
Abstract
Background
Incisional hernias are among the most frequent complications following abdominal surgery and cause substantial morbidity, impaired health-related quality of life and costs. Despite improvements in abdominal wall closure techniques, the risk for developing an incisional hernia is reported to be between 10 and 30% following midline laparotomies. There have been two recent innovations with promising results to reduce hernia risks, namely the small stitches technique and the placement of a prophylactic mesh. So far, these two techniques have not been evaluated in combination.
Methods
The HULC trial is a multicentre, randomized controlled, observer- and patient-blinded surgical effectiveness trial with two parallel study groups. A total of 812 patients scheduled for elective abdominal surgery via a midline laparotomy will be randomized in 12 centres after informed consent. Patients will be randomly assigned to the control group receiving closure of the midline incision with a slowly absorbable monofilament suture in the small stitches technique or to the intervention group, who will receive a small stitches closure followed by augmentation with a light-weight polypropylene mesh in the onlay technique. The primary endpoint will be the occurrence of incisional hernias, as defined by the European Hernia Society, within 24 months after surgery. Further perioperative parameters, as well as patient-reported outcomes, will be analysed as secondary outcomes.
Discussion
The HULC trial will address the yet unanswered question of whether a combination of small stitched fascial closure and onlay mesh augmentation after elective midline laparotomies reduces the risk of incisional hernias. The HULC trial marks the logical and innovative next step in the development of a safe abdominal closure technique.
Trial registration
German Clinical Trials Register, DRKS00017517. Registered on 24th June 2019.
Similar content being viewed by others


Background
Incisional hernias (IHs) are among the most frequent complications following open abdominal surgery [1]. IHs cause substantial morbidity and costs and reduce health-related quality of life (HRQoL) [2]. In recent years several randomized controlled trials (RCTs) have been conducted comparing different techniques of abdominal wall closure. These trials have shown convincingly that abdominal wall closure with a continuous running suture is superior to interrupted suture techniques, at least in the elective setting [3,4,5,6]. Similarly, slowly absorbable suture material creates less IHs than rapidly absorbable sutures [3, 4, 7, 8]. These results were summarized in a meta-analysis [9]. Despite these advances, IH risks of 10–30% are regularly reported in RCTs following abdominal wall closure [3,4,5,6, 10] and increase to 36% in certain subgroups [11,12,13,14]. Furthermore, IH risks increase with extended time of follow-up [15].
Since the INLINE meta-analysis in 2010 [9], there have been two recent innovations in the field of abdominal wall closure aiming at a reduction of incisional hernias: the small stitches technique (SST) [10, 16] and prophylactic mesh placement [17, 18]. SST abdominal closure using a slowly absorbable suture and an increased suture-length to wound-length ratio of ≥4 significantly reduced IH in a pseudorandomized trial by Millbourn et al. [10]. A recent multicentre RCT has verified the superiority of this technique in terms of reduced IH frequency in comparison to standard abdominal wall closure [1]. However, even under these optimized conditions, 13% of patients developed an IH after 12 months [1].
Regarding prophylactic mesh placement, several RCTs have been performed in specific subsets of patients, with encouraging results [11, 19,20,21,22,23]. A meta-analysis confirmed a significantly lower risk of IHs in the mesh group [17]. Most importantly, other wound complications like surgical site infections (SSIs) were not increased in the mesh groups [17]. This is in line with the results of a multicentre RCT comparing prophylactic mesh placement to primary closure (PRIMA trial) that confirmed the superiority of prophylactic mesh placement [18, 24]. PRIMA compared onlay mesh augmentation (OMA) or sublay mesh augmentation (SMA) vs. primary suture closure of the abdominal incision in high-risk patients and identified significantly fewer patients with an IH in the OMA group (13%) than in the primary suture group (30%) after 2 years (odds ratio [OR] 0.37; 95% confidence interval [CI] 0.27 to 0.77; p = 0.0016). Comparing the SMA group with primary suture, the results just failed statistical significance (SMA vs. primary suture 18% vs. 30%; OR 0.55; 95% -CI 0.30 to 1.00; p = 0.05).
However, in none of these trials was prophylactic mesh placement combined with SST. A combination of these two techniques, which showed effectiveness as single interventions, is considered to be the logical consequence and may result in an additional reduction of IH formation.
Methods/design
Trial rationale
The objective of the HULC trial is to investigate whether prophylactic OMA in addition to abdominal wall closure in SST reduces the risk of IH formation in patients undergoing elective midline laparotomy compared to SST alone.
Trial design
HULC is a multicentre, randomized controlled, observer- and patient-blinded surgical effectiveness trial with two parallel study groups.
Patients and trial centres
To enrol the required number of patients in the planned recruitment period, 12 trial sites of the Clinical Trials Network of the German Surgical Society (CHIR-Net) will participate in this trial (www.chir-net.de). These 12 centres will be high-volume centres committing to include at least 50 patients each. To improve recruitment at all centres, brochures and flyers for patients including information about the trial will be available.
