical principles based on the Declaration of Helsinki [18] and the ICH E6 R2 guidelines for good clinical practice. The trial was approved by the ethics committee Hamburg (Ethikkommission der Ärztekammer Hamburg, Hamburg, Germany, registration number 2022–100879-BO-ff) acting as the primary ethics committee for this trial. Secondary ethics committee approvals will be obtained for each trial site before patient recruitment starts. The trial was registered at ClinicalTrials.gov (NCT05416944) on June 14, 2022. This article adheres to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) statement (Additional file 1) [19].
IMPROVE-multi will be a multicenter randomized controlled blinded (participating patients, outcome adjudicators, and data analysts) clinical superiority trial in 1272 high-risk patients having elective major abdominal surgery. We plan to enroll patients at approximately 16 German university medical centers. The trial coordinating center will be the University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Patients
We will include consenting patients ≥ 45 years scheduled for elective major abdominal surgery with general anesthesia that is expected to last ≥ 90 min. Patients must fulfill at least one of the following high-risk criteria: exercise tolerance < 4 metabolic equivalents as defined by the guidelines of the American College of Cardiology/American Heart Association; renal impairment (serum creatinine ≥ 1.3 mg/dL or estimated glomerular filtration rate < 90 mL/min/1.73 m2 within the last 6 months); coronary artery disease (any stage); chronic heart failure (New York Heart Association Functional Classification ≥ II); valvular heart disease (moderate or severe); history of stroke; peripheral arterial occlusive disease (any stage); chronic obstructive pulmonary disease (any stage) or pulmonary fibrosis (any stage); diabetes mellitus requiring oral hypoglycemic agent or insulin; immunodeficiency due to a disease (e.g., HIV, leukemia, multiple myeloma, solid organ cancer) or therapy (e.g., immunosuppressants, chemotherapy, radiation, steroids [above Cushing threshold]); liver cirrhosis (any Child–Pugh class); body mass index ≥ 30 kg/m2; current smoking or 15 pack-year history of smoking; age ≥ 65 years; expected anesthesia duration ≥ 180 min; B-type natriuretic peptide > 80 ng/L or N-terminal B-type natriuretic peptide > 200 ng/L within the last 6 months.
We will not include patients having emergency surgery, nephrectomy, and liver or kidney transplantation; patients who had kidney, liver, heart, or lung transplantation; patients having sepsis (according to current Sepsis-3 definition); pregnant women; and patients in whom preoperative automated blood pressure monitoring is not possible.
Protocol
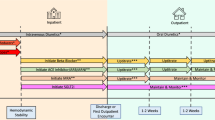
The trial flow and measures for each participating patient are provided in Fig. 1 and Table 1.

Flowchart illustrating patient screening, enrollment, randomization, trial intervention, and the primary outcome
Written informed consent will be obtained by dedicated trial personnel. Baseline demographic and medical characteristics will be recorded and entered in an electronic Case Report Form (eCRF). Preoperative automated blood pressure monitoring will be performed either at home or in the hospital for one night using upper arm cuff oscillometry at 30-min intervals. After excluding artifacts, we will calculate the mean of the nighttime mean arterial pressures. We will define nighttime as 00:00 to 06:00 h [20, 21].
Patients will be randomized in a 1:1 ratio using central block-wise randomization with variable block length stratified by centers with an electronic tool to personalized blood pressure management or to routine blood pressure management. The randomization will be performed directly before the induction of general anesthesia.
In patients assigned to personalized management, intraoperative mean arterial pressure will be maintained at least at the mean of the nighttime mean arterial pressures. If the mean of the nighttime mean arterial pressures will be lower than 65 mmHg, the intraoperative mean arterial pressure will be maintained at least at 65 mmHg; if the mean of the nighttime mean arterial pressures will be higher than 110 mmHg, the intraoperative mean arterial pressure will be maintained only at least at 110 mmHg. The intervention period will start at the induction of general anesthesia and will end 2 h after the end of surgery.
