
Abstract
Despite thousands of genetic loci identified to date, a large proportion of genetic variation predisposing to complex disease and traits remains unaccounted for. Advances in sequencing technology enable focused explorations on the contribution of low-frequency and rare variants to human traits. Here we review experimental approaches and current knowledge on the contribution of these genetic variants in complex disease and discuss challenges and opportunities for personalised medicine.
Similar content being viewed by others

Introduction
Genetic research has played an instrumental role in the discovery of new biological pathways underpinning complex human disease and the evaluation of new targets for therapeutic development. The past decade has seen an exponential increase in the number of known genetic loci predisposing to complex disease, enabled by large-scale meta-analyses based on genome-wide single-nucleotide polymorphism (SNP) arrays imputed into reference haplotype panels [1]. These efforts have identified thousands of (mostly common) genetic loci associated with disease biomarkers and disease endpoints [2], with some initial examples of how these genetic findings can be used to inform disease prediction [3], identification of causal mechanisms of disease [4, 5] and the prioritisation of new biological targets in drug discovery programmes [6,7,8].
Many challenges continue to exist in both the discovery and interpretation of findings from genome-wide association studies (GWASs). Highly successful international collaborative efforts have enabled association studies to reach unprecedented sizes of thousands to hundreds of thousands of study participants [9,10,11,12]. Despite the increases in statistical power afforded by these large-scale studies, for the majority of human traits genetic associations discovered account for a fraction of disease or trait heritability (the “missing heritability” paradigm). Genetic variants that are outside the reach of the most statistically powered association studies [13] are thought to contribute to the missing heritability of many human traits, including common variants (here denoted by minor allele frequency [MAF] >5%) of very weak effect, low-frequency (MAF 1–5%) and rare variants (MAF <1%) of small to modest effect, or a combination of both, with several possible scenarios all deemed plausible in simulation studies [14].
Empirical studies attempting to understand the impact of rare or less common variation on human complex diseases and traits remain to date relatively limited [15, 16], but some lessons on their properties are beginning to emerge from exome-wide and genome-wide sequencing studies. For most traits, these studies have demonstrated an inverse relationship between the variant’s “regression effect size” (or disease odds ratio) and its frequency in the population, as predicted by population genetic models [17]. Differential selective pressures acting on variants across the allele frequency spectrum underpin the observed shape of this relationship in different human traits. Such a relationship tends to be skewed in favour of rare variants for traits most strongly influenced by natural selection, compared with quantitative phenotypes or late-onset diseases [17]. Mendelian diseases are at the extreme end of the spectrum because of the high impact of selection on transmission of rare variants to subsequent generations. Initial evidence for complex diseases suggests that autism spectrum disorders may be skewed towards rarer susceptibility variants [18] compared with diseases such as type 2 diabetes [19], age-related macular degeneration [15] and schizophrenia [20], and quantitative cardiometabolic traits [21, 22]. Further efforts to discover associations driven by low-frequency and rare variants through genome sequencing and large-scale imputation efforts allow continuous refinements of the proportion of trait heritability explained by variants across the frequency spectrum [23]. Finally, it is worth noting that estimates of missing heritability from genome-wide variants are strongly dependent on assumptions on linkage disequilibrium, allele frequency and genotype certainty [13, 24]. Rare SNPs have been estimated to contribute substantial fractions of heritability (half the heritability of common SNPs [25]), but these early estimates will likely be revised as data continue to be accrued.
Another important challenge for complex disease genetics is the identification and functional characterisation of causal variants, or mutations in relevant genes, responsible for association signals detected through GWASs [26]. Common risk variants map overwhelmingly to regulatory regions [12], where inference of the underlying causative genes is difficult. Recent developments in cellular and functional genomics provide effective strategies to annotate the clinical and phenotypic consequences of genome sequence variation [27]. These approaches, which investigate a range of processes such as transcription, translation and epigenetic regulation at the organismal, physiological or cellular level [28], are a necessary step towards our understanding of the complex relationship between genotype and phenotype on a global (genome-wide) scale. Even in the presence of expansive datasets for annotation, however, the interpretation of the precise functional consequence of each variant requires rigorous and often painstaking evaluation of many genes in different possible cellular and environmental contexts [29]. On the other hand, rare variants in or near gene targets display larger average effects on phenotype compared with both regulatory variants of comparable allele frequencies and common genetic variants [21, 30]. The discovery of these variants through focused sequencing explorations of protein-coding regions is expected to greatly facilitate the task of annotating genes underpinning genetic associations with complex disease and describing the functional consequences of human sequence variation. There are, therefore, compelling arguments to accelerate efforts to identify variants within these regions because of the relative ease with which these discoveries can be turned into biological insights.
Here we review the current state of knowledge from rare variant association studies (RVASs) of complex traits and review approaches for discovering and testing associations for rare variants. Further, we discuss the growing body of literature documenting examples of highly clinically informative genetic variants identified through bespoke genotyping arrays, imputation and population-scale whole-exome and whole-genome sequencing.
Genomic tools for assessing low-frequency and rare variants
Three broad strategies are available to access low-frequency and rare variants: genotype imputation, the use of custom genotyping arrays and the use of whole-exome or whole-genome sequencing.