Patient inclusion criteria
All patients scheduled for elective clean or clean-contaminated [25] abdominal surgery as defined by the Centers for Disease Control and Prevention (CDC) via a midline laparotomy for any indication will be screened consecutively for eligibility and will be informed about the HULC trial. All subjects must be able to understand the nature and extent of the trial, and only adult patients (> 18 years of age) with a life expectancy of at least 2 years who provide written informed consent will be included in the trial. The participant informed consent form includes general information about the trial (indication, clinical data about incisional hernias) as well as details about the experimental and control intervention, randomization, benefits and risks of participation in the trial and trial visits. Furthermore, it includes general study information like the voluntariness of participation, possibility of termination of the trial, organization and financing of the trial, data protection and important contact details for further questions. As the intervention of the trial includes two well-established surgical standard therapies for abdominal wall closure, there is no increased risk of any harm to be expected through participation in the trial. Thus, no additional compensation for any postoperative complications or harm is planned. Patients will be insured against travel accidents for their follow-up visits.
Patient exclusion criteria
Patients with planned re-laparotomy via the midline incision within 2 years after trial intervention, midline laparotomy within the last 60 days prior to trial intervention or previous IHs or fascial dehiscences will be excluded from the trial. Moreover, patients with concurrent abdominal wall infections will not be included in the trial, in order to reduce the risk of SSI and potential mesh infections. Furthermore, patients with an American Society of Anesthesiologists (ASA) grade > 3 classification, pregnant or lactating women and patients who participate in another intervention trial with interference of the intervention and/or outcome of the HULC trial will be excluded.
Patient withdrawal criteria
Patients are free to stop their trial participation at any time and without giving reasons for their decision. When a trial participant withdraws his/her informed consent, he/she is asked to decide whether his/her data captured so far may be analysed or if it should be discarded. In addition, if, in the surgeon’s opinion at the end of the operation, the trial intervention will be detrimental to the subject’s well-being, the trial participation can be stopped for this patient. In this case, the patient will not be randomized, and the reason for screening failure must be recorded in the screening log. All randomized patients, including those with premature trial termination, will be included in the final analysis.
Control intervention
Patients in both groups will receive closure of the midline incision with a slowly absorbable monofilament suture (USP 2-0, PDS Plus, Ethicon, Somerville, NJ, USA) in SST as in previous trials [1, 10]. Tissue bites of 5 mm and intersuture spacing of 5 mm are applied exclusively to the fascia within the linea alba (omitting subcutaneous fat and muscle tissue). Suturing will be initiated at both ends of the median laparotomy towards the centre. An overlap of up to 2 cm may be created. Both sutures should be knotted independently. The suture-length to wound-length ratio (SL:WL) must be ≥4:1. The SL:WL ratio is recorded intraoperatively and is calculated as follows [16]: SL:WL = (A-(B + C)) / D (A = total length of suture used; B = length of suture remnants at starting knots; C = length of suture remnants at finishing knots; D = length of fascial incision (all in centimetres)).
Experimental intervention
In the experimental group, but not in the control group, the abdominal wall closure is augmented with a light-weight polypropylene mesh in onlay technique (OMA). To this end an anterior plane will be created between the anterior rectus fascia and the subcutis. The mesh should overlap the fascial midline incision by 3–4 cm on all sides [18, 24] and must be fixed to the fascia tension-free with USP 2-0 Prolene single knots (Ethicon, Somerville, NJ, USA). The mesh material will be standardized and an Optilene® Mesh (B. Braun, Melsungen, Germany) will be used. The interventional procedure will prolong the operation by approximately 20 min.
The materials and surgical technique will be standardized. HULC will use the same materials and surgical technique as the previous PRIMA and STITCH trials [1, 24] to ensure comparability of results and to avoid potential bias.
Closure technique of the skin and the subcutaneous tissue will be the same in both groups and will be standardized to reduce dead space and seroma formation. The subcutaneous tissue should be closed with monofilament or polyfilament absorbable sutures. No subcutaneous drains should be placed. The subcutaneous sutures in the experimental group will include the mesh in the midline in order to reduce seroma formation, as the latter was increased in previous OMA trials without subcutaneous sutures [18, 24] but not in OMA trials with subcutaneous sutures [19]. The skin will be closed using staples.
Assignment of intervention and randomization
In order to ensure equal distribution of patient characteristics, randomization will be used. Allocation of treatments will be performed using a web-based randomization tool (www.randomizer.at) by means of block-wise randomization. Randomization will be performed intraoperatively at the end of surgery, after closure of the fascia. This prevents potential bias by different intraoperative techniques. Before randomization, the surgeon needs to confirm a clean or clean-contaminated operation according to CDC definition [25]. Randomization will be stratified by centre and by IH risk (low-risk vs. high-risk patients, defined as patients with body mass index (BMI) ≥ 27 and/or those having surgery for abdominal aortic aneurysm). The surgeon who will perform the closing technique must be chosen before abdominal wall closure. As randomization is performed after closure of the facia in SST, surgeons call their respective clinical trial centre to have the online randomization performed and to receive the results by phone. Alternatively, the online randomization can be performed in the operating room by a third person, if the necessary equipment and internet access is available.