In patients assigned to routine management, intraoperative blood pressure will be managed per routine which usually includes keeping mean arterial pressure ≥ 65 mmHg [5, 10]. Anesthesiologists caring for patients assigned to routine management will not be informed of nighttime blood pressures.
In both groups, the interventions used to achieve the mean arterial pressure target will be at the discretion of treating anesthesiologists and will include intravenous fluid administration, vasoactive medications, adjusting anesthetic depth, and modifying patient position.
In all patients, perioperative blood pressure will be routinely monitored using intraarterial blood pressure monitoring (arterial catheter) or upper arm cuff oscillometry. All other anesthesiologic and surgical procedures will be performed according to routine care and the discretion of the attending anesthesiologists.
Outcomes
Primary outcome
The primary outcome will be a composite of acute kidney injury, acute myocardial injury, non-fatal cardiac arrest, and death within 7 days after surgery.
Secondary outcomes
Secondary outcomes will be as follows:
-
Incidence of the composite primary outcome within 3 days after surgery and the incidences of each of the individual components of the primary outcome within 3 and 7 days after surgery
-
Incidence of a composite outcome of need for renal replacement therapy, myocardial infarction, non-fatal cardiac arrest, and death within 30 and 90 days after surgery and the incidences of each of the individual components of this composite outcome within 30 and 90 days after surgery
-
Incidence of a composite outcome of infectious complications within 7 days after surgery and the incidences of each of the individual components of this composite outcome within 7 days after surgery
-
Time-to-event outcome with the event “transfer from intensive care unit to normal ward” within 90 days after surgery
-
Time-to-event outcome with the event “hospital discharge” within 90 days after surgery
-
Incidence of unplanned hospital re-admission within 30 days after surgery
Outcome definitions and measures
Acute kidney injury will be defined as an increase in serum creatinine of ≥ 50% from baseline or need for renal replacement therapy within the first 7 postoperative days [22].
Acute myocardial injury will be defined as an increase in high-sensitivity troponin concentration within the first 7 postoperative days according to the definition of “myocardial injury and infarction associated with non-cardiac procedures” set forth in the Fourth Universal Definition of Myocardial Infarction (2018) [23].
Serum creatinine and high-sensitivity troponin I or T (whichever is used in each center) will be measured before surgery (baseline; within the last 30 preoperative days) and on postoperative days 1, 2, 3, and 7. When patients are discharged earlier, serum creatinine and high-sensitivity troponin will be measured on the day of discharge. Additional serum creatinine and high-sensitivity troponin concentrations will be recorded during the initial 30 postoperative days on an as-available basis.
Non-fatal cardiac arrest will be defined as successful resuscitation from ventricular fibrillation, sustained ventricular tachycardia, asystole, or pulseless electrical activity requiring cardiopulmonary resuscitation, pharmacological therapy, or cardiac defibrillation.
Infectious complications will be defined according to the “Standardised Endpoints in Perioperative Medicine (StEP)” initiative [24] and include fever, respiratory infection, neurological infection, urinary system infection, colitis or infection with Clostridium difficile, endometritis, surgical site infection, deep incisional surgical site infection, organ or space surgical site infection, unknown infection with pathogenic organisms in tissue or fluid, and sepsis.
Data for the outcome assessment will be collected using medical records and telephone interviews on postoperative days 3, 7, 30, and 90.
Serious adverse events will be documented during the trial and will be reported to the primary ethics committee.
Statistical considerations and methods
Sample size
The sample size was estimated from the anticipated incidence of the composite primary outcome of at least one severe complication or death within 7 days after surgery. We expected an incidence of 25% in the routine management group (adapted from own data and literature [2, 25, 26]) and 17% in the personalized management group. The assumed absolute risk reduction of 8% is a conservative estimate based on a previous trial investigating the effect of individualized blood pressure management compared to routine blood pressure management on postoperative organ failure [13].
