
Abstract
Background
Cesarean delivery has already become a very common method of delivery around the world, especially in low-income countries. Hypertrophic scars and wound infections have affected younger mothers and frustrated obstetricians for a long time. Mesenchymal stem cells (MSCs) have strong potential for self-renewal and differentiation to multilineage cells. Previous studies have demonstrated that MSCs are involved in enhancing diabetic wound healing. Therefore, this study is designed to investigate the safety and efficacy of using MSCs in the treatment of Cesarean section skin scars.
Methods
This trial is a prospective, randomized, double-blind, placebo-controlled, single-center trial with three parallel groups. Ninety eligible participants will be randomly allocated to placebo, low-dose (transdermal hydrogel MSCs; 3 × 106 cells) or high-dose (transdermal hydrogel MSCs; 6 × 106 cells) groups at a 1:1:1 allocation ratio according to a randomization list, once a day for six consecutive days. Study duration will last for 6 months, comprising a 1 week run-in period and 24 weeks of follow-up. The primary aim of this trial is to compare the difference in Vancouver Scar Scale rating among the three groups at the 6th month. Adverse events, including severe and slight signs or symptoms, will be documented in case report forms. The study will be conducted at the Department of Obstetric of Southern Medical University Affiliated Maternal & Child Health Hospital of Foshan.
Discussion
This trial is the first investigation of the potential for therapeutic use of MSCs for the management of women’s skin scar after Cesarean delivery. The results will give us an effective therapeutic strategy to combat Cesarean section skin scars, even with uterine scarring.
Trial registration
ClinicalTrials.gov, NCT02772289. Registered on 10 May 2016.
Similar content being viewed by others

Background
Over the past decades, Cesarean delivery has already become a very common method of delivery around the world, especially in low-income countries [1]. In China, although the Cesarean rate has decreased in individual megacities, the overall annual rate has still increased, reaching 34.9% in 2014, from 28.8% in 2008 [2]. Hypertrophic scars and wound infections have affected younger mothers and frustrated obstetricians for a long time. As one of the top three hospital-acquired infections, wound infections can prolong hospitalization and greatly increase the rates of hospital readmission, risk of death, and overall costs of healthcare [3]. It is reported that the incidence of wound infections ranges from 3% to 20% in women after Cesarean delivery [4]. In addition, hypertrophic scars and psychological stress are causes of dissatisfaction in women after Cesarean delivery.
The cutaneous wound healing process is very complex. It requires a variety of cells to collaborate, such as resident cells of the skin, hematopoietic cells, and immune cells [5]. Many complex factors, such as abnormal macrophage polarization, abnormal keratinocyte and fibroblast migration, proliferation, differentiation and apoptosis, impaired recruitment of mesenchymal stem cells (MSCs) and endothelial progenitor cells, and decreased vascularization may contribute to an abnormal wound healing process [6,7,8]. It is also reported that enhanced and prolonged expression of tumor necrosis factor-alpha contributes to abnormal wound healing processes [9, 10].
Hypertrophic scarring is a fibrotic disease, arising from fibroproliferation disorder, which occurs after the damage of the deep dermis by deep skin injury, surgical procedure, or burns [11]. Overproduction of extracellular matrix and collagens are considered to be the main pathological characteristics of hypertrophic scars [12, 13]. Key cell subpopulations, including deep dermal fibroblasts, myofibroblasts, fibrocytes, and T-helper cells, could both modify and interact with the extracellular matrix of the wound, ultimately forming a hypertrophic scar [14]. Abnormal wound healing processes can also cause bacterial proliferation. In some instances, the bacterial load increases sufficiently for infection to ensue [15]. Therefore, an abnormal wound healing process could increase the risk of hypertrophic scarring and wound infection. In turn, continuous inflammation and bacterial proliferation also increase the risk of abnormal wound healing processes.
