
Abstract
Background
Sciatica is a common condition reported to affect over 3% of the UK population at any time and is often caused by a prolapsed intervertebral disc (PID). Although the duration and severity of symptoms can vary, pain persisting beyond 6 weeks is unlikely to recover spontaneously and may require investigation and treatment. Currently, there is no specific care pathway for sciatica in the National Health Service (NHS), and no direct comparison exists between surgical microdiscectomy and transforaminal epidural steroid injection (TFESI). The NERVES (NErve Root block VErsus Surgery) trial aims to address this by comparing clinical and cost-effectiveness of surgical microdiscectomy and TFESI to treat sciatica secondary to a PID.
Methods/design
A total of 163 patients were recruited from NHS out-patient clinics across the UK and randomised to either microdiscectomy or TFESI. Adult patients (aged 16–65 years) with sciatic pain endured for between 6 weeks and 12 months are eligible if their symptoms have not been improved by at least one form of conservative (non-operative) treatment and they are willing to provide consent. Patients will be excluded if they present with neurological deficit or have had previous surgery at the same level. The primary outcome is patient-reported disability measured using the Oswestry Disability Questionnaire (ODQ) score at 18 weeks post randomisation and secondary outcomes include disability and pain scales using numerical pain ratings, modified Roland-Morris and Core Outcome Measures Index at 12-weekly intervals, and patient satisfaction at 54 weeks. Cost-effectiveness and quality of life (QOL) will be assessed using the EQ-5D-5 L and self-report cost data at 12-weekly intervals and Hospital Episode Statistics (HES) data. Adverse event data will be collected. Analysis will follow the principle of intention-to-treat.
Discussion
NERVES is the first trial to evaluate the comparative clinical and cost-effectiveness of microdiscectomy to local anaesthetic and steroid administered via TFESI. The results of this research may facilitate the development of an evidence-based treatment strategy for patients with sciatica.
Trial registration
ISRCTN, ID: ISRCTN04820368. Registered on 5 June 2014.
EudraCT EudraCT2014–002751-25. Registered on 8 October 2014.
Similar content being viewed by others
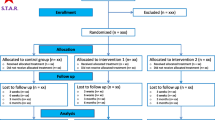


Background
Sciatica is broadly defined as leg pain in the distribution of a lumbosacral nerve root [1]. It is a common condition affecting over 3% of the population at any one time and over 90% of sciatica is due to a prolapsed intervertebral disc (PID) [2]. Patients affected are typically young, working adults and it can be helpful to consider three categories of sciatica: (1) acute sciatica – lasts less than 6 weeks and may be self-limiting with little or no impact on the patient’s ability to perform usual activities; (2) chronic sciatica – persists beyond 6 weeks and has a tremendous impact upon the patient’s working ability and (3) resistant sciatica – persists beyond 12 months [3]. Although the duration of pain may vary considerably, and the natural history of sciatica is favourable within 1 year, many patients have pain that persists beyond 6 weeks which could have considerable impact upon the employment market and patients’ lives [4]. It is generally accepted that pain persisting beyond 6 weeks is unlikely to get better imminently and requires further investigation and treatment [1,2,3,4]. There is no current accepted treatment paradigm for sciatica within the UK [1]. Treatment options are largely uproven but include analgesic drugs of various categories including antiepileptics and antidepressants, injections of drug combinations into the spine and surgical techniques to remove the prolapsed disc [1] Recent evidence has suggested that the commonly used neuromodulator drug pregabalin may not have a strong benefit in the treatment of sciatica in the community [5]. Epidural steroid injections (ESI) are another treatment modality for sciatica and involve the administration of a mixture of local anaesthetic and steroid into the spine via one of three main routes; through the base of the spine (caudal epidural), through the back of the spine (inter-laminar) or through the nerve tunnel directly adjacent to the prolapsed disc (transforaminal epidural steroid injection (TFESI) [6]. Randomised controlled trials (RCTs) have looked at ESI for acute sciatica but these have not included comparisons between TFESI and inter-laminar ESI [7,8,9,10]. However, prospective and case control studies have compared these and