Blinding
Patients, observers and data analysts will be blinded to the intervention in order to guarantee unbiased assessment of the primary outcome. The person performing randomization and the surgical team conducting the control/experimental intervention (“unblinded” study members) will be documented and will not be part of further outcome assessment. Moreover, neither the operation report nor the discharge letter will contain information regarding group allocation. In any case of an emergency including possible re-operation or a clinical situation that necessitates the knowledge of the trial group of the participant, patients can be unblinded.
Other methods against bias
To minimize performance bias, the intervention will be standardized in both groups and the suture-length to wound-length ratio must be recorded intraoperatively and will be monitored. Furthermore, to minimize training effects, all participating surgeons must pass an obligatory eLearning tutorial demonstrating the SST before participation in the trial. Only surgeons having performed a minimum of 10 SST abdominal wall closures are allowed to perform interventions in the HULC trial. In addition, only centres committing to include at least 50 patients will participate in the trial.
Primary endpoint
The primary outcome measure of the trial will be the occurrence of IHs within 24 months after surgery as defined by the European Hernia Society (EHS) [26]. Consequently, “any abdominal wall gap with or without a bulge in the area of a postoperative scar perceptible or palpable by clinical examination or imaging” is regarded as an IH. Occurrence of a burst abdomen will not be counted as a primary endpoint, but as a secondary endpoint by consensus [1, 3]. Follow-up time will be 24 months, as has been recommended by the EHS [26] since IH incidence increases over time [15]. Patients will be assessed for the primary endpoint at 6, 12 and 24 months after trial intervention. At these time points, patients will be examined by a clinician blinded for the trial intervention and by a radiologic examination performed by a blinded assessor. Radiologic exams allowed in the trial are sonography, computed tomography (CT) or magnetic resonance imaging (MRI) scans. In case of conflicting results between clinical and radiologic exams, the radiologic imaging is decisive to increase sensitivity [26]. If only one of the two examinations is performed (i.e. either clinical or imaging), the result of this assessment will be used for analysis. Possible results are listed in Table 1. As many patients included in this trial are expected to have an oncological indication for laparotomy and the included centres perform their oncological follow-up themselves, the loss to follow-up of patients is expected to be low. For patients who are unable or unwilling to attend the follow-up visits, a telephone follow-up is incorporated. The patient-reported outcome questionnaire developed by Jairam et al. [27] will be used, as it exhibits a high reliability. It will be used as a screening tool; i.e. patients who are suspected to have an IH based on the questionnaire might be convinced to attend an outpatient visit, even if they were reluctant to do so before.
Primary estimand
In the recently released addendum to the ICH E9 guideline (draft version) [28], the estimands framework is recommended as a clear and transparent definition of “what needs to be estimated to address a specific scientific question of interest”. Such an estimand can be defined through the population of interest, variable of interest, specification of how intercurrent events are handled, and summary measure. The specification of how intercurrent events are handled is referred to here as intervention effect:
-
Population. The population is defined as all patients fulfilling all the inclusion and none of the exclusion criteria.
-
Variable. The variable is the occurrence of IHs as defined by the EHS within 24 months after intervention.
-
Intervention effect. Possible intercurrent events and the strategies to handle them are as follows: missing values due to death, drop-out, loss to follow-up and re-laparotomy will be replaced by using multiple imputation. Since re-laparotomy changes the probability of occurrence of an IH, information of occurrence or non-occurrence of IH after re-laparotomy will not be considered for primary analysis. This represents a hypothetical strategy for the post-randomization events re-laparotomy, drop-out, loss to follow-up and death. Except for these events, other post-randomization events will not be considered, thus reflecting a treatment policy approach, which means that the effect of randomized treatment is estimated irrespectively of other post-randomization events not captured in the primary endpoint definition.
-
Summary measure. The summary measure is the odds ratio (OR). The OR will be calculated by a two-level binary logistic regression analysis including the fixed factors treatment group and IH risk (low vs. high), the latter being deemed as the most important confounder by far and being also used for stratification in the randomization procedure, and the random factor centre. Confounding by other less important prognostic and predictive factors can assumed to be controlled by the randomized study design. The model will be fitted using a variance-components covariance matrix. The level of significance is set to 5% (two-sided). The p value for judging the primary hypothesis will result from the two-level binary logistic regression model, where the coefficient of the factor treatment effect is tested against zero using the Wald test.
Additionally, sensitivity and supplementary estimands will be considered, but are not described in further detail in this publication.
Key secondary endpoints
The secondary measurements chosen in the HULC trial have been proposed by international guidelines [26]. For an adequate evaluation of the secondary endpoints, follow-up visits on postoperative days 5 to 7, 10 to 14 and 30 to 35 will be performed (see Table 2) in addition to the follow-up visits for the primary endpoint described above (at 6, 12 and 24 months postoperatively). The key secondary endpoints are as follows:
-
1.
Risk of superficial and deep surgical site infections (SSIs) within 1 year in both groups [25]. SSIs will be assessed by clinical examination as defined by the CDC [25]. Organ-space SSIs are excluded in this measurement as they are independent of abdominal wall closure technique, but rather depend on the underlying surgery. Consequently, organ-space SSIs will be recorded in the overall complication rate and as serious adverse events (SAEs) if applicable. Follow-up for SSI is 1 year in line with CDC guidelines, as patients in the experimental group undergo implantation of alloplastic material (mesh).