Post-Cesarean wound infection has been ascribed to many factors, including obstetrician-related factors, such as skin incision type, time of operation, suture technique, or intraoperative blood loss, and maternal factors, such as intrauterine infection prior to delivery, presence of comorbidities, or body mass index [16, 17]. Antibiotics, wound exploration, and debridement are mainstays in the medical care of post-Cesarean wound infections at present [18]. Many therapeutic approaches, such as laser therapy, radiation, cryotherapy, cryosurgery, or intralesional injections of corticosteroids, have also been reported in the management of hypertrophic scars [19]. However, many of them are involved in high rates of recurrence, and many are also expensive and painful.
Mesenchymal stem cells are a population of pluripotent stem cells. They possess high potencies of self-renewal and differentiation into canonical cells of the mesenchyme [20]. They are initially discovered in bone marrow and are subsequently found in almost every type of tissue, including the endometrium, placenta, umbilical cord, adipose tissue, and gingiva [21]. They have been reported as an effective and successful treatment for many diseases, including rheumatoid arthritis [22], experimental autoimmune encephalomyelitis [23], bone regeneration [24], and ischemic cardiomyopathy [25]. As a treatment modality, MSCs have demonstrated great potential value.
Through their migratory, anti-inflammatory, and trophic properties, MSCs exert numerous functions that may be of relevance for restoring skin tissue function and enhancing healing [26]. Using a rabbit model, researchers found that human MSCs can regulate inflammation and prevent the formation of hypertrophic scars [27]. Previous results of clinical trials also demonstrated the benefits derived by the employment of MSCs in wound healing [28, 29]. Falanga et al. [30] indicated that MSCs can be safely and effectively delivered to wounds using a fibrin spray system. Another clinical trial showed that directly applied bone marrow-derived cells can lead to dermal rebuilding and closure of nonhealing chronic wounds [31]. Yoshikawa et al. [32] demonstrated that MSCs are therapeutically effective in patients with intractable dermatopathies. In addition, Dash et al. [33] showed that autologous implantation of bone marrow-derived MSCs in nonhealing ulcers accelerated the healing process and significantly improved clinical parameters.
In this trial, we hypothesize that MSCs can reduce hypertrophic scars and decrease wound infection after Cesarean delivery. Therefore, we undertake a Phase II clinical trial to evaluate the safety and efficacy of MSCs in the treatment of Cesarean section skin scars in a prospective, randomized, double-blind, placebo-controlled, single-center study.
Methods
Study design
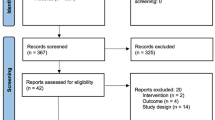
This study protocol conforms to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines (see Fig. 1 and Additional file 1). The trial is intended to target primiparous women between the 37th and 42nd weeks of gestation. Trained out-patient doctors will introduce the details of the trial to each potential participant during a clinic visit. The trial coordinator will contact the interested participant by mobile phone and WeChat, a very popular social networking app in China [34]. Eligible participants will be introduced in the trial. The inclusion and exclusion criteria and trial flow are shown in Table 1 and Fig. 2, respectively. The selection, information process, and randomization will be implemented as soon as we know that the primiparous woman is going to have a programmed Cesarean delivery. There is about 1 day between selection and delivery. The Cesarean delivery will be programmed for a gestational age ≥ 37 weeks and < 42 weeks. This trial will be a prospective, randomized, double-blind, placebo-controlled with three parallel groups. The trial will be conducted at the Department of Obstetric of Southern Medical University Affiliated Maternal & Child Health Hospital of Foshan.

SPIRIT figure

Trial schema
Interventions
Eligible participants, who have signed informed consent forms, will be randomized to placebo, low-dose (3 × 106 cells) or high-dose (6 × 106 cells) groups, receiving transdermal hydrogel MSCs or placebo once a day for six consecutive days. Each pump dispenses 1.0 ml gel (including 1 × 106 cells or none). The justification of doses is based on previous studies for other indications [35, 36] and our preliminary experiment (unpublished). Each pump, either MSCs or placebo, has the same external characteristics except for a digital tag, to allow blinding of the intervention. After suturing the skin incision as usual, the first intervention will take place on the operating table and the remaining five interventions will take place in the postnatal ward. Participants in the placebo group will receive placebo hydrogel once a day for 6 days; those in the low-dose group will receive one dose of hydrogel with cells once a day for three consecutive days and then placebo hydrogel for the next three consecutive days; those in the high-dose group will receive cells for 6 days.