Imputation
Genotype imputation provides a cost-effective strategy for expanding the SNP content of genome-wide genotyping arrays. It relies on the availability of reference panels of phased haplotypes that can be used to impute genotypes into sparse datasets generated by commercial genotyping arrays [31, 32]. Multiple different reference panels have been generated since 2005, enabled by expanding collections of polymorphisms in human populations. The first two widely used reference panels generated by the HapMap project included 269 samples and just over one million SNPs (phase I) [33] and 3.1 million SNPs (phase II) [34], respectively. The ascertainment of these early panels was strongly skewed towards common variants (MAF >5%) found near human genes, thus limiting the representation of low-frequency and rare variants in early GWASs [35]. HapMap phase III included 1.6 million SNPs in 1184 individuals from 11 populations, ascertained by common SNP repositories and from targeted resequencing of ten 100-kb regions in 692 of these individuals. Compared with previous reference panels, the authors demonstrated gains in imputation accuracy particularly for low-frequency and rare variants [36].
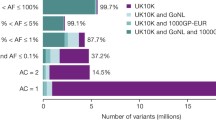
Further improvements in imputation panels were enabled by large-scale whole-genome sequencing (WGS) efforts in reference human populations, and particularly the 1000 Genomes Project (pilot, phase I and phase III). In the first phase of the project (phase I), a combination of low read depth WGS (2–4×) and targeted deep (50–100×) exome sequencing was used to characterise 38 million single-nucleotide variants (SNVs) and 1.4 million short insertion-deletions (INDELs) in 1092 individuals from 14 populations. The authors further showed that individuals from the various populations display different profiles of rare and common variants with considerable geographic differentiation [37]. The data set was expanded in phase III where the genomes of 2504 individuals from 26 populations were reconstructed by applying a combination of low-read-depth WGS, deep exome sequencing and dense microarray genotyping. This resulted in over 88 million variants which were phased onto high-quality haplotypes. The authors estimated that this resource includes >99% of SNVs with a frequency of >1% [38].
In addition to the 1000 Genomes Project, which comprises samples from all over the world, other panels based on WGS have been generated in individual populations. One of these efforts was the UK10K Cohorts Project, which carried out low-read-depth (approximately 7×) WGS in 3781 individuals of British ancestry from two population-based cohorts. Overall, the project identified over 42 million SNVs and 3.5 million INDELs, of which about 80% were rare and about 5% were low frequency, and in total 24 million were novel variants. The UK10K WGS imputation reference panel was shown to increase coverage and accuracy in European populations, especially for low-frequency and rare variants, when compared with the 1000 Genomes Project phase I (1000GP) reference panel (where the European sample comprises only about 10% of the UK10K sample size) [39]. Zheng and co-authors demonstrated the value of using a combined UK10K/1000 Genomes Project reference panel to discover low-frequency variants associated with bone mineral density [40]. Other sequencing studies, such as Genome of the Netherlands (GoNL) [41], SardiNIA [42, 43] and HELIC-MANOLIS [44], also reported the usefulness of population-specific samples for the characterisation of rare variants.
Finally, efforts are now in place to combine publicly available WGS datasets to create a single reference panel with increased depth of low-frequency and rare haplotypes. To date, the Haplotype Reference Consortium has combined low-read-depth WGS data (4–8×) from 20 studies of mainly European ancestry. The relative panel contains 64,976 haplotypes from 39,235,157 SNVs with minor allele count ≥5, and the large number of samples and variant sites increases the accuracy of the genotype imputation, especially at low-frequency variants down to 0.1% MAF and allows efficient phasing and imputation on existing servers with the aim to carry out imputation in a more streamlined manner [45, 46]. The Haplotype Reference Consortium panel will continue to incorporate samples from worldwide populations, which is important; since rare variants are, on average, younger than common variants, they show more geographical clustering and they are more difficult to impute. In order to provide a comprehensive imputation reference panel, it is important to combine many samples and to include samples from the geographical area of interest [47]. Additional advances to current reference panels are likely to emerge from large-scale sequencing studies such as the Trans-Omics for Precision Medicine (TOPMed) Program [48] or the 100,000 Genomes Project in the UK [49].
Custom genotyping arrays
An alternative strategy to imputation to survey low-frequency and rare variants in association studies takes advantage of bespoke genotyping arrays. These arrays are often disease focused and aim to enrich standard haplotype tagging SNP panels with variants of interest identified through sequencing and fine-mapping efforts. One such array was Immunochip, designed in 2009 by investigators of 11 distinct autoimmune and inflammatory diseases to assay 195,806 SNPs and 718 small INDELs. It included the top 2000 independent variants for each disease that showed evidence for an association, as well as SNPs from the 1000 Genomes Project and resequencing data to densely cover 186 different disease loci, including the major histocompatibility complex (MHC) and the killer immunoglobulin-like receptor (KIR) loci. The coverage of the low-frequency and rare variant spectrum is incomplete since the array was designed using early 1000 Genomes Pilot data (February 2010 release). Another limitation of the Immunochip is that the design is based on studies of European samples, and thus non-European variation is under-represented in this array [50].