We will use a group sequential-design with one unblinded interim analysis using the O’Brien-Fleming spending function [27]. A total sample size of 1144, i.e., 572 patients per group will provide 90% power to detect a difference of 8% between the groups at a significance level of 0.05 using a two-sided test of proportions with continuity correction including an unblinded interim analysis after 50% of patients are recruited. Allowing a total drop-out rate of 10% (assuming an 8% drop-out rate before randomization and 2% after randomization), we plan to recruit 1272 patients. We assume that all patients will remain in their allocated treatment group throughout the observation period. PASS version 16.0.3 was used for the sample size calculation.
Statistical analyses
Descriptive analysis will be performed to describe the patient characteristics and clinical data. The primary and secondary outcomes will be analyzed according to the intention-to-treat principle. The primary outcome analyses (interim as well as final analysis) will be conducted using a two-sided test of proportions with continuity correction. When a total of 636 patients will have been observed for 7 days, an unblinded interim analysis will be performed. If the resulting p-value is < 0.0015 one-sided, the null hypothesis will be rejected, and the trial will be stopped for efficacy. Otherwise, recruitment will be continued. At the end of the trial, the statistical test for the primary outcome will be performed at a significance level of 0.049 two-sided.
In secondary outcome analyses, the incidence of each of the individual components of the primary outcome and of the additional secondary outcomes will be analyzed using a two-sided test of proportions with continuity correction. In additional analyses, the primary outcome and its individual components will be treated as time-to-event outcomes. Due to the competing event death, all time-to-event outcomes will be assessed using Aalen-Johansen estimators and accompanied by effect estimates based on cause-specific Cox regressions. In further analyses, the primary and secondary outcome analyses will be repeated in the per-protocol population. As a sensitivity analysis, the primary outcome will be analyzed using a mixed logistic regression model including a fixed effect for the random group and a random effect for the center to account for the cluster structure in the data. In further sensitivity analyses, regression models (logistic and cause-specific Cox regression) including potential prognostic baseline variables will be used.
If more than 5% of values are missing for the primary outcome, we will use multiple imputation in a sensitivity analysis. The number of imputations for multiple imputation will be chosen depending on the proportion of missing data according to White et al. [28].
A full statistical analysis plan will be developed before any data are evaluated.
Methods against bias
Screening logs will be collected to assess the risk for selection bias in patient recruitment. Randomization codes will be provided by an independent biostatistician to ensure blinding as long as practical. Participating patients, outcome adjudicators, and data analysts will be blinded to group assignment. Furthermore, a blinded outcome committee will be implemented to avoid detection bias. Anesthesiologists treating patients assigned to personalized management cannot be blinded as the results of preoperative automated blood pressure monitoring are necessary for perioperative personalized blood pressure management.
Data management and monitoring
Patient data will be documented by dedicated trial personnel and managed in an eCRF using a trial management software. Data will be entered via an encrypted connection (HTTPS) in web browser input masks. Each patient will be given an identification number to ensure pseudonymized data analysis. Each change in the data, e.g., due to resolved queries, will be documented by an automated audit trail. Pseudonymized data will be stored in accordance with local data protection laws.
Trial site monitoring will be conducted to ensure that the rights and well-being of patients are protected; that the reported trial data are accurate, complete, and verifiable; and that the conduct of the trial follows the currently approved protocol/amendment(s), with good clinical practice and with applicable regulatory requirements. The monitoring comprises the following tasks: training of the trial personnel prior to trial start (site initiation visit), regular on-site monitoring visits, and a close-out visit at the end of the trial. Central elements of the monitoring are the verification of consent and inclusion criteria, along with trial conduct.
Data Safety Monitoring Board
Three independent experts—including a biostatistician—will be the members of the Data Safety Monitoring Board (DSMB). DSMB members will meet before trial initiation, 3 months after the inclusion of the first patient, thereafter every 6 months, and after the interim analysis—and more often as they deem necessary or advisable. DSMB members will consider by-group results on a blinded basis. If DSMB members identify safety issues, they will have the prerogative to request unblinded outcomes. DSMB members will advise whether to continue, modify, or stop the trial. DSMB members will act independently from the sponsor and competing interests.