The government has been committed to promoting cooperation between governments, academia, pharmaceutical companies, biotechnology firms, and private investors to research and evaluate MSC therapies, since they are advanced therapies [37, 38]. The Health-Biotech Pharmaceutical Company (Beijing, China) manufactures both MSCs and the placebo hydrogel. The products will be provided without charge. The MSCs are extracted from the umbilical cord, which is donated by a healthy donor who has provided informed consent. Detailed descriptions of the method have been given in a previous article [39].
The study duration will last for 6 months, including a 1 week run-in period and 6 months of follow-up. All participants will be assessed at 0.5, 1, 3, and 6 months during the follow-up period. Table 2 provides the time points and specific measurements of data. Cosmetic use may affect wound function. If participants use cosmetics in the follow-up period, they will be withdrawn from the study.
Outcomes
Primary outcome
The primary aim of this study is to compare the difference in Vancouver Scar Scale (VSS) rating among the three groups in the 6th month. The VSS rates vascularity (normal, pink, red, or purple), pigmentation (normal, hypopigmented, mixed, or hyperpigmented), height (flat, < 2 mm, 2–5 mm, or > 5 mm), and pliability (normal, supple, yielding, firm, ropes, or contracture). The Chinese version of the VSS has been shown to have good intraclass correlations and Cronbach’s α measures [40, 41]. All scars will be assessed independently by two observers (SW and SY) on the same day when the participants are lying in a supine position with the scar exposed in bright light. If the data varies, another researcher (DF) will be required to assess the scar at the same day and the results with the highest frequency will be recorded.
Secondary outcomes
The secondary outcome measures are as follows:
-
1)
The VSS at 1 and 3 months after treatment
The VSS will be also evaluated at 1 and 3 months after treatment. The differences in the 1st and 3rd month will also be compared among the three groups.
-
2)
Wound healing
Wound healing status will be assessed 14 days after surgery using the REEDA scale. The REEDA scale contains five variables: redness, edema, ecchymosis, discharge, and the approximation of wound edges [19].
-
3)
Erythema and pigmentation
These will be measured using a narrowband reflectance spectrophotometer (Mexameter MX18) at 1, 3, and 6 months after treatment.
-
4)
Scar thickness and area
The scar thickness and area will be measured using a high definition ultrasound device at 1, 3, and 6 months after treatment.
-
5)
Meanwhile, the mother’s milk will be collected at each visit and immunoglobulins (IgG, IgA, IgM) and complements (C3, C4) will be detected by the transmission immune turbidity method using an automatic biochemical analyzer.
-
6)
Participants’ satisfaction of the treatment will be measured using a satisfaction scale, including five categories, namely, none, slight, moderate, good, and very good.
-
7)
The number of hypertrophic scars and wound infections during the 6 months.
-
8)
Safety and tolerability of the intervention.
All adverse events, including side effects and other ailments, will be recorded in a case report form. The researcher will report all adverse events to the ethics committee. For all participants, additional services arising from all adverse events will be provided free of charge.
Sample size
Our sample size calculation is based on previous results from Professor Yu-Chen Huang’s preliminary trial in Taipei Medical University WanFang Hospital [42]. These results found that the mean VSS was 4.50 ± 1.68 in healthy pregnant women. The ratio of the MSC and control group sample size sets was 2:1. We will use the conventional values α = 0.05 and β = 0.1 for two-sided tests of probability. Meanwhile, we hypothesize that the difference in mean VSS at 6 months after treatment between the MSCs and placebo group will be δ = 1.5. The total sample size is estimated to be 74. Considering a dropout and other potential influencing factors, the final participant is estimated to be about 90 (n = 30 in each group).
Randomization, concealment, and blinding
A computerized random number generator will be used to produce a randomization schedule employing simple randomization by an independent clinical researcher, who is not involved in the recruitment, intervention, assessment, or statistical analysis. During the study period, the independent clinical researcher will retain the randomization list. The randomization sequences will be concealed in lightproof, sealed envelopes. After signing informed consent forms, eligible participants will be randomly allocated to the three parallel groups at a 1:1:1 allocation ratio, according to the randomization list.