The Metabochip custom array interrogates nearly 200,000 SNP markers of 257 genome-wide significant association signals for metabolic diseases (type 2 diabetes, coronary artery disease, myocardial infarction) and quantitative traits (body mass index, glucose and insulin levels, lipid levels and blood pressure). This array, similar to Immunochip, was very cost-effective, meaning more samples could be genotyped and its uniformity enabled direct comparison across phenotypes [51]. Metabochip SNPs were selected from International HapMap [34] and 1000 Genomes Projects [52] repositories to include SNPs across a wide range of allele frequencies. Metabochip SNPs focus on trait-associated loci (1.5% of the genome) by increasing their SNP resolution by fine-mapping. Imputation accuracy in fine-mapping regions is increased compared to traditional SNP arrays, as 54.4% of European SNPs from 1000GP phase I are tagged with r 2 ≥ 0.8 [51].
More recently, custom genotyping arrays have been developed to enhance representation of low-frequency and rare variants genome-wide. The UK Biobank Axiom Array contains 820,967 genetic variants, targeting specifically disease-specific and rare coding variants [53]. The Illumina HumanExome BeadChip (ExomeChip) comprises 247,870 variants (of which about 75% have MAF <0.5%) discovered through exome sequencing in approximately 12,000 individuals, including high-confidence non-synonymous and protein-altering variants (splice-site and stop gain or loss codons). Additionally, the exome chip includes common variants found through GWAS, ancestry informative markers (for African and Native Americans), mitochondrial variants, randomly selected synonymous variants, HLA tag variants and Y chromosome variants. The widespread application of the ExomeChip array has resulted in relatively few novel discoveries, including the identification of novel associations of a low-frequency coding variant in GLP1R with fasting glucose and type 2 diabetes [54], a number of novel low-frequency lipid signals at previously known loci [55, 56] and a large set of 32 rare and 51 low-frequency coding variants associated with height [57].
Exome or whole-genome sequencing
Historically, candidate gene sequencing studies have been used to explore sequence variation through relatively small-scale sequencing efforts. These were based mainly on capillary (Sanger) sequencing, typically focused on small numbers of patients and healthy controls and on genes with a strong a priori biological candidacy or importance for a given trait of disease [58,59,60,61,62,63,64]. Studies based on whole-exome sequencing (WES) and WGS have been increasingly used to systematically assess the properties and associations of rare variants, enabled by decreases in sequencing costs and increases in sequencing throughput [65]. WES probes only approximately 1.2% of the genome, and is thus cheaper relative to WGS, but limits investigations to variants in protein-coding regions of the genome. An enrichment analysis in the UK10K Project used functional and regulatory features, such as genic annotations, chromatin states, DNaseI hypersensitive sites, transcription factor binding sites, conservation scores and histone modifications, to assess the relative contribution of low-frequency and common variants to associations. The results showed that low-frequency variants in exonic regions displayed the strongest degree of enrichment (25-fold, compared with fivefold for common variants), which is compatible with the signatures of purifying selection, such as a negative correlation between functionally important variants and allele frequency [66]. However, non-coding low-frequency alleles were shown to also contribute to phenotypic trait variation: both common and low-frequency variants had comparably strong levels of functional enrichment for several non-coding domains (i.e. transcription start sites, DNase I hotspots and 3′ UTRs of genes) [21]. Additionally, it has been suggested that the quality and the calling of coding SNVs and INDELs is comparable if not better in WGS, i.e. an estimated 3% of coding variants were found by WGS but not called by WES [67]. We review later results of recent exome- and genome-sequencing studies of complex disease.
Optimal methods for association analysis with low-frequency and rare variants
Approaches typically used for testing associations of genetic variants with phenotype based on simple regression models are underpowered for rare variants [68]. Moreover, many more rare independent variants are found throughout the genome compared with common variants, increasing the multiple testing penalty for these studies. To overcome both of these issues, several statistical methods have been proposed to increase statistical power in association studies, typically by seeking to combine information across multiple rare variants within a specific genomic functional unit (e.g. gene, exon). Rare variant region-based methods can be grouped in four broad categories (Table 1).
Burden tests
Burden tests (ARIEL test [69], RWAS [70], CAST [71], CMC method [72], MZ Test [73], WSS [74], aSum [75], Step-up [76], EREC test [77], VT [78], KBAC method [79], RBT [80]) collapse information for genetic variants within a predefined functional unit into a single score and then regress this score against the trait of interest. The various burden tests differ in how this information is summarised. For example, the simplest form of burden test counts the number of minor alleles across all variants in the set producing a genetic score for each individual [69]. The cohort allelic sums test (CAST) [71] sets the genetics score to 0 or 1 based on the presence or absence, respectively, of at least one rare variant in the region tested. A more sophisticated weighting function was proposed by Madsen and Browning [74] with the weighted sum statistic (WSS) that takes into account all the variants’ frequencies without the need to set a fixed threshold to define rare and common variant as in CAST. Moreover, WSS considers other information on functional annotation of variants in its weighting method. Other kinds of burden tests have been developed to combine the collapsing methods with a multivariate test, such as the combined multivariate and collapsing (CMC) method [72]. Main limitations of burden tests are the strong assumption that the variants tested within the functional unit are all causal and associated with the trait with the same direction and magnitude of effect. This assumption is violated most of the time due to the highly variable and unknown allelic architecture of complex traits. For example, the PCSK9 gene carries alleles with both loss and gain function effects on LDL cholesterol [81, 82].
Variance-component tests
Varience-component tests (C-Alpha test [83], SKAT [84], SSU test [85], KBAT [86]) have been developed to consider the particular scenario where both risk and protective alleles may be found within a given gene or functional unit, testing for the distributions of genetic effects within a set of variants. This approach is flexible and allows for a mixture of effects in the rare variant set. The sequence kernel association test (SKAT) is one of the most widely used approaches, can take into account weightings of rare variants, family structure and covariates and is primarily designed for quantitative traits. Other tests (C-alpha [a special case of SKAT], WSS and CMC) can be applied only in case–control studies [84].