-
2.
Postoperative 30-day morbidity. Complications will be recorded and classified according to the Dindo-Clavien classification [30].
-
3.
Occurrence of non-infectious wound complications (hematoma, seroma) within 30 days. Seroma is defined as a collection of serous fluid in a dead space, which can either be in situ or leaking through a wound. Hematoma is defined as an accumulation of blood in the wound area, which warrants (bedside) surgical exploration and intervention.
-
4.
Occurrence of postoperative burst abdomen within 30 days. Postoperative burst abdomen will be defined as missing continuity of the fascia in combination with wound dehiscence with consecutive re-operation.
-
5.
Postoperative wound pain at rest and during movement. Assessment will be performed using the well-established numeric pain rating scale. Pain is an important patient-reported outcome measure and is influenced by hernia occurrence and by surgery. Pain will be assessed at visits 3–5 (see Table 2), and the mean pain will be compared between the groups at these time points.
-
6.
HRQoL measured with the Short Form Health Survey (SF-36) and EuroQoL five dimensions (EQ-5D) questionnaires. As HRQoL is an important patient-reported outcome measure and is influenced by hernia occurrence and by surgery [2], it will be recorded both preoperatively (visit 1) and during follow-up (visits 6–8) (see Table 2). The median HRQoL will be compared at these time points between the groups and also in terms of the change from baseline.
-
7.
Length of primary hospital stay in days from index operation.
Patient timeline and trial visits
Patients scheduled for elective abdominal surgery via a midline incision are screened preoperatively at day 0 (visit 1). Patients are enrolled given their ability to understand the extent and nature of the trial and their provision of written informed consent after detailed patient information. All inclusion criteria and no exclusion criteria must be fulfilled. Baseline data are collected during the screening/baseline visit. The duration of visit 1 will be approximately 25 min. Included patients are randomized during surgery (visit 2) after closure of the fascia in SST. Follow-up visits will be on postoperative days 5 to 7, 10 to 14 and 30 to 35 (visits 3–5) for evaluation of secondary endpoints (time expenditure approximately 15 min). In addition, 6, 12 and 24 months (visits 6–8) after surgery, patients are planned for follow-up to evaluate primary and secondary outcome parameters. The expenditure of time for each visit will be approximately 30 min per patient. An overview of trial visits and items captured during the trial visits is presented in Table 2 according to the guidelines of the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) [29].
Data management
All protocol-required information collected during the trial must be entered by the investigator, or designated representative, in an electronic case report form (eCRF) implemented in the Research Electronic Data Capture (REDCap™) system [31] (www.project-redcap.org). An explanation should be given for all missing data. The completed eCRF must be reviewed and signed by the investigator named in the trial protocol or by a designated sub-investigator. The Institute of Medical Biometry and Informatics of the University of Heidelberg (IMBI) is responsible for data management within the trial. To assure a safe and secure environment for any data acquired, data transmission is encrypted with Secure Sockets Layer (SSL) technology. Only authorized users are able to enter or edit data, and access is restricted to patients’ data in the respective centre. All changes to data are logged with a computerized timestamp in an audit trail. All data will be pseudonymized. Completeness, validity and plausibility of data will be checked in time of data entry (edit checks) and with the use of validating programs, which will generate queries. If no further corrections are to be made in the database, eCRF data will be locked. All data management procedures will be conducted according to written defined standard operating procedures (SOPs) of the IMBI that guarantee an efficient conduct complying with good clinical practice (GCP).
Sample size calculation
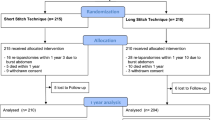
The sample size calculation is based on the primary efficacy endpoint (IH risk) within 24 months after surgery. Based on the assumption that the percentage of patients developing an IH after midline laparotomy in a general surgical population closed with the SST is approximately 15% for the control group, we hypothesize a reduction of 7% in the intervention arm based on previous RCTs [13, 20]. Consequently, a sample size per group of 325 patients is needed for the between-group comparison by the chi-squared test to achieve 80% power in detecting this difference in IH risk at a two-sided level of significance of 5%. It is assumed that using a two-level logistic regression model adjusting for the random factor centre and the fixed factor IH risk (low-risk vs. high-risk patients: BMI ≥ 27 and/or surgery for abdominal aortic aneurysm) [20] in the primary analysis will lead to less unexplained variance and thus to an increased power. Assuming a drop-out rate of up to 20% based on previous trials [1, 3, 10, 18], a total of 812 patients (406 per group) will be randomized in the study (Fig. 1). The potential occurrence of missing values for the primary outcome is partially addressed by the predefined multiple imputation strategy. Sample size calculation was performed using ADDPLAN v6.1.