Dissemination plans
The results of this trial will be published in an international peer-reviewed medical journal. Both positive and negative results will be reported. The principal investigator is responsible for the preparation of the final report and therefore will have access to all available data. The full protocol, anonymized data, and the statistical code used for the analysis will be available from the corresponding author upon reasonable request. Authorships will be based on the recommendations of the International Committee of Medical Journal Editors (http://www.icmje.org/).
Discussion
In this trial, we will test whether personalized perioperative blood pressure management—based on preoperative automated blood pressure monitoring—reduces the incidence of a composite outcome of acute kidney injury, acute myocardial injury, non-fatal cardiac arrest, and death within 7 days after surgery compared to routine blood pressure management in high-risk patients having major abdominal surgery.
At some level, intraoperative hypotension surely causes organ injury—but the harm threshold for individual patients remains unclear. Perioperative blood pressure targets are thus a matter of current research. In a single-center trial including 451 high-risk patients having major non-cardiac surgery, universally targeting mean arterial pressure ≥ 75 mmHg—compared to ≥ 60 mmHg—did not reduce postoperative major adverse cardiovascular events [12]. Interestingly, the authors of this trial conclude that we should move away from population-based to individualized definitions of hypotension, “which could open the door to a paradigm of personalized intraoperative blood pressure targets” [12]. Using a population harm threshold of 65 mmHg or universally targeting higher pressures for all patients indeed ignores the fact that normal blood pressure varies considerably among individuals [15]. From a physiologic perspective, it is thus reasonable to assume that optimal intraoperative blood pressure depends on each individual patient’s normal blood pressure physiology.
There is only one randomized trial on individualized blood pressure management [13]. The INPRESS trial tested the hypothesis that targeting individual preoperative resting blood pressures reduces the incidence of postoperative systemic inflammation and organ dysfunction within 1 week after surgery compared to routine blood pressure management in 292 high-risk patients having major surgery [13]. Individualized blood pressure management reduced the risk of postoperative systemic inflammation and organ dysfunction compared to routine blood pressure management [13]. However, this small trial had limitations that limit its internal validity. First, this trial not only compared two different blood pressure management strategies but also used two different vasopressors (norepinephrine vs. ephedrine) in study and control group patients. Second, the individual intraoperative blood pressure target was defined based on a single preoperative blood pressure measurement taken in the preoperative evaluation clinic or on the surgical ward the day before surgery. However, single preoperative blood pressure measurements do not adequately reflect individual blood pressure profiles [14,15,16]. We, therefore, will perform preoperative automated blood pressure monitoring for one night to define individual intraoperative blood pressure targets.
Automated blood pressure monitoring is the clinical reference method to assess blood pressure profiles [29] and an international consensus group recently defined it as “the optimal method to establish baseline values” before surgery [17]. We will define nighttime as 00:00 to 06:00 h [20, 21] and thus exclude retiring and rising periods (evening 21:00 to 00:00 h; morning 06:00 to 09:00 h) which are subject to considerable variation [20, 21].
We will include high-risk patients having major abdominal surgery to ensure that the studied population is broad and representative of the patient population benefiting from hypotension avoidance [30]. Participating trial centers will be mainly large university medical centers with many high-risk patients having different types of major abdominal surgery procedures.
Our chosen outcomes are clinically relevant and are recommended by an expert consensus panel of the StEP initiative [31, 32]. Standardized and precisely defined outcomes ensure valid comparisons between different trials [33]. The primary composite outcome includes major perfusion-related complications within 7 days after surgery. Considering complications within the first 7 postoperative days minimizes the risk of complications being confounded by postoperative events rather than intraoperative blood pressure management. In addition, most patients are still hospitalized during the first 7 postoperative days, which allows a more accurate outcome assessment. We will also assess the incidence of a composite outcome of need for renal replacement therapy, myocardial infarction, non-fatal cardiac arrest, and death within 30 and 90 days after surgery.