Combined tests
Combined tests (SKAT-O [87], EMMPAT [88], Fisher method [89], MiST [90]) have been developed to maximise power in a broad range of allelic architecture scenarios. In fact, this is the more realistic assumption and there are a number of statistical approaches to combine p values from two or more complementary tests. Among these approaches Fisher’s method [89] has been extensively used. More recently Lee and colleagues proposed an optimisation of the SKAT test (SKAT-O) that combines the burden and SKAT tests considering their best linear combination [87, 91].
Other tests
Other tests have been developed to account for signal sparsity across the tested region and include least absolute shrinkage and selection operator (LASSO) and the exponential combination (EC) test [92, 93]. Also Bayesian approaches have been proposed, but due to the computational time they are not as widely used as the aforementioned frequentist approaches [94]. A critical problem is to account for sequence quality, especially in next-generation sequencing data with relatively low coverage per individual. Two previous approaches are able to incorporate weights based on genotype uncertainty metrics for imputed genetic variants or for sequencing-derived variants [95], outperforming some pre-existing models [96].
Power, replication and confounding affecting rare variant association tests
An ongoing challenge is to systematically evaluate the relative merit, assumptions, implementation and statistical power of different analyses. Attempts to systematically evaluate the power of different methods for different allelic predisposition scenarios have been carried out using both simulations and empirical data [68,98,, 69, 97–99]. They have shown that gene-based tests are sensitive to variables such as the choice of analysis unit (e.g. exon versus whole gene), the number of variants tested within an aggregation unit and also the choice of particular functional classes of variants (e.g. loss-of-function, non-synonymous, etc.) or the magnitude of linkage disequilibrium between variants. As an example, Moutsianas and colleagues carried out a comprehensive study based on simulated data of similar size to current next-generation sequencing (NGS)-based association studies (3000 case–control individuals) [68]. The authors assessed power to detect associations using the main gene-based rare variant tests and for six different architecture scenarios informed by an empirical study of type 2 diabetes (T2D) (described in [68]). They showed that power to discover associations was low (<20%, for type I error (α) = 2.5 × 10–6), and even with sample sizes more than triple those of current empirical studies (about 10,000 case–control individuals) the power remained modest (on average about 60%). The authors further showed that combined tests (e.g. SKAT-O and MiST) had marginally greater power to detect associations across the number of simulated allelic architectures. This suggests that the application of these tests may be preferable in the context of genome-wide explorations in order to capture the widest possible range of allelic scenarios at different genes. Burden tests were shown to have more power to identify associations for deleterious variants, especially when neutral variation is filtered out. However, it is still unclear to what extent the simulations used in this and other studies may reflect the true allelic architecture of traits, highlighting the importance of implementing flexible testing scenarios in RVASs.
Other strategies for increasing statistical power are also liable to potential problems. For instance, the benefits of increases in sample size that are achieved through combining different sequencing studies can potentially be outweighed by issues of heterogeneity in disease state or in environmental exposures, or even differences in allele frequency between studies. Furthermore, studies focusing solely on certain categories of variants (e.g. loss of function variants) could on one hand increase the power by only considering variants with strong effect on phenotype. On the other hand, it has been suggested that removing flanking variants could potentially decrease the overall power to detect an association signal [100]. To address these issues, Liu et al. [101] developed a new method to meta-analyse rare variants that instead of using p values combines score statistics for each individual variant and employs a covariance matrix between variants reflecting the linkage disequilibrium structure inside the tested region.
Another challenge for RVASs is to achieve robust replication of signals, particularly in the instances where associations present allelic and locus heterogeneity [102]. For rare variants identified through single variant association tests, replication can be achieved by genotyping the identified variant in replication cohorts, provided obviously that the variant is indeed polymorphic in that cohort. For variants identified through aggregation methods, replication may be achieved by genotyping all the variants within the functional units discovered or direct sequencing of all the functional units [103]. Advances in sequencing and target-capture technologies reduce the cost of resequencing and, although it is more expensive than genotyping, resequencing can potentially identify new variants inside the functional unit that the discovery cohorts were not able to pinpoint [104, 105].
Finally, population stratification poses unique challenges in RVASs. In fact, systematic differences in allele frequencies due to differences in ancestry are more pronounced for rare variants [37]. Moreover, strong patterns of population stratification are predicted to arise in the presence of sharp spatial distributions for non-genetic risk of disease [106]. Adjusting for population stratification using traditional methods such as principal component analysis (PCA) and linear mixed effect models may, in most of the cases, not be suitable for rare variant tests [106,107,108,109]. Alternatives to reduce the confounding effects of population stratification in rare variant tests are using family-based designs or including spatial/geographical information [21, 106]. Moreover, calculating principal components using all or only common variants has shown to be more effective than using only rare variants [110]. Babron et al. [111] reported differences in population stratification patterns between rare and common variants in the UK population.