Each participant will receive a unique randomized number. Meanwhile, each participant’s pump will be labeled with a unique randomization number, which will become the participant’s number. Direct participants and investigators, including the outcome assessors and statisticians, will be blinded to the allocation status throughout the study. Once participants have been allotted randomized numbers, a reasonable effort will be made by investigators to avoid missing data.
Data collection and management
All research investigators will be trained uniformly in standard operating procedures. A regular monitoring scheme will be set up to collect accurate and valid data. Non-numeric data will be converted into numbers for storage. All laboratory specimens will be identified only by a coded identification number to maintain participant confidentiality. To ensure confidentiality, data dispersed to project team members will be blinded to any identifying participant information. After verification of the content, two research investigators will independently input the data into a database. All participant information will be kept safely under confidential conditions and archived for 10 years. The project principal investigator could access the full data. Participant recruitment is currently in progress. The first participant was recruited on 14 September 2016.
Meanwhile, a data monitoring committee will be established, which will be independent of the study organizers and will periodically review the accumulating data and determine whether the trial should be modified or discontinued. The committee members will perform independent review of trial processes every 2 months.
Statistical analysis
All analyses will be conducted on the intention-to-treat and per-protocol principles. Regardless of whether participants received the randomized treatment, the intention-to-treat principle considers all of them as randomized. A complete case analysis will be performed if missing data for the randomized participants accounts for less than 5% of total data. Multiple imputations will be used if missing data is more than 5%. Dropouts will be included in the analysis by multiple imputations for missing data. The effect that the per-protocol participants and missing data might have on results will be assessed via sensitivity analysis. Subgroup analysis will be performed based on age and gestational age.
The primary comparison, both doses vs. placebo, will be analyzed using a superiority analysis. We will use the t test for the continuous outcome. Meanwhile, high dose vs. placebo and low dose vs. placebo will also be analyzed using the t test. Other outcomes will be analyzed using noninferiority analysis. The mean (and standard deviation) will be expressed for continuous variables, and numbers (percentages) will be expressed for categorical variables. Group variances will be compared using Leven’s test at the 0.05 significance level. Analysis of variance (ANOVA) will be used to analyze continuous data, and the chi-square test or Fisher’s exact test will be used for categorical data. To investigate the effects of treatment and time course, repeated measures ANOVA will be applied to determine changes in the continuous outcome data at each visit. R 3.1 software will be used for analysis.
Discussion
Mesenchymal stem cells have a strong potential for self-renewal and differentiation to multilineage cells. They can secrete several extracellular matrix molecules, growth factors, and cytokines that play a pivotal role in the regulation of angiogenesis and immune and inflammatory responses [19, 43]. Currently, several clinical trials have found potential value in using MSCs in healing chronic and acute wounds and scar remodeling [44].
To assess the efficacy and feasibility of autologous bone marrow-derived MSCs in the treatment of chronic nonhealing ulcers, researchers designed a randomized control study on a series of 24 participants with a history of nonhealing leg ulcer [33]. After 12 weeks, compared with control participants, the treatment participants had significant improvement in reduction in ulcer size. These results indicated that autologous implantation of bone marrow-derived MSCs in nonhealing ulcers accelerated the healing process. A single-arm clinical trial also indicated that autologous MSCs were shown to be therapeutically effective in patients with skin wounds [32]. To identify better cells for the treatment of diabetic foot ulcers, a randomized controlled trial was conducted on a sample of 41 type 2 diabetic patients with bilateral foot ulcer [45]. All patients were injected intramuscularly with bone marrow MSCs, bone marrow-derived mononuclear cells, or normal saline as placebo. They found that bone marrow mesenchymal stem cell therapy might be more effective than bone marrow-derived mononuclear cell therapy in promoting foot ulcer healing in diabetic patients. Another similar randomized controlled trial also obtained a similar result [46]. The frequency of major limb amputation was lower in a group treated with autologous bone marrow stem cells than in a group receiving standard medical care. Meanwhile, a meta-analysis demonstrated that autologous stem cell transplantation can be considered a safe and effective approach for treatment of many patients with diabetes mellitus [47].