demonstrated a superior efficacy of TFESI [6,7,8,9]. One recent study of TFESI ([9]; n = 238) reported that 65% of injections were effective at follow-up greater than 6 months (based on patient-reported measures) suggesting that the administration of drug closer to the disc prolapse may improve efficacy when compared to other methods of administration. Moreover, efficacy is improved if symptom duration is less than 6 months prior to injection [9]. Only one trial [10]; n = 100) has directly compared inter-laminar ESI to surgery for sciatica secondary to PID and suggested that ESI could prevent 50% of surgical interventions. Although this specific use of steroid is outside the marketing authorisation (off-label) it is commonly used and a widely accepted treatment for sciatica. Of the surgical techniques, microdiscectomy to remove the prolapsed disc is considered the ‘gold standard’ with reported success rates of 90% [11]. As sciatica has a good natural history there is potential that the treatment administered in the form of injection may render surgery as excessive, but results from other studies have shown that ESI only have a small short-term effect on leg pain and disability compared with placebo, and no effect in the long term [12]. These poor medium- to long-term results have given ESI poor perceived efficacy and hence they are widely ignored in the treatment of acute sciatica [13]. Perhaps because of this at the time of trial conception no care pathway in the National Health Service (NHS) suggests any particular treatment over another [1]. No direct comparison exists between surgical microdiscectomy to treat sciatica secondary to lumber disc prolapse and nerve root blocks such as TFESI. In the UK in 2010/2011 over 25,000 therapeutic ESIs were administered and over 9000 surgical procedures were performed to remove herniated lumbar disc prolapses for sciatica (HES data). The costs to the NHS in the United Kingdom (UK) are £600 per ESI and approximately £4000 for surgical microdiscectomy (which requires an average of two nights in hospital per patient) [14].
The NERVES (NErve Root block VErsus Surgery) trial is funded to compare surgical microdiscectomy to local steroid and anaesthetic administered accurately to the source of leg pain in terms against various clinical and quality of life (QOL) outcomes to determine if there should be a recommended treatment pathway for patients with sciatica secondary to a PID. Surgical microdiscectomy and TFESI will be performed as per local NHS policy. Given the cost differential between the interventions being evaluated, and the potential for differences in clinical benefit and health outcomes, an economic evaluation will be conducted alongside the trial to determine which treatment option is the best use of health-care resources.The primary objective is to compare the clinical effectiveness of TFESI and surgical microdiscectomy for sciatica secondary to PID. Secondary objectives are to compare the cost-effectiveness of TFESI and microdiscectomy for the treatment of sciatica secondary to PID and to compare QOL outcomes for both treatments.
Methods/design
Study design and setting
NERVES is a two-arm, multi-centre, phase III, randomised trial comparing TFESI to surgical microdiscectomy for sciatic pain secondary to a PID. A Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) flowchart summarising the study protocol is presented in Fig. 1 (see Additional file 1 for the SPIRIT Checklist). Recruitment will occur in NHS out-patient neurosurgical, pain and orthopaedic clinics. Sites have been selected pragmatically, prior to opening a site suitability assessment including screening for the required patient volume was completed. Eligible patients who provided consent were randomised to TFESI and microdiscectomy in a ratio of 1:1 using an online computerised service. The schedule will be generated by an independent statistician, stratified by site, using permuted blocks of random sizes. Due to the nature of the procedures involved it was not possible to blind the participants.

Schematic of trial design
Patients will be followed up for 54 weeks from the date of randomisation with follow-up visits scheduled at 18 weeks, 30 weeks, 42 weeks and 54 weeks (12-week intervals assuming treatment at 6 weeks).
Patients who have additional treatment after receiving their randomised treatment or, who do not receive their randomised treatment and instead crossover and receive the other treatment will continue with the scheduled follow-up visits, they will not be withdrawn from the trial.
All participants will complete trial assessments as shown in Fig. 2.

Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) Figure. Trial assessments
Internal pilot
The original trial design includes an internal pilot phase allowing analysis of 6 months of recruitment data from two sites before progressing to full trial. The criteria for progression to full trial are:
-
At least 30 patients recruited
-
Consent rate of 40% or more
-
Fewer than 10% of patients unhappy with allocation and receive the alternative treatment
-
Fewer than 50% of patients in the injection group proceed to surgery
Study population
The trial is open to adult patients with sciatica secondary to a PID who meet the eligibility criteria listed in Table 1. Contraindications for both arms of treatment are to be assessed on a case-by-case basis by the healthcare team as per routine NHS practice and according to local policy. Written informed consent will be obtained for all patients before any study-specific assessments are conducted. Only patients who have suffered from leg pain for more than 6 weeks of duration and who have tried at least one non-invasive treatment form were selected.
Trial interventions
After baseline assessments have been completed and eligibility confirmed, patients were randomised to receive either an injection or surgery (1:1). Treatment was given within 6 weeks of randomisation where possible and should occur within 12 weeks of randomisation to ensure valid collection of primary outcome data at the 18-week follow-up. During the course of follow-up participants may require further intervention for acute sciatica as per routine NHS practice.
Transforaminal epidural steroid injection
TFESI is a standard nerve root blockade that will be performed as per local policy using the lateral foraminal portal of entry and guided fluoroscopically (i.e. computerised tomography (CT) or x-ray) to identify the correct level. As this is a pragmatic trial the agents used are expected to be obtained and prescribed via normal NHS routes. To minimise variability across the participating sites it is expected that the following injection regimen will be followed where possible: steroid: 20–60 mg triamcinolone, e.g. Kenalog; local anaesthetic: 0.25% levobupivacaine (2 ml), e.g. Chirocaine. Information on exact dosage and agents used, the level of injection and whether the block was preganglionic (at the level of the index disc) or postganglionic (the level below the disc) will be collected. All patients randomised to TFESI will receive at least one therapeutic injection, but may also be offered additional injections if there was partial or short-term benefit from the first injection. TFESI is an off-label use of steroid, but is commonly accepted practice within the NHS and in the wider medical field.
Surgical microdiscectomy
Standard microdiscectomy will be performed as per local treatment protocols. Sites will identify the correct side (left or right) and level prior to treatment with level localisation advised as per local treatment protocols. Information on site and spinal level of surgery will be collected.
Outcomes
Primary outcome
The primary outcome is the Oswestry Disability Questionnaire (ODQ) at 18 weeks after randomisation (approximately 12 weeks after intervention).
Secondary outcomes
The secondary outcomes include the following:
-
1.
ODQ score at 30, 42 and 54 weeks after randomisation
-
2.
Numerical rating scores for leg and back pain at baseline, and at 18, 30, 42 and 54 weeks after randomisation
-
3.
To assess patient treatment satisfaction at 54 weeks after randomisation
-
4.
Modified Roland-Morris outcome score for sciatica at baseline, and at 18, 30, 42 and 54 weeks after randomisation
-
5.
Core Outcomes Measures Index (COMI) at baseline, and at 18, 30, 42 and 54 weeks after randomisation
-
6.
Work status (return to work and work days lost if applicable) at 18 and 54 weeks after randomisation
-
7.
Cost-effectiveness, expressed as the incremental cost per quality-adjusted life-year (QALY) based on the EuroQol five-dimension, five-level quality of life questionnaire (EQ-5D-5 L) at baseline, and at 18, 30, 42 and 54 weeks after randomisation
Monitoring, safety and quality control
Data will be collected using paper Case Report Forms (CRFs) and patient-completed questionnaires during the 54-week follow-up period. Data capture will be monitored in accordance with the Clinical Trials Research Centre’s standard operating procedures to ensure compliance with the International Conference on Harmonisation, Good Clinical Practice and the Research Governance Framework 2005. Adverse events (AEs) are defined by the Medicines for Human Use (Clinical Trials) Regulations 2004 (SI 2004/1031). AE data will be collected throughout follow-up; the AE reporting period begins as soon as the study intervention is received and ends 30 days after the treatment visit. Serious adverse events (SAEs) and suspected unexpected serious adverse reactions (SUSARs) will be reported as per regulatory requirements. Safety and other relevant data will be reviewed throughout the trial by an Independent Data and Safety Monitoring Committee and further oversight is provided by a Trial Steering Committee.