Study designs for enriching or prioritising rare variants
Study designs exploiting unique characteristics of different populations have been used to boost power in association studies of rare and low-frequency alleles. One notable example is population isolates, which provide powerful study designs for medical genetics due to a number of advantageous characteristics. For example, variants of medical importance that are rare in outbred populations might be found at higher frequencies in isolated populations due to past bottleneck events, genetic drift or adaptation and selection [43, 112], increasing power to detect associations with medically important phenotypes [113, 114].
A particularly interesting case of rare variation is variants that lead to inactivation of the corresponding protein. Such so-called loss-of-function (LoF) variants include variants predicted to lead to premature termination of the protein (stop-gain variants or protein-truncating variants) and insertion or deletion polymorphisms that affect the overall codon sequence of the protein (frameshift INDELS) or alter pre-mRNA splicing of essential exons (essential splice-site variants). LoF variants provide powerful tools to understand the impact of “knocking out” human genes, akin to gene knockout experiments commonly conducted in model organisms [115]. Understanding the phenotypic and clinical consequences of carrying LoF alleles, particularly when they are carried in the homozygous (i.e. complete knockout) state, has been shown to provide crucial insights into the identification of new disease genes and druggable pathways [116,117,118]. Further, studies of LoF variants in established drug targets, when carried by an otherwise healthy individual, provide evidence for safety of modulating that particular target to reduce disease risk. The data set of 60,706 individuals collated by the Exome Aggregation Consortium (ExAC) can assist in filtering of candidate disease-causing variants and in the discovery of human “knockout” variants in protein-coding genes [119].
Efforts to discover these mutations are boosted in populations with high rates of homozygosity, for example in populations with a tradition of consanguineous marriage, and where such variants occur more often in a homozygous state. Analysing samples from the PROMIS study, it was found that 961 genes were completely inactivated in at least one participant. Combined with rich phenotype information, this enabled the discovery of genotype–phenotype associations of clinical importance, such as the association of APOC3 with absent plasma apolipoprotein C-III levels [120]. Another study predicted LoF in 781 genes after analysing 3222 British Pakistani heritage adults with high parental relatedness [121]. The whole genomes of 2636 Icelanders together with imputing additional 101,584 chip-genotyped and phased Icelanders has begun to enable studies of rare complete human gene knockouts in the Icelandic population. The authors are also planning to characterise most homozygous LoF variants in the Icelandic population and to carry out bespoke phenotyping of the carriers [122]. A caveat of this approach is that the functional consequences of sequence variants are typically bioinformatically annotated as based on generic transcript annotations (for instance based on the most deleterious consequence among all annotated transcripts). LoF variants may therefore not lead to protein inactivation in a biologically relevant context, which could be due to gene redundancy, or to heterozygosity, or to genuine variants that do not actually disrupt gene function, or to variants that are only active in certain tissue-specific (or rare) isoforms [112, 115]. Thus, extensive and painstaking follow-up efforts are required to validate the predicted consequences of these variants.
Initial results from associations from large-scale sequencing projects
A growing number of studies have explored properties of low-frequency and rare variants and their relevance for complex traits and disease (Fig. 1, Tables 2, 3, Additional file 1). A first exploration based on exome-sequencing in 200 individuals from Denmark identified an excess of low-frequency deleterious, non-synonymous SNVs compared with synonymous SNVs [123]. In another study 15,585 human protein-coding genes were sequenced to an average median depth of 111× in 2440 individuals of European and African ancestry. The majority of the SNVs were rare (MAF <0.5%), previously unknown and population-specific. It was estimated that 2.3% of the 13,595 SNVs each person carried were predicted to affect protein function of about 313 genes per genome and most of the variants that affected the protein function were rare [66].

The allele frequency spectrum for a genome-wide association study variants (Additional file 1) and b sequenced variants that were associated with a variety of traits (Table 3 and Additional file 1). There is a clear shift to lower allele frequencies for variants discovered in sequencing studies. c The effect size versus allele frequency for sequenced variants; i.e. to detect associations that involve variants with lower allele frequencies, higher effect sizes are needed or large sample sizes. Effect size is usually measured as “beta” for quantitative traits and as “odds ratio” for dichotomous traits
A study by the UK10K Project exploited low-read-depth WGS and focused on 64 different quantitative cardiometabolic traits in the general UK population [21, 39]. While yielding initial discoveries of rare informative alleles [22,125,, 124–126], these initial efforts have highlighted a clear need to increase the statistical power of studies of complex human disease, particularly to target the contribution of rare variation. Further, they showed that highly penetrant alleles contributing to phenotypic variance of cardiometabolic traits are likely to be found at frequencies well below 1% in the general European population, but are poorly tagged by imputation reference panels, suggesting that direct assessment through genome sequencing will be required to comprehensively access this frequency range for complex traits.
deCODE gathered genotypic and medical data of more than half of the Icelandic population [127]. They generated a population-specific reference imputation panel based on WGS data for approximately 2000 study participants. They then applied imputation not only to the approximately 90,000 participants with genome-wide SNP arrays available, but also to over 250,000 participants where genotypes could be inferred from comprehensive genealogical records; this led to novel discoveries for a range of different complex traits and diseases. As one example, Styrkarsdottir et al. [128] identified a nonsense variant in LGR4 associated with low bone mineral density (osteoporosis). The study included 4931 individuals with low bone mineral density and 69,034 individuals as control group. Steinthorsdottir et al. [129] discovered four previously unreported rare and low-frequency variants in CCND2, PAM and PDX1 genes affecting risk of T2D. Helgason et al. [130] found a rare variant in the C3 gene associated with age-related macular degeneration. Also, rare variants in TREM2 and APP genes were associated with Alzheimer’s disease [131, 132]. Further, this project identified 6795 autosomal LoF SNPs and INDELs in 4924 genes of which 7.7% were homozygotes or compound heterozygotes with a MAF below 2% [122], boosting further effort to study gene inactivation in humans. Recently, a rare variant in ASGR1 gene was found to lower the risk of a heart attack by more than one-third in Icelanders [133]. The function of this gene needs still to be elucidated, but possibly it could be protective against heart disease with an alternative mechanism rather than acting on blood lipids, making it a potentially promising drug target to prevent heart disease.