Although previous results of trials with MSCs in wound healing have been reported, the application of MSCs on Cesarean section skin scars has not yet been investigated. In this prospective, randomized, double-blind, placebo-controlled study, we aim to investigate the possible effects of MSCs in women’s skin scars after Cesarean delivery. We hypothesize that MSCs can enhance wound healing, reduce hypertrophic skin scars, and decrease wound infection. To our knowledge, this trial is the first to investigate the potential of the therapeutic use of MSCs for the management of women’s skin scars after Cesarean delivery. The outcomes from this trial will help to determine the efficacy and safety of MSC treatment in Cesarean section skin scars. The results will also identify a therapeutically effective dose of MSCs in preventing hypertrophic scars and wound infections risk factors. It will give us an effective therapeutic strategy to combat Cesarean section skin scars, even with uterine scarring.
A limitation of this trial is that this is a single-center clinical trial, which will limit the extrapolation of results. Meanwhile, loss of participants at follow-up is possible, especially for nonresponders in the prospective trial study. Notwithstanding its limitations, this trial will suggest whether MSC can be a safe and effective in the treatment of Cesarean section skin scars.
Trial status
Participant recruitment is currently in progress. The first participant was recruited on 14 September 2016. We hope to complete enrolment for the trial by March 2018 with all 6 month follow-up data expected by September 2018.
Abbreviations
- ANOVA:
-
Analysis of variance
- MSCs:
-
Mesenchymal stem cells
- REEDA:
-
Redness, Edema, Ecchymosis, Discharge, and Approximation
- VSS:
-
Vancouver Scar Scale
References
Lumbiganon P, Laopaiboon M, Gulmezoglu AM, Souza JP, Taneepanichskul S, Ruyan P, et al. Method of delivery and pregnancy outcomes in Asia: The WHO global survey on maternal and perinatal health 2007–08. Lancet. 2010;375:490–9.
Li HT, Luo S, Trasande L, Hellerstein S, Kang C, Li JX, et al. Geographic variations and temporal trends in Cesarean delivery rates in China, 2008–2014. JAMA. 2017;317:69–76.
Stapleton H, Team DT. Wound healing in obese women following Caesarean section. Aust Nurs Midwifery J. 2015;23:34.
Owens SM, Brozanski BS, Meyn LA, Wiesenfeld HC. Antimicrobial prophylaxis for Cesarean delivery before skin incision. Obstet Gynecol. 2009;114:573–9.
Sun BK, Siprashvili Z, Khavari PA. Advances in skin grafting and treatment of cutaneous wounds. Science. 2014;346:941–5.
Fu X, Li H. Mesenchymal stem cells and skin wound repair and regeneration: Possibilities and questions. Cell Tissue Res. 2009;335:317–21.
Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5:e9539.
Xu F, Zhang C, Graves DT. Abnormal cell responses and role of TNF-α in impaired diabetic wound healing. Biomed Res Int. 2013;2013:754802.
Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N, et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-α dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia. 2010;53:378–88.
Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J Cell Sci. 2001;114:131–9.
Yang QQ, Yang SS, Tan JL, Luo GX, He WF, Wu J. Process of hypertrophic scar formation: Expression of eukaryotic initiation factor 6. Chin Med J (Engl). 2015;128:2787–91.
Xue M, Jackson CJ. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv Wound Care (New Rochelle). 2015;4:119–36.
Ray S, Ju X, Sun H, Finnerty CC, Herndon DN, Brasier AR. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J Invest Dermatol. 2013;133:1212–20.
Kwan PO, Tredget EE. Biological principles of scar and contracture. Hand Clin. 2017;33:277–92.
Withycombe C, Purdy KJ, Maddocks SE. Micro-management: Curbing chronic wound infection. Mol Oral Microbiol. 2017;32:263–74.
Connery SA, Downes KL, Young C. A retrospective study evaluating silver-impregnated dressings on Cesarean wound healing. Adv Skin Wound Care. 2012;25:414–9.
Zuarez-Easton S, Zafran N, Garmi G, Salim R. Postcesarean wound infection: Prevalence, impact, prevention, and management challenges. Int J Womens Health. 2017;9:81–8.