Trial flow chart
Analysis variables and statistical methods
The primary efficacy analysis will be based on the full analysis set (FAS) built according to the intention-to-treat (ITT) principle, thus reflecting the recommendations given in guidelines [32]. As a sensitivity analysis, an evaluation based on the per-protocol (PP) population (based on those patients without major protocol violation and excluding patients who receive a fascial closure not predefined in the randomization scheme) will be performed. The risk of IH will be analysed via a two-level binary logistic regression model including the fixed factors treatment group and IH risk (high vs. low), the latter being deemed as the most important confounder by far and being also used for stratification in the randomization procedure, and the random factor centre. Confounding by other less important prognostic and predictive factors can assumed to be controlled by the randomized study design. The model will be fitted using a variance-components covariance matrix. The level of significance is set to 5% (two-sided). All secondary outcomes will be evaluated descriptively, and descriptive p values will be reported together with 95% CIs for the corresponding effects.
Further analyses
Interim analyses during the trial are not predefined or planned, but depending on the frequency of SAEs in both groups, safety analyses can be performed as recommended by the independent Data Safety Monitoring Board (DSMB). Further sensitivity analyses will be performed with the PP set, and the results will be compared with those of the ITT analysis. Moreover, for missing data in the ITT population set, further sensitivity analyses will be conducted by a worst-case scenario for the intervention, a minimal and a maximal IH risk imputation and by another alternative method of dealing with missing data as described by Higgins et al. [33]. Furthermore, a time-to-event analysis for the outcome “time from randomization to occurrence of IH” will be performed in the ITT population according to Kaplan-Meier. Additionally, prespecified subgroup analyses will be performed in the ITT population for the risk of incisional hernias in the subgroups of different types of surgery (colorectal, small bowel, hepatobiliary-pancreatic, upper GI (oesophageal and gastric), vascular, others), patients who are adipose vs. non-adipose and the presence or absence of neoadjuvant therapy, previous laparotomy or chronic obstructive pulmonary disease. All secondary outcomes will be evaluated descriptively, and descriptive p values will be reported together with 95% CIs for the corresponding effects.
Safety analysis
The assessment of safety will be based on the frequency of SAEs in both groups, which will be analysed via descriptive statistical methods in the study population. For comparisons of frequencies between groups, the chi-squared test will be used. All analyses will be done using SAS version 9.4 or higher.
Clinical data monitoring
Clinical monitoring will be performed by independent monitors of the Study Centre of the German Surgical Society (SDGC) according to its standard operating procedures in line with the ICH-GCP guideline (E6) [34]. A risk-based monitoring strategy will be conducted based on patient safety, patient rights, protocol adherence and data. The frequency of monitoring visits will be determined depending on recruitment numbers and individual performance of each centre based on feedback from project and data management.
Premature closure of the trial
The trial may be prematurely closed by the coordinating investigator in consultation with the Steering Committee including the responsible biometrician. If termination of the trial becomes necessary, the Steering Committee of the trial will discuss this issue with the independent DSMB. Similarly, the DSMB can recommend closing the trial based on the safety reports; however, the decision remains with the Steering Committee. Reasons that may necessitate termination of the trial include the incidence or severity of SAEs, morbidity or complications in this trial that indicate a potential health hazard caused by the study treatment. Furthermore, the trial should be terminated if it appears that patients’ enrolment is unsatisfactory with respect to quality and/or quantity or data recording is severely inaccurate and/or incomplete. Another case in which termination of the trial becomes necessary is if external evidence demands a termination of the trial.
Discussion
Despite the rise of laparoscopic surgery, open abdominal surgery by a midline laparotomy is still the most performed approach in abdominal surgery today [35]. Regardless of improvements in the surgical techniques of abdominal wall closure, IH incidence remains high and causes substantial morbidity and costs [24]. In the USA approximately 348,000 IH repairs leading to more than US$3.2 billion in healthcare expenditure are performed annually [36]. Similar per capita numbers have been reported in Germany, where more than 51,000 IH repairs are performed each year, making it one of the most frequently performed operations [37]. Total costs for IH repair were estimated to be 6451 € per patient in France [38]. Thus, reducing the IH risk by 5% was calculated to result in an annual cost savings of 4 million € [38]. Furthermore, IH-related reduction of the HRQoL is an important patient-reported outcome, as has been shown in recent trials [2].
Primary prevention of IH is of utmost importance, since recurrence and re-recurrence risks reach 40% [39, 40] and a considerable decrease of HRQoL [41]. Thus, prevention of IHs would have a significant impact on the patient’s well-being and the whole healthcare system by reducing complications, avoiding additional interventions and increasing the HRQoL of affected patients.