The Genome of the Netherland (GoNL) project used WGS to characterise DNA sequence variation in the Dutch population, focusing on a representative sample consisting of 250 trio-families from all provinces in the Netherlands [41, 134]. Significant improvement in the imputation quality for rare variants (MAF 0.05–0.5%) compared with the 1000GP were demonstrated for the Dutch population, illustrating the value of using large, population-specific reference panels for imputing rare variants [135]. Further, use of this panel led to the identification of a rare deleterious missense variant in ABCA6 associated with LDL-C and TC in the Dutch population [136].
Similarly, the African Genome Variation Project, consisting of dense genotypes from 1481 individuals and whole-genome sequences from 320 individuals across sub-Saharan Africa, demonstrates the importance of adding population specific cohorts to existing reference panels to improve imputation accuracy [137] to account for the greater genetic diversity in these regions compared with the other populations who have expanded out of Africa.
The SardiNIA project is a longitudinal study including genetic and phenotypic data for 1257 multigenerational families from four villages in the Lanusei valley in Sardinia, Italy. In a recent study, WGS was performed in a total of 2120 participants [43], discovering 76,000 variants that were common in the SardiNIA study (frequency >5%) but rare elsewhere (<0.5% in the 1000GP). This study identified 14 associations for lipid levels (including two major new loci) and 19 for inflammatory markers (including two novel loci). In a companion study [138], the authors also identified five variants regulating haemoglobin levels at previously undetected loci (MPHOSPH9, PLTP-PCIF1, ZFPM1 (FOG1), NFIX and CCND3), highlighting the importance of sequencing isolated populations in finding variants that may be very rare and possibly not present in other populations.
The Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE) design includes five prospective cohort studies from the USA and Europe: the Age, Gene/Environment Susceptibility—Reykjavik Study, the Atherosclerosis Risk in Communities Study, the Cardiovascular Health Study, the Framingham Heart Study and the Rotterdam Study [139]. Among the studies published by this project (Table 2), one for instance identified rare variants with large effects associated with HDL-C levels through WGS of individuals sampled from the tails of the phenotypic distribution, some of which overlap with previously identified variants in Mendelian disorders [140].
ENGAGE was a successful consortium effort bringing together data from large-scale research in genetic and genomic epidemiology from population cohorts to be translated into information relevant for future clinical applications [141]. In a recent study based on imputation using the 1000GP, 15 loci with low-frequency and ten loci with missense lead-SNPs and two loci with an accumulation of rare variants were found to be associated with lipid levels, and were also found to increase the proportion of variance explained for LDL-C and TC [142].
As part of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project, Emond et al. [105] identified missense variants in DCTN4 that are associated with resistance to Pseudomonas aeruginosa infections. This study was conducted using an extreme phenotype design in which WES was carried out on patients with cystic fibrosis (n = 91). A large WES study (n = 2005), also part of the Exome Sequencing Project, identified a novel gene, PNPL5, affecting LDL-C levels [143]. Do et al. [144] found rare variants in LDLR and APOA5, increasing risk for myocardial infarction. In another study, rare and common variants were found to be associated with von Willebrand disease and factor VIII levels in African Americans [145]. Finally, analysis of whole exome sequences of 3734 participants of European or African ancestry identified rare mutations disrupting APOC3 function associated with lower levels of plasma triglycerides and a reduced risk of coronary heart disease for carriers of these mutations [104].
A large-scale sequencing study by the GoT2D and T2D-GENES consortia [19] investigated lower frequency variants discovered from WGS of 2657 European individuals with and without T2D and WES of 12,940 individuals from five ancestry groups. The variants discovered were not sufficient to explain the large fraction of heritability missed from previous GWASs.
Extending to neuropsychiatric disorders, a recent study identified rare LoF variants in the SETD1A gene to be associated with schizophrenia. The WES study of 4264 schizophrenia cases, 9343 controls and 1077 trios identified three de novo mutations and seven LoF variants found in cases in the discovery cohort but none in controls. Two analytical approaches, one based on Fisher’s method to combine de novo and case–control p values and the other using the transmission and de novo association (TADA) model, were used in the study [146].
Finally, cancer such as breast cancer has a high incidence worldwide with 5–10% of cases associated with highly penetrant germline susceptibility alleles. BRCA1 and BRCA2 are the first genes found to be associated with a higher predisposition to breast cancer [147]. Most BRCA1 and BRCA2 pathogenic variants are predicted to produce a truncated protein product and thus loss of protein function [148]. However, the prevalence of BRCA1 and BRCA2 mutations is only approximately 24% [149, 150]. Recently, exome sequencing has uncovered substantial locus heterogeneity among affected families without BRCA1 or BRCA2 mutations [151, 152]. The new pathogenic variants are rare, posing challenges to estimation of risk attribution through patient cohorts. Among these, rare monoallelic LoF variations within the PALB2 gene (partner and localiser of BRCA2) are associated with breast cancer at a risk two to four times that among non-mutation carriers [153].