Fitzwater JL, Tita AT. Prevention and management of Cesarean wound infection. Obstet Gynecol Clin North Am. 2014;41:671–89.
Samadi S, Khadivzadeh T, Emami A, Moosavi NS, Tafaghodi M, Behnam HR. The effect of Hypericum perforatum on the wound healing and scar of Cesarean. J Altern Complement Med. 2010;16:113–7.
Chen X, Yang X, Wu R, Chen W, Xie H, Qian X, et al. Therapeutic effects of Wharton jelly-derived mesenchymal stem cells on rat abortion models. J Obstet Gynaecol Res. 2016;42(8):972–82.
Fan L, Hu C, Chen J, Cen P, Wang J, Li L. Interaction between mesenchymal stem cells and B-cells. Int J Mol Sci. 2016;17(5):650.
Hu J, Li H, Chi G, Yang Z, Zhao Y, Liu W, et al. IL-1RA gene-transfected bone marrow-derived mesenchymal stem cells in APA microcapsules could alleviate rheumatoid arthritis. Int J Clin Exp Med. 2015;8:706–13.
Glenn JD, Smith MD, Calabresi PA, Whartenby KA. Mesenchymal stem cells differentially modulate effector CD8+ T cell subsets and exacerbate experimental autoimmune encephalomyelitis. Stem Cells. 2014;32:2744–55.
Yoshioka M, Tanimoto K, Tanne Y, Sumi K, Awada T, Oki N, et al. Bone regeneration in artificial jaw cleft by use of carbonated hydroxyapatite particles and mesenchymal stem cells derived from iliac bone. Int J Dent. 2012;2012:352510.
Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY, et al. Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: The POSEIDON randomized trial. JAMA. 2012;308:2369–79.
Domergue S, Bony C, Maumus M, Toupet K, Frouin E, Rigau V, et al. Comparison between stromal vascular fraction and adipose mesenchymal stem cells in remodeling hypertrophic scars. PLoS One. 2016;11:e0156161.
Liu S, Jiang L, Li H, Shi H, Luo H, Zhang Y, et al. Mesenchymal stem cells prevent hypertrophic scar formation via inflammatory regulation when undergoing apoptosis. J Invest Dermatol. 2014;134:2648–57.
Gu C, Huang S, Gao D, Wu Y, Li J, Ma K, et al. Angiogenic effect of mesenchymal stem cells as a therapeutic target for enhancing diabetic wound healing. Int J Low Extrem Wounds. 2014;13:88–93.
Isakson M, de Blacam C, Whelan D, McArdle A, Clover AJ. Mesenchymal stem cells and cutaneous wound healing: Current evidence and future potential. Stem Cells Int. 2015;2015:831095.
Falanga V, Iwamoto S, Chartier M, Yufit T, Butmarc J, Kouttab N, et al. Autologous bone marrow-derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Eng. 2007;13:1299–312.
Badiavas EV, Falanga V. Treatment of chronic wounds with bone marrow-derived cells. Arch Dermatol. 2003;139:510–6.
Yoshikawa T, Mitsuno H, Nonaka I, Sen Y, Kawanishi K, Inada Y, et al. Wound therapy by marrow mesenchymal cell transplantation. Plast Reconstr Surg. 2008;121:860–77.
Dash NR, Dash SN, Routray P, Mohapatra S, Mohapatra PC. Targeting nonhealing ulcers of lower extremity in human through autologous bone marrow-derived mesenchymal stem cells. Rejuvenation Res. 2009;12:359–66.
Hou J, Ndasauka Y, Jiang Y, Ye Z, Wang Y, Yang L, et al. Excessive use of WeChat, social interaction and locus of control among college students in China. PLoS One. 2017;12:e0183633.
Liang J, Zhang H, Hua B, Wang H, Wang J, Han Z, et al. Allogeneic mesenchymal stem cells transplantation in treatment of multiple sclerosis. Mult Scler. 2009;15:644–6.
Liu M, Han ZC. Mesenchymal stem cells: Biology and clinical potential in type 1 diabetes therapy. J Cell Mol Med. 2008;12:1155–68.