Among earlier techniques to optimize the abdominal wall closure and therefore reduce the occurrence of IHs, the SST and OMA are the two most recent and promising ones. So far there has been no RCT combining these two techniques of abdominal wall closure. The HULC trial will be the first RCT to fill this gap of evidence and will combine the usage of SST and the prophylactic mesh augmentation. The strengths of the HULC trial will be its randomized and blinded study design and the adhesion to the most recent evidence of abdominal wall closure regarding the control and intervention techniques. The HULC trial will be a multicentre trial including 12 high-volume centres in abdominal surgery to minimize selection bias. All centres will be trained to standardize the surgical techniques and outcome assessment as much as possible, leading to an expected low performance bias. Furthermore, as recommended recently [42], the HULC trial will perform blinding as far as practicable by blinding patients and outcome assessors to reduce performance and detection bias. For the control and intervention group of the HULC trial, the most promising techniques of recent studies will be used, including a slowly absorbable suture material in continuous suture technique in SST in both groups [1]. The intervention group will receive an additional non-absorbable mesh in the onlay position, as absorbable meshes have failed to show a reduction in IH risk [43] in contrast to non-absorbable ones, and the technique of OMA has been identified as superior to SMA recently [24]. For safety reasons regarding the development of SSI after receiving the OMA treatment in the intervention group, the HULC trial will perform continuous follow-up visits including the evaluation of SSI during the first year after surgery according to the recommendations of the CDC [25].
As earlier trials [6, 9] have shown an increase of IH risk even after 12 months, and a minimum follow-up period of 24 months has been proposed by the EHS for future trials, the HULC trial will follow this recommendation. The trial will include a large sample size with an adequate drop-out rate measured by a properly conducted sample size calculation based on the reliable results of earlier trials. The HULC trial will enable an adequate risk-benefit assessment due to secondary endpoints including all relevant intervention-related adverse events. It will also include an extensive HRQoL assessment as a secondary outcome, as the reduction in quality of life through IHs has been shown to be of great importance for patients [2].
In summary, the results of the HULC trial will influence future guidelines and surgical practice concerning abdominal wall closure.
Trial status
This manuscript was written according to the most current version of the study protocol (version 1.1, last updated on June 25, 2019). Recruitment of patients for the HULC trial will start in August 2019. The clinical phase of the trial (last patient out) is expected to be completed in in August 2023.
Availability of data and materials
After completion of the trial, the data obtained by the trial will be summarized and analysed according to this protocol and hereafter published in a peer-reviewed journal to be accessible by any healthcare professional, participant or the public. An anonymized minimal data set underlying the results of the trial will be made available upon publication of the final results as a supplement in line with national and international data protection rules.
Abbreviations
- ASA:
-
American Society of Anesthesiologists
- CDC:
-
Centers for Disease Control and Prevention of Surgical Site Infections
- DFG:
-
German Research Foundation
- DSMB:
-
Data Safety Monitoring Board
- eCRF:
-
Electronic case report form
- EHS:
-
European Hernia Society
- FAS:
-
Full analysis set
- GCP:
-
Good clinical practice
- HRQoL:
-
Health-related quality of life
- IEC:
-
Independent ethics committee
- IH:
-
Incisional hernia
- IMBI:
-
Institute of Medical Biometry and Informatics of the University of Heidelberg
- ITT:
-
Intention-to-treat
- OMA:
-
Onlay mesh augmentation
- PP:
-
Per-protocol
- RCT:
-
Randomized controlled trial
- SAE:
-
Serious adverse event
- SDGC:
-
Study Centre of the German Surgical Society
- SL:
-
Suture length
- SOP:
-
Standard operating procedure
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
- SSI:
-
Surgical site infection
- SSL:
-
Secure Sockets Layer
- SST:
-
Small stitches technique
- WL:
-
Wound length
References
Deerenberg EB, Harlaar JJ, Steyerberg EW, Lont HE, van Doorn HC, Heisterkamp J, et al. Small bites versus large bites for closure of abdominal midline incisions (STITCH): a double-blind, multicentre, randomised controlled trial. Lancet. 2015;386(10000):1254–60.
van Ramshorst GH, Eker HH, Hop WC, Jeekel J, Lange JF. Impact of incisional hernia on health-related quality of life and body image: a prospective cohort study. Am J Surg. 2012;204(2):144–50.
Seiler CM, Bruckner T, Diener MK, Papyan A, Golcher H, Seidlmayer C, et al. Interrupted or continuous slowly absorbable sutures for closure of primary elective midline abdominal incisions: a multicenter randomized trial (INSECT: ISRCTN24023541). Ann Surg. 2009;249(4):576–82.
Trimbos JB, Smit IB, Holm JP, Hermans J. A randomized clinical trial comparing two methods of fascia closure following midline laparotomy. Arch Surg. 1992;127(10):1232–4.
Brolin RE. Prospective, randomized evaluation of midline fascial closure in gastric bariatric operations. Am J Surg. 1996;172(4):328–31.
Colombo M, Maggioni A, Parma G, Scalambrino S, Milani R. A randomized comparison of continuous versus interrupted mass closure of midline incisions in patients with gynecologic cancer. Obstet Gynecol. 1997;89(5 Pt 1):684–9.
Bresler L, Courbey PJ, Feldman L, Bilweiss J, Tortuyaux JM, Rauch P, et al. Results of a controlled trial comparing 3 suture threads at slow resorption for the closure of supra-umbilical midline laparotomies. Ann Chir. 1995;49(6):544–8.
Diener MK, Knebel P, Kieser M, Schuler P, Schiergens TS, Atanassov V, et al. Effectiveness of triclosan-coated PDS Plus versus uncoated PDS II sutures for prevention of surgical site infection after abdominal wall closure: the randomised controlled PROUD trial. Lancet. 2014;384(9938):142–52.