These and other examples illustrate the value of different designs, including sequencing population-specific cohorts to enhance the imputation quality of rare and low-frequency variants, exploiting population isolates, and sequencing of extremes of phenotypic traits. Despite limitations of power and resolution, rare variant association studies are becoming increasingly mature. The majority of associations with low-frequency and rare variants demonstrate relatively small effects on complex traits and disease. Interestingly, a study conducted by Wood et al. [154] in an Italian cohort (InCHIANTI) specifically compared phenotypic effects of low-frequency and rare variants to those of common variants. While some low-frequency variants with larger effect sizes (and similarly phenotypic variance explained) were detected, these represented a very small proportion of all association. This suggests that, particularly for outbred populations, greater sample sizes will be necessary to realise the potential of RVASs to identifying new genes involved in human disease pathways and biology.
Future prospects
Despite the success of GWASs in identifying thousands of robust associations with complex diseases and traits, few examples of these results have been successfully translated into clinical use [118, 155, 156]. Nevertheless, GWAS loci have been shown to increase the therapeutic validity of selected targets by twofold compared with previous target selection [157]. Substantial decreases in sequencing costs, coupled with increases in throughput afforded by massively parallel sequencing, offer the promise to greatly boost the discovery of highly informative rare and low-frequency genetic variants through WES and WGS. Advances in phenotyping (including multivariate measures of traditional disease risk factors, disease-relevant endpoints derived from electronic health records or molecular traits driven by advances in functional and cellular genomics) will further boost the power of these genomic approaches. Multiple areas of research will benefit from these enhancements. First, they will lead to discoveries of highly informative rare alleles, including LoF mutations, associated with risk of disease. Second, they will provide more powerful genetic tools to assess the causal contribution of novel biological pathways to disease risk through Mendelian randomisation approaches. Finally, they will enable efforts to dissect and refine understanding of causal regulatory variants through genome-scale molecular and cellular assays. Thus, the discovery of associations driven by low-frequency and rare variants are expected to contribute to efforts to validate therapeutic targets, for instance by identifying alleles that mimic the effect of modulating drug target genes, which can inform the likelihood of success in treating disease by modulating biological pathways through novel and existing drugs. These approaches thus offer great promise for reducing the attrition rate in drug development by identifying new drugs with higher efficacy and by informing repositioning of existing drugs towards new disease indications.
Abbreviations
- CAST:
-
Cohort allelic sums test
- CMC:
-
Combined multivariate and collapsing
- GWAS:
-
Genome-wide association study
- INDEL:
-
Insertion-deletion
- MAF:
-
Minor allele frequency
- RVAS:
-
Rare variant association study
- SKAT:
-
Sequence kernel association test
- SNP:
-
Single-nucleotide polymorphism
- SNV:
-
Single-nucleotide variant
- T2D:
-
Type 2 diabetes
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole-genome sequencing
References
International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–96.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–7.
Clayton DG. Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 2009;5:e1000540.
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80.
Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013;9:e1003444.
López M, Lage R, Saha AK, Pérez-Tilve D, Vázquez MJ, Varela L, et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 2008;7:389–99.
Chatenoud L, Warncke K, Ziegler A-G. Clinical immunologic interventions for the treatment of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:a007716.
Hitomi Y, Cirulli ET, Fellay J, McHutchison JG, Thompson AJ, Gumbs CE, et al. Inosine triphosphate protects against ribavirin-induced adenosine triphosphate loss by adenylosuccinate synthase function. Gastroenterology. 2011;140:1314–21.
Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–86.
Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167:1415–29. e19.
Speed D, Hemani G, Johnson MR, Balding DJ. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet. 2012;91:1011–21.
Agarwala V, Flannick J, Sunyaev S, Go TDC, Altshuler D. Evaluating empirical bounds on complex disease genetic architecture. Nat Genet. 2013;45:1418–27.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53.
Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–25.
Park JH, Gail MH, Weinberg CR, Carroll RJ, Chung CC, Wang Z, et al. Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc Natl Acad Sci U S A. 2011;108:18026–31.
Griswold AJ, Dueker ND, Van Booven D, Rantus JA, Jaworski JM, Slifer SH, et al. Targeted massively parallel sequencing of autism spectrum disorder-associated genes in a case control cohort reveals rare loss-of-function risk variants. Mol Autism. 2015;6:43.
Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–7.
Lee SH, DeCandia TR, Ripke S, Yang J, Schizophrenia Psychiatric Genome-Wide Association Study Consortium, International Schizophrenia Consortium, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet. 2012;44:247–50.
UK10K Consortium, Walter K, Min JL, Huang J, Crooks L, Memari Y, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90.
Iotchkova V, Huang J, Morris JA, Jain D, Barbieri C, Walter K, et al. Discovery and refinement of genetic loci associated with cardiometabolic risk using dense imputation maps. Nat Genet. 2016;48:1303–12.
Kryukov GV, Shpunt A, Stamatoyannopoulos JA, Sunyaev SR. Power of deep, all-exon resequencing for discovery of human trait genes. Proc Natl Acad Sci U S A. 2009;106:3871–6.
Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2006;2:e41.