Statistics
Statistical analysis
The ODQ is recommended as part of core outcome measures for low back research [15,16,17,18]. The scale ranges from 100 (extreme disability) to 0 (extreme ability) [16]. A change of 10 points has been widely accepted in the academic literature as the minimum clinical significance [17]. In order to detect a difference between two groups of 10 points on the ODQ at 5% significance level with 90% power, a total of 172 participants are required. This assumes a standard deviation (SD) of 20 points based on a similar population in previously published trials [13,14,15,16,17,18]. Baseline ODQ data collected on 11 potentially eligible patients from the ‘fast-track sciatica clinic’ at The Walton Centre generated an SD of 14.4, under the assumed value. We therefore initially aimed to recruit a target of 200 patients to allow for a 10% rate of missing outcome data. Randomisation was stratified by site. The primary outcome (ODQ score at 18 weeks post randomisation) will be compared between groups using a linear regression model, adjusted for the stratification variable centre, baseline ODQ score, and possibly other (specified in advance) variables considered to be potential confounders. Analysis of secondary outcomes will use similar methods, or logistic regression analyses, where appropriate. The intention-to-treat principle will be applied as far as is practically possible. The analysis set for the primary outcome will include all participants with an ODQ score at 18 weeks. Reasons for missing primary outcome data will be assessed, blind to treatment allocation, as to whether they are informative of likely outcome. Participants with non-informative reasons for missingness will be excluded from the primary analysis set. Sensitivity analyses will be carried out using multiple imputation to assess the robustness of the analysis to missing primary outcome data.
Revision of sample size calculation
Due to recruitment difficulties early in the trial, the sample size calculation was revisited. The original calculation did not assume any correlation between baseline and follow-up ODQ scores, as no data was available to estimate this. Based on a blinded analysis of the correlation between baseline and follow-up ODQ scores in the first 47 trial participants to have outcome data available, this correlation was estimated to be 0.49. Using this estimate, the revised sample size to achieve 90% power was found to be 66 per group. Allowing for 10% loss to follow-up gives a revised target of 74 per group (148 total). The trial was extended by the funder for a further 12 months and Steering Committees decided to continue recruitment until the end of the extension provided recruitment did not exceed 200 subjects. The trial has now stopped recruitment and 163 subjects were recruited.
Health economic analysis
The health economic analysis will adopt the perspective of the NHS and Personal Social Services (PSS) and additionally consider indirect costs such as time off work (secondary analysis).
Resource use associated with secondary care will be obtained from patient-level Hospital Episode Statistics (HES) data obtained from NHS Digital. Patients’ use of primary care services, personal social services, non-scheduled clinic attendance, out-of-pocket expenditures and indirect costs will be collected at baseline, treatment visit and at 18, 30, 42, 54 weeks post randomisation using a resource use questionnaire. Unit cost data will be obtained from standard sources (NHS reference costs and PSSRU Costs of Health and Social Care).
The health outcome measure will be the QALY, estimated by administering the EQ-5D-5 L at each follow-up point. The number of QALYs experienced by each patient will be calculated as the area under the curve, using the trapezoidal rule, applying the UK tariffs and corrected for baseline utility score.
Total costs will be combined with QALYs to calculate the incremental cost-utility ratio which will be compared with the £20,000 to £30,000 per QALY threshold of cost-effectiveness specified by the National Institute for Health and Care Excellence. Where appropriate, missing resource use or health outcome data will be imputed. A range of one-way sensitivity analyses will be conducted to assess the robustness of the analysis, and multivariate sensitivity analyses will be applied where interaction effects are suspected. The joint uncertainty in costs and benefits will be considered through the application of bootstrapping (10,000 replicates) and the estimation of cost-effectiveness acceptability curves. We will also employ simple parametric approaches for analysing cost and QALY data that assume normal distributions. Should the data indicate otherwise, we will develop a generalised linear model to deal with problems such as skewness.