The 13th five-year plan for health and health science and technology innovation. http://www.most.gov.cn/tztg/201706/t20170613_133484.htm.
Lu SB, Wu ZZ, Fu XB, Guo QY, Cheng J, Zhao SC, et al. Strategic research on the industrial development of innovative technology in cell technology-related regenerative medicine in China. Eng Sci. 2017;19:95–9.
Lu LL, Liu YJ, Yang SG, Zhao QJ, Wang X, Gong W, et al. Isolation and characterization of human umbilical cord mesenchymal stem cells with hematopoiesis-supportive function and other potentials. Haematologica. 2006;91:1017–26.
Li X, Li J, Ju X, Chen X, Wu X. Abdominal scar characteristics as a predictor of cervical stenosis after abdominal radical trachelectomy. Oncotarget. 2016;7(25):37755–61.
Liu HB, Tang D, Cao HY, Li KC. Reliability of Vancouver Scar Scale. Chin J Rehabili Med. 2006;21:240–2.
Chiang YY, Huang YC. Fractional CO2 Laser in the treatment for Cesarian scar. Taipei Medical University WanFang Hospital. 2012. https://clinicaltrials.gov/ct2/show/NCT01654406?term=%22caesarean+scar%22&rank=5.
Zhao Q, Ren H, Li X, Chen Z, Zhang X, Gong W, et al. Differentiation of human umbilical cord mesenchymal stromal cells into low immunogenic hepatocyte-like cells. Cytotherapy. 2009;11:414–26.
Zahorec P, Koller J, Danisovic L, Bohac M. Mesenchymal stem cells for chronic wounds therapy. Cell Tissue Bank. 2015;16:19–26.
Lu D, Chen B, Liang Z, Deng W, Jiang Y, Li S, et al. Comparison of bone marrow mesenchymal stem cells with bone marrow-derived mononuclear cells for treatment of diabetic critical limb ischemia and foot ulcer: A double-blind, randomized, controlled trial. Diabetes Res Clin Pract. 2011;92:26–36.
Prochazka V, Gumulec J, Jaluvka F, Salounova D, Jonszta T, Czerny D, et al. Cell therapy, a new standard in management of chronic critical limb ischemia and foot ulcer. Cell Transplant. 2010;19:1413–24.
El-Badawy A, El-Badri N. Clinical efficacy of stem cell therapy for diabetes mellitus: A meta-analysis. PLoS One. 2016;11:e0151938.
Acknowledgements
Not applicable.
Funding
Funding was provided by Health-Biotech Pharmaceutical Co., Ltd., Beijing, China (no. 201607001). The funding body has had no role in designing the trial, will not be involved in collection, management, analysis, interpretation of data, manuscript preparation and submission, or the final report and its publication, and does not have ultimate authority over any of these actions.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
ZL is the principal investigator of this trial. DF participated in the conception and design of the trial and wrote the first draft. QX and LL provided methodological guidance. SW, SY, WW, and XG reviewed the draft for clinical content. All the authors conceived this review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocol will be carried out in accordance with the Declaration of Helsinki, and has been approved by the ethics committee of the Southern Medical University Affiliated Maternal & Child Health Hospital of Foshan. It was registered with the Clinical Trial Registry (http://clinicaltrials.gov, registration number NCT02772289) on 10 May 2016. Before recruitment, the purpose of the trial, benefits, and risks will be described in detail to each participant. Moreover, all participants must sign an inform consent form before participation in the trial. They will be introduced to the main aspects of the study by trained research investigators. The information will be entered in a case report form.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests. The experimental product with MSCs and the placebo with hydrogel are provided without charge by Health-Biotech Pharmaceutical Co., Ltd., Beijing, China, which has had no role in designing the trial, and will not be involved in collection, management, analysis, interpretation of data, manuscript preparation and submission, or the final report and its publication, and does not have ultimate authority over any of these actions.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT checklist. (DOCX 43 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fan, D., Xia, Q., Wu, S. et al. Mesenchymal stem cells in the treatment of Cesarean section skin scars: study protocol for a randomized, controlled trial. Trials 19, 155 (2018). https://doi.org/10.1186/s13063-018-2478-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2478-x