In summary, we will perform a randomized trial to test the hypothesis that personalized perioperative blood pressure management—based on preoperative automated blood pressure monitoring—reduces the incidence of a composite outcome of acute kidney injury, acute myocardial injury, non-fatal cardiac arrest, and death within 7 days after surgery compared to routine blood pressure management in high-risk patients having major abdominal surgery.
Trials status
Patient recruitment for this trial is scheduled to start in November 2022 and is estimated to be completed in May 2024. This article is based on the most recent version of the study protocol (i.e., version 1.0, July 2022).
Availability of data and materials
Not applicable.
Change history
10 February 2023
Missing Open Access funding information has been added in the Funding Note.
Abbreviations
- DSMB:
-
Data Safety Monitoring Board
- eCRF:
-
electronic Case Report Form
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
- StEP:
-
Standardised Endpoints in Perioperative Medicine
References
Pearse RM, Moreno RP, Bauer P, Pelosi P, Metnitz P, Spies C, et al. Mortality after surgery in Europe: a 7 day cohort study. Lancet. 2012;380:1059–65.
Spence J, LeManach Y, Chan MTV, Wang CY, Sigamani A, Xavier D, et al. Association between complications and death within 30 days after noncardiac surgery. CMAJ. 2019;191:E830–7.
Nepogodiev D, Martin J, Biccard B, Makupe A, Bhangu A. Global burden of postoperative death. Lancet. 2019;393:401.
Walsh M, Devereaux PJ, Garg AX, Kurz A, Turan A, Rodseth RN, et al. Relationship between intraoperative mean arterial pressure and clinical outcomes after noncardiac surgery: toward an empirical definition of hypotension. Anesthesiology. 2013;119:507–15.
Salmasi V, Maheshwari K, Yang D, Mascha EJ, Singh A, Sessler DI, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126:47–65.
Gregory A, Stapelfeldt WH, Khanna AK, Smischney NJ, Boero IJ, Chen Q, et al. Intraoperative hypotension is associated with adverse clinical outcomes after noncardiac surgery. Anesth Analg. 2021;132:1654–65.
Mascha EJ, Yang D, Weiss S, Sessler DI. Intraoperative mean arterial pressure variability and 30-day mortality in patients having noncardiac surgery. Anesthesiology. 2015;123:79–91.
Wesselink EM, Kappen TH, Torn HM, Slooter AJC, van Klei WA. Intraoperative hypotension and the risk of postoperative adverse outcomes: a systematic review. Br J Anaesth. 2018;121:706–21.
Ahuja S, Mascha EJ, Yang D, Maheshwari K, Cohen B, Khanna AK, et al. Associations of intraoperative radial arterial systolic, diastolic, mean, and pulse pressures with myocardial and acute kidney injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2020;132:291–306.
Sessler DI, Bloomstone JA, Aronson S, Berry C, Gan TJ, Kellum JA, et al. Perioperative Quality Initiative consensus statement on intraoperative blood pressure, risk and outcomes for elective surgery. Br J Anaesth. 2019;122:563–74.
Saugel B, Sessler DI. Perioperative blood pressure management. Anesthesiology. 2021;134:250–61.
Wanner PM, Wulff DU, Djurdjevic M, Korte W, Schnider TW, Filipovic M. Targeting higher intraoperative blood pressures does not reduce adverse cardiovascular events following noncardiac surgery. J Am Coll Cardiol. 2021;78:1753–64.
Futier E, Lefrant JY, Guinot PG, Godet T, Lorne E, Cuvillon P, et al. Effect of individualized vs standard blood pressure management strategies on postoperative organ dysfunction among high-risk patients undergoing major surgery: a randomized clinical trial. JAMA. 2017;318:1346–57.
Kallioinen N, Hill A, Horswill MS, Ward HE, Watson MO. Sources of inaccuracy in the measurement of adult patients’ resting blood pressure in clinical settings: a systematic review. J Hypertens. 2017;35:421–41.