Dissemination of results
Study findings will be presented in conference abstracts, poster presentations and scientific publications in medical journals. The chief investigator will work with the Trial Management Group and other principal investigators to generate manuscripts for publications.
Discussion
Surgical microdiscectomy for removal of PIDs has been shown to successfully relieve symptoms in the majority of patients [6, 11]. Disadvantages of surgery are the resource implications for the NHS due to the requirement for hospitalisation and the high skill level required of the treating physician. Surgery also carries the highest level of risk of all treatments for sciatica. Injections are relatively cheap and low risk in comparison to sciatica; they are delivered as a day-case procedure and the range of treatment providers is large, ranging from radiologists to surgeons or pain physicians. There is potential that treatment administered in the form of an injection may circumvent the need for surgery. However, the true success rate of spinal injections is largely unknown and there is no evidence comparing TFESI to surgical microdiscectomy that could be used to advise one treatment pathway over another. This protocol describes the design of a RCT to evaluate the clinical effectiveness and cost-effectiveness of TFESI to surgical microdiscectomy to treat sciatica secondary to a PID. It is the first randomised trial to address this issue to date.
Trial status
At the time of submission, the NERVES trial was closed to recruitment. Twelve participating sites had recruited 163 patients (first patient randomised on 6 March 2015). An internal pilot had been completed at two trial sites (The Walton Centre, Liverpool, and Salford Royal Hospital) as part of an initial feasibility study and followed the same study procedures as for the main trial. The decision to progress to full trial was based on the pre-defined internal pilot criteria for progression to a full trial. No between-group statistical comparisons were carried out after the internal pilot. Only the four criteria specified for progression to the full trial were to be considered after the internal pilot.
Abbreviations
- AE:
-
Adverse event
- COMI:
-
Core Outcomes Measures Index
- CRF:
-
Case Report Form
- CT:
-
Computerised tomography
- ESI:
-
Epidural steroid injection
- HES:
-
Hospital Episode Statistics
- MRI:
-
Magnetic resonance imagery
- NHS:
-
National Health Service
- NIHR:
-
National Institute of Health Research
- ODQ:
-
Oswestry Disability Questionnaire
- PID:
-
Prolapsed intervertebral disc
- QALY:
-
Quality-adjusted life-year
- QOL:
-
Quality of life
- SAE:
-
Serious adverse event
- TFESI:
-
Transforaminal epidural steroid injection
References
Low back pain and sciatica in over 16s: assessment and management NICE guideline [NG59] Published date: November 2016. www.nice.org.uk/guidance/ng59
Konstantinou K, Dunn KM. Sciatica: review of epidemiological studies and prevalence estimates. Spine (Phila Pa 1976). 2008;33(22):2464–72.
Van Tulder M, Peul W, Koes B. Sciatica: what the rheumatologist needs to know. Nat Rev Rheumatol 2010. 2010;6(3):139–45.
Peul WC, et al. Prolonged conservative care versus early surgery in patients with sciatica caused by lumbar disc herniation: two year results of a randomised controlled trial. BMJ. 2008;336(7657):1355–8.
Mathieson S, et al. Trial of pregabalin for acute and chronic sciatica. N Engl J Med. 2017;376:1111–20.
Cohen SP, et al. Epidural steroids: a comprehensive, evidence-based review. Reg Anesth Pain Med. 2013;38(3):175–200.
Lutz GE, Vad VB, Wisneski RJ. Fluoroscopic transforaminal lumbar epidural steroids: an outcome study. Arch Phys Med Rehabil. 1998;79(11):1362–6.
Rados I, et al. Efficacy of interlaminar vs transforaminal epidural steroid injection for the treatment of chronic unilateral radicular pain: prospective, randomized study. Pain Med. 2011;12(9):1316–21.