Diener MK, Voss S, Jensen K, Buchler MW, Seiler CM. Elective midline laparotomy closure: the INLINE systematic review and meta-analysis. Ann Surg. 2010;251(5):843–56.
Millbourn D, Cengiz Y, Israelsson LA. Effect of stitch length on wound complications after closure of midline incisions: a randomized controlled trial. Arch Surg. 2009;144(11):1056–9.
Bevis PM, Windhaber RA, Lear PA, Poskitt KR, Earnshaw JJ, Mitchell DC. Randomized clinical trial of mesh versus sutured wound closure after open abdominal aortic aneurysm surgery. Br J Surg. 2010;97(10):1497–502.
Sugerman HJ, Kellum JM Jr, Reines HD, DeMaria EJ, Newsome HH, Lowry JW. Greater risk of incisional hernia with morbidly obese than steroid-dependent patients and low recurrence with prefascial polypropylene mesh. Am J Surg. 1996;171(1):80–4.
Fassiadis N, Roidl M, Hennig M, South LM, Andrews SM. Randomized clinical trial of vertical or transverse laparotomy for abdominal aortic aneurysm repair. Br J Surg. 2005;92(10):1208–11.
Musella M, Milone F, Chello M, Angelini P, Jovino R. Magnetic resonance imaging and abdominal wall hernias in aortic surgery. J Am Coll Surg. 2001;193(4):392–5.
Fink C, Baumann P, Wente MN, Knebel P, Bruckner T, Ulrich A, et al. Incisional hernia rate 3 years after midline laparotomy. Br J Surg. 2014;101(2):51–4.
Israelsson LA, Millbourn D. Closing midline abdominal incisions. Langenbeck's Arch Surg. 2012;397(8):1201–7.
Timmermans L, de Goede B, Eker HH, van Kempen BJ, Jeekel J, Lange JF. Meta-analysis of primary mesh augmentation as prophylactic measure to prevent incisional hernia. Dig Surg. 2013;30(4–6):401–9.
Timmermans L, Eker HH, Steyerberg EW, Jairam A, de Jong D, Pierik EG, et al. Short-term results of a randomized controlled trial comparing primary suture with primary glued mesh augmentation to prevent incisional hernia. Ann Surg. 2015;261(2):276–81.
Gutierrez de la Pena C, Medina Achirica C, Dominguez-Adame E, Medina Diez J. Primary closure of laparotomies with high risk of incisional hernia using prosthetic material: analysis of usefulness. Hernia. 2003;7(3):134–6.
Strzelczyk JM, Szymanski D, Nowicki ME, Wilczynski W, Gaszynski T, Czupryniak L. Randomized clinical trial of postoperative hernia prophylaxis in open bariatric surgery. Br J Surg. 2006;93(11):1347–50.
El-Khadrawy OH, Moussa G, Mansour O, Hashish MS. Prophylactic prosthetic reinforcement of midline abdominal incisions in high-risk patients. Hernia. 2009;13(3):267–74.
Garcia-Urena MA, Lopez-Monclus J, Hernando LA, Montes DM. Valle de Lersundi AR, Pavon CC, et al. Randomized controlled trial of the use of a large-pore polypropylene mesh to prevent incisional hernia in colorectal surgery. Ann Surg. 2015;261(5):876–81.
Muysoms FE, Detry O, Vierendeels T, Huyghe M, Miserez M, Ruppert M, et al. Prevention of incisional hernias by prophylactic mesh-augmented reinforcement of midline laparotomies for abdominal aortic aneurysm treatment: a randomized controlled trial. Ann Surg. 2016;263(4):638–45.
Jairam AP, Timmermans L, Eker HH, Pierik R, van Klaveren D, Steyerberg EW, et al. Prevention of incisional hernia with prophylactic onlay and sublay mesh reinforcement versus primary suture only in midline laparotomies (PRIMA): 2-year follow-up of a multicentre, double-blind, randomised controlled trial. Lancet. 2017;390(10094):567–76.
Mangram AJ, Horan TC, Pearson ML, Silver LC, Jarvis WR. Guideline for Prevention of Surgical Site Infection, 1999. Centers for Disease Control and Prevention (CDC) Hospital Infection Control Practices Advisory Committee. Am J Infect Control. 1999;27(2):97–132 quiz 3-4; discussion 96.
Muysoms FE, Antoniou SA, Bury K, Campanelli G, Conze J, Cuccurullo D, et al. European Hernia Society guidelines on the closure of abdominal wall incisions. Hernia. 2015;19(1):1–24.
Jairam AP, Wilson M, Steyerberg EW, Jeekel J, Lange JF. Patient reported outcome measurements in the diagnosis of incisional hernia: PROMIS questionnaire, a pilot study. J Surg Res. 2016;203(2):378–82.
Mehrotra DV, Hemmings RJ, Russek-Cohen E, Group IEREW. Seeking harmony: estimands and sensitivity analyses for confirmatory clinical trials. Clin Trials. 2016;13(4):456–8.
Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza-Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Dindo D, Demartines N, Clavien PA. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann Surg. 2004;240(2):205–13.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–81.
ICH Harmonised Tripartite Guideline. Statistical principles for clinical trials. International Conference on Harmonisation E9 Expert Working Group. Stat Med. 1999;18(15):1905–42.
Higgins JP, White IR, Wood AM. Imputation methods for missing outcome data in meta-analysis of clinical trials. Clin Trials. 2008;5(3):225–39.
International Council for Harmonisation of technical requirements for pharmaceuticals for human use (ICH): Integrated addendum to ICH E6 (R1): Guideline for Good Clinical Practice E6 (R2) 2016. https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf. Accessed 7 Dec 2019.
Patel SV, Paskar DD, Nelson RL, Vedula SS, Steele SR. Closure methods for laparotomy incisions for preventing incisional hernias and other wound complications. Cochrane Database Syst Rev. 2017;11:CD005661.
Poulose BK, Shelton J, Phillips S, Moore D, Nealon W, Penson D, et al. Epidemiology and cost of ventral hernia repair: making the case for hernia research. Hernia. 2012;16(2):179–83.
Statistisches Bundesamt. Fallpauschalenbezogene Krankenhausstatistik (DRG-Statistik) Operationen und Prozeduren der vollstationären Patientinnen und Patienten in Krankenhäusern: DeStatis; 2012. https://www.destatis.de/DE/Publikationen/Thematisch/Gesundheit/Krankenhaeuser/OperationenProzeduren.html
Gillion JF, Sanders D, Miserez M, Muysoms F. The economic burden of incisional ventral hernia repair: a multicentric cost analysis. Hernia. 2016;20(6):819–30.
Flum DR, Horvath K, Koepsell T. Have outcomes of incisional hernia repair improved with time? A population-based analysis. Ann Surg. 2003;237(1):129–35.
Luijendijk RW, Hop WC, van den Tol MP, de Lange DC, Braaksma MM, Ijzermans JN, et al. A comparison of suture repair with mesh repair for incisional hernia. N Engl J Med. 2000;343(6):392–8.
Juvany M, Hoyuela C, Carvajal F, Trias M, Martrat A, Ardid J. Long-term follow-up (at 5 years) of midline incisional hernia repairs using a primary closure and prosthetic onlay technique: recurrence and quality of life. Hernia. 2018;22(2):319–24.
Probst P, Zaschke S, Heger P, Harnoss JC, Huttner FJ, Mihaljevic AL, et al. Evidence-based recommendations for blinding in surgical trials. Langenbeck's Arch Surg. 2019;404(3):273–84.
Muysoms FE, Jairam A, Lopez-Cano M, Smietanski M, Woeste G, Kyle-Leinhase I, et al. Prevention of incisional hernias with biological mesh: a systematic review of the literature. Front Surg. 2016;3:53.
World Medical Association. Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects 2013. http://www.wma.net/en/30publications/10policies/b3.
Schulz KF, Altman DG, Moher D, Group C. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med. 2010;152(11):726–32.
Acknowledgements
None.
Funding
The HULC trial is funded by the German Research Foundation (DFG) under project number 410901776.
Author information
Authors and Affiliations
Contributions
ALM, PH, MF, CDH, ST and MKD are responsible for the study design, the definition of endpoints and the preparation of the protocol. JK, MF and CK are the study’s statisticians and are responsible for the sample size calculation and statistical design of the study. MWB, as the head of the surgical department, provided general support from a clinical perspective and support concerning technical aspects. All authors revised the manuscript critically, and all authors read and approved the final manuscript. As all authors are planned to substantially contribute to the conduction, data collection and interpretation of the trial, they will all be considered for authorship of the final publication with the addition of the substantial contributors at the specific centres. The authorship criteria of the International Committee of Medical Journal Editors (ICMJE) will be applied. No professional writer will be used for the final publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The HULC trial is conducted according to the Medical Association’s professional code (Berufsordnung der Bundesärztekammer) §15. To ensure patient’s rights and safety, the responsible investigator will ensure that the trial will be conducted according to the ethical principles laid out in the declaration of Helsinki [44]. Before participation in the HULC trial, written informed consent will be obtained from all study participants. This protocol is designed to ensure that the trial will be conducted and analysed in accordance with ICH-GCP E6 [34]. The protocol has already been approved by the independent ethics committee (IEC) of the medical faculty of the University of Heidelberg, and secondary approval of the corresponding ethical bodies of all other participating centres has been or will be obtained. The trial protocol has been formulated in accordance with the recommendations of the Consolidated Standards of Reporting Trials (CONSORT) and SPIRIT guidelines [29, 45]. The SPIRIT checklist is provided as Additional file 1.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
SPIRIT 2013 checklist: recommended items to address in a clinical trial protocol and related documents.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Heger, P., Feißt, M., Krisam, J. et al. Hernia reduction following laparotomy using small stitch abdominal wall closure with and without mesh augmentation (the HULC trial): study protocol for a randomized controlled trial. Trials 20, 738 (2019). https://doi.org/10.1186/s13063-019-3921-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-019-3921-3