Saugel B, Reese PC, Sessler DI, Burfeindt C, Nicklas JY, Pinnschmidt HO, et al. Automated ambulatory blood pressure measurements and intraoperative hypotension in patients having noncardiac surgery with general anesthesia: a prospective observational study. Anesthesiology. 2019;131:74–83.
van Klei WA, van Waes JA, Pasma W, Kappen TH, van Wolfswinkel L, Peelen LM, et al. Relationship between preoperative evaluation blood pressure and preinduction blood pressure: a cohort study in patients undergoing general anesthesia. Anesth Analg. 2017;124:431–7.
Ackland GL, Brudney CS, Cecconi M, Ince C, Irwin MG, Lacey J, et al. Perioperative Quality Initiative consensus statement on the physiology of arterial blood pressure control in perioperative medicine. Br J Anaesth. 2019;122:542–51.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–4.
Chan A-W, Tetzlaff JM, Gøtzsche PC, Altman DG, Mann H, Berlin JA, et al. SPIRIT 2013 Explanation and Elaboration: guidance for protocols of clinical trials. BMJ. 2013;346:e7586.
Parati G, Stergiou G, O’Brien E, Asmar R, Beilin L, Bilo G, et al. European Society of Hypertension practice guidelines for ambulatory blood pressure monitoring. J Hypertens. 2014;32:1359–66.
O’Brien E, Parati G, Stergiou G, Asmar R, Beilin L, Bilo G, et al. European Society of Hypertension position paper on ambulatory blood pressure monitoring. J Hypertens. 2013;31:1731–68.
McIlroy DR, Bellomo R, Billings FT 4th, Karkouti K, Prowle JR, Shaw AD, et al. Systematic review and consensus definitions for the Standardised Endpoints in Perioperative Medicine (StEP) initiative: renal endpoints. Br J Anaesth. 2018;121:1013–24.
Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol. 2018;72:2231–64.
Barnes J, Hunter J, Harris S, Shankar-Hari M, Diouf E, Jammer I, et al. Systematic review and consensus definitions for the Standardised Endpoints in Perioperative Medicine (StEP) initiative: infection and sepsis. Br J Anaesth. 2019;122:500–8.
Gameiro J, Fonseca JA, Neves M, Jorge S, Lopes JA. Acute kidney injury in major abdominal surgery: incidence, risk factors, pathogenesis and outcomes. Ann Intensive Care. 2018;8:22.
Smilowitz NR, Redel-Traub G, Hausvater A, Armanious A, Nicholson J, Puelacher C, et al. Myocardial injury after noncardiac surgery: a systematic review and meta-analysis. Cardiol Rev. 2019;27:267–73.
O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35:549–56.
White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med. 2011;30:377–99.
Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll of Cardiol. 2018;71:e127-248.
Mathis MR, Naik BI, Freundlich RE, Shanks AM, Heung M, Kim M, et al. Preoperative risk and the association between hypotension and postoperative acute kidney injury. Anesthesiology. 2020;132:461–75.
Beattie WS, Lalu M, Bocock M, Feng S, Wijeysundera DN, Nagele P, et al. Systematic review and consensus definitions for the Standardized Endpoints in Perioperative Medicine (StEP) initiative: cardiovascular outcomes. Br J Anaesth. 2021;126:56–66.
McIlroy DR, Shaw AD, Myles PS. Standardized renal endpoints for perioperative clinical trials: the standardized endpoints in Perioperative Medicine Initiative. Nephron. 2017;137:302–5.
Boney O, Moonesinghe SR, Myles PS, Grocott MP. Standardizing endpoints in perioperative research. Can J Anaesth. 2016;63:159–68.
Acknowledgements
We acknowledge the tremendous contribution of the staff of the Center for Clinical Studies Jena (Zentrum für Klinische Studien Jena, ZKS).