Jeong HS, et al. Effectiveness of transforaminal epidural steroid injection by using a preganglionic approach: a prospective randomized controlled study 1. Radiology. 2007;245(2):584–90.
Buttermann GR. Treatment of lumbar disc herniation: epidural steroid injection compared with discectomy. A prospective, randomized study. J Bone Joint Surg Am. 2004;86-a(4):670–9.
Peul WC, et al. Surgery versus prolonged conservative treatment for sciatica. N Engl J Med. 2007;356(22):2245–56.
Pinto RZ, et al. Epidural corticosteroid injections in the management of sciatica: a systematic review and meta-analysis. Ann Intern Med. 2012;157(12):865–77.
Price C, et al. Cost-effectiveness and safety of epidural steroids in the management of sciatica. Health Technol Assess. 2005;9(33):1–58. iii.
Commissioning Spinal Services – Getting the Service Back on Track; A Guide for Commissioners of Spinal Services NHS National Spinal Taskforce Report January 2013. www.nationalspinetaskforce.co.uk/
Deyo RA, et al. Outcome measures for low back pain research. A proposal for standardized use. Spine (Phila Pa 1976). 1998;23(18):2003–13.
Fairbank JC, et al. The Oswestry Low Back Pain Disability Questionnaire. Physiotherapy. 1980;66(8):271–3.
Davidson M, Keating JL. A comparison of five low back disability questionnaires: reliability and responsiveness. Phys Ther. 2002;82(1):8–24.
Devogelaer JP, et al. Guidelines for clinical studies assessing the efficacy of drugs for the management of acute low back pain. Clin Exp Rheumatol. 2003;21(6):691–4.
Acknowledgements
This study was developed with the specific support of the Clinical Trial Research Centre (CTRC), part of the Liverpool Trials Collaborative based at the University of Liverpool. The authors would like to thank all of the principal investigators, neurosurgeons, pain specialists, physiotherapists, research nurses and all other staff involved in the NERVES trial at hospital sites. The authors would also like to thank all members of the NERVES Trial Management Group. The trial is sponsored by The Walton Centre NHS Foundation Trust and supported by the NIHR Clinical Research Network: Neurological Disorders (https://www.nihr.ac.uk/nihr-in-your-area/neurological/).
Funding
The NERVES trial is funded by the National Institute for Health Research’s Health Technology Assessment Programme (project reference number: 12/201/10). The views expressed are those of the authors and not necessarily those of the NIHR. The funders played no role in the study design; collection, analysis and interpretation of data; writing of the report; or the decision to submit the article for publication. Researchers were independent of influence from study funders.
Author information
Authors and Affiliations
Contributions
MW is the chief investigator and has led all stages of study design with support from MS and SC. CH, EB, SH, GB, AM and PW participated in the writing of the protocol, the design of the CRFs and the preparation and submission of regulatory applications and amendments. DH designed the health economic analysis and data collection questionnaire. GB and BC are the statisticians for the study. CH prepared the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study is conducted in accordance with the principles of the Declaration of Helsinki, the Medicine for Human Use Regulations 2004 and Amendment Regulations 2006 and subsequent amendments, the Good Clinical Practice guidelines, and the Research Governance Framework for Health and Social Care 2005. Centralised full ethical approval was granted by the North West – Liverpool Central Research Ethics Committee (reference: 14/NW/1219) on 8 October 2014. Written informed consent will be obtained from all patients before recruitment by a trained researcher and patients are free to withdraw at any time. All data will be kept confidential and will be anonymised for analyses.
Consent for publication
The Patient Information Sheet and Consent Form (approved by the Research Ethics Committee) provides information that the results will be submitted for publication in a scientific journal and a final report written. Participants will not be identified in any reports or publications.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) Checklist. (DOC 127 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wilby, M.J., Hopkins, C., Bedson, E. et al. Nerve root block versus surgery (NERVES) for the treatment of radicular pain secondary to a prolapsed intervertebral disc herniation: study protocol for a multi-centre randomised controlled trial. Trials 19, 475 (2018). https://doi.org/10.1186/s13063-018-2677-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2677-5