Funding
This trial is an investigator-initiated trial that is funded by the German Research Foundation (“Deutsche Forschungsgemeinschaft,” Kennedyallee 40, 53175 Bonn, Germany; project number: 445158321). IMRPOVE-multi underwent a full external peer review as part of the funding process with the German Research Foundation. Open Access funding enabled and organized by Projekt DEAL. The German Research Foundation does not have a role in the collection, analysis, and interpretation of the data or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
AB, LK, KK, and BS: drafting of the manuscript. AZap, FMB, KK, and BS: development of the study design and study protocol. ASM, MF, EK, AZap, PM, KZ, AZar, and DIS: editing and critical review. BS: principal investigator. KK: coordinating investigator. AZap, FMB, PM, KZ, and AZar: members of the Executive Committee. LK, EV, and AZap: trial statisticians. DIS: scientific advisor of the trial. The authors critically read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics committee approval is obtained for each site prior to patient documentation. All participating patients need to provide written informed consent (obtained by dedicated trial personnel) before trial inclusion.
Consent for publication
Not applicable.
Competing interests
AB, ASM, LK, EV, AZap, FMB, PM, and AZar declare that they have no competing interests. MF has received institutional restricted research grants and honoraria for giving lectures and consulting from CNSystems Medizintechnik GmbH (Graz, Austria). MF is a consultant for Edwards Lifesciences Inc. (Irvine, CA, USA). The Department of Anaesthesiology, Intensive Care Medicine & Pain Therapy of the University Hospital Frankfurt, Goethe University, received support from B. Braun Melsungen, CSL Behring, Fresenius Kabi, and Vifor Pharma for the implementation of Frankfurt’s Patient Blood Management program. KZ has received honoraria for participation in advisory board meetings for Haemonetics and Vifor and received speaker fees from CSL Behring, Masimo, Pharmacosmos, Boston Scientific, Salus, iSEP, Edwards, and GE Healthcare. He is the principal investigator of the EU-Horizon 2020 project ENVISION (intelligent plug-and-play digital tool for real-time surveillance of COVID-19 patients and smart decision-making in intensive care units) and Horizon Europe 2021 project COVend (biomarker and AI-supported FX06 therapy to prevent progression from mild and moderate to severe stages of COVID-19). DIS is a consultant for Edwards Lifesciences (Irvine, CA, USA) and has received research funding from the company. He has equity interests in Sensifree (Cupertino, CA, USA) and Perceptive Medical (Newport Beach, CA, USA). KK is a consultant for Edwards Lifesciences Inc. (Irvine, CA, USA) and for Vygon (Aachen, Germany). BS is a consultant for and has received institutional restricted research grants and honoraria for giving lectures from Edwards Lifesciences Inc. (Irvine, CA, USA). BS is a consultant for and has received institutional restricted research grants and honoraria for giving lectures from Pulsion Medical Systems SE (Feldkirchen, Germany). BS has received honoraria for giving lectures from Getinge AB (Gothenburg, Sweden). BS has received institutional restricted research grants and honoraria for giving lectures from CNSystems Medizintechnik GmbH (Graz, Austria). BS is a consultant for and has received honoraria for giving lectures from Philips Medizin Systeme Böblingen GmbH (Böblingen, Germany). BS is a consultant for and has received institutional restricted research grants and honoraria for giving lectures from GE Healthcare (Chicago, IL, USA). BS is a consultant for and has received honoraria for giving lectures from Vygon (Aachen, Germany). BS is a consultant for and has received honoraria for giving lectures from Baxter (Deerfield, IL, USA). BS is a consultant for and has received institutional-restricted research grants from Retia Medical LLC (Valhalla, NY, USA). BS has received institutional restricted research grants from Osypka Medical (Berlin, Germany). BS was a consultant for and has received institutional restricted research grants from Tensys Medical Inc. (San Diego, CA, USA).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
SPIRIT checklist.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bergholz, A., Meidert, A.S., Flick, M. et al. Effect of personalized perioperative blood pressure management on postoperative complications and mortality in high-risk patients having major abdominal surgery: protocol for a multicenter randomized trial (IMPROVE-multi). Trials 23, 946 (2022). https://doi.org/10.1186/s13063-022-06854-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-022-06854-0