Speed D, Cai N, The Ucleb Consortium, Johnson M, Nejentsev S, Balding D. Re-evaluation of SNP heritability in complex human traits. bioRxiv. 2016. doi: https://doi.org/10.1101/074310.
Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wagele B, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60.
Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214–20.
Cheng Y, Ma Z, Kim B-H, Wu W, Cayting P, Boyle AP, et al. Principles of regulatory information conservation between mouse and human. Nature. 2014;515:371–5.
Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373:895–907.
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proença C, Sparsø T, Holmkvist J, Marchand M, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. 2009;41:89–94.
Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13.
Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511.
International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320.
International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61.
Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14:549–58.
International HapMap 3 Consortium, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8.
1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL, et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat Commun. 2015;6:8111.
Zheng H-F, Forgetta V, Hsu Y-H, Estrada K, Rosello‐Diez A, Leo PJ, et al. Whole‐genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature. 2015;526:112–7.
Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–25.
Pistis G, Porcu E, Vrieze SI, Sidore C, Steri M, Danjou F, et al. Rare variant genotype imputation with thousands of study-specific whole-genome sequences: implications for cost-effective study designs. Eur J Hum Genet. 2015;23:975–83.
Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47:1272–81.
Gilly A, Ritchie GR, Southam L, Farmaki A-E, Tsafantakis E, Dedoussis G, et al. Very low-depth sequencing in a founder population identifies a cardioprotective APOC3 signal missed by genome-wide imputation. Hum Mol Genet. 2016;24:2360–5.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
The Haplotype Reference Consortium. http://www.haplotype-reference-consortium.org/participating-cohorts. Accessed 30 Mar 2017.
Surakka I, Kristiansson K, Anttila V, Inouye M, Barnes C, Moutsianas L, et al. Founder population-specific HapMap panel increases power in GWA studies through improved imputation accuracy and CNV tagging. Genome Res. 2010;20:1344–51.
Trans-Omics for Precision Medicine (TOPMed) Program. https://www.nhlbi.nih.gov/research/resources/nhlbi-precision-medicine-initiative/topmed. Accessed 30 Mar 2017.
Genomics England: The 100,000 Genomes Project. https://www.genomicsengland.co.uk/the-100000-genomes-project. Accessed 30 Mar 2017.
Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14:661–73.
Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793.
1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73.
UkBiobank. http://www.ukbiobank.ac.uk/. Accessed 30 Mar 2017.
Wessel J, Chu AY, Willems SM, Wang S, Yaghootkar H, Brody JA, et al. Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat Commun. 2015;6:5897.
Kanoni S, Masca NG, Stirrups KE, Varga TV, Warren HR, et al. Analysis with the exome array identifies multiple new independent variants in lipid loci. Hum Mol Genet. 2016;25:4094–106.
Exome Chip Design. http://genome.sph.umich.edu/wiki/Exome_Chip_Design. Accessed 30 Mar 2017.
Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. 2017;542:186–90.
Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science. 1996;274:1185–8.
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–7.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6.
Cohen JC, Pertsemlidis A, Fahmi S, Esmail S, Vega GL, Grundy SM, et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A. 2006;103:1810–5.
Cohen J, Kiss R, Pertsemlidis A, Marcel Y, McPherson R, Hobbs H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–72.
Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–8.
NIH. The cost of sequencing a human genome. https://www.genome.gov/27565109/the-cost-of-sequencing-a-human-genome/. Accessed 30 Mar 2017.
Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–9.
Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112:5473–8.
Moutsianas L, Agarwala V, Fuchsberger C, Flannick J, Rivas MA, Gaulton KJ, et al. The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS Genet. 2015;11:e1005165.
Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutat Res. 2007;615:28–56.
Sul JH, Han B, He D, Eskin E. An optimal weighted aggregated association test for identification of rare variants involved in common diseases. Genetics. 2011;188:181–8.
Asimit JL, Day-Williams AG, Morris AP, Zeggini E. ARIEL and AMELIA: testing for an accumulation of rare variants using next-generation sequencing data. Hum Hered. 2012;73:84–94.
Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21.
Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–93.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384.
Han F, Pan W. A data-adaptive sum test for disease association with multiple common or rare variants. Hum Hered. 2010;70:42–54.
Hoffmann TJ, Marini NJ, Witte JS. Comprehensive approach to analyzing rare genetic variants. PLoS One. 2010;5:e13584.
Lin D-Y, Tang Z-Z. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–67.
Price AL, Kryukov GV, de Bakker PIW, Purcell SM, Staples J, Wei L-J, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86:832–8.
Liu DJ, Leal SM. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010;6:e1001156.
Ionita-Laza I, Buxbaum JD, Laird NM, Lange C. A new testing strategy to identify rare variants with either risk or protective effect on disease. PLoS Genet. 2011;7:e1001289.
Cohen JC, Boerwinkle E, Mosley Jr TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–9.
Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93.
Pan W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet Epidemiol. 2009;33:497–507.
Mukhopadhyay I, Feingold E, Weeks DE, Thalamuthu A. Association tests using kernel-based measures of multi-locus genotype similarity between individuals. Genet Epidemiol. 2010;34:213–21.
Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case–control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–37.
King CR, Rathouz PJ, Nicolae DL. An evolutionary framework for association testing in resequencing studies. PLoS Genet. 2010;6:e1001202.
Derkach A, Lawless JF, Sun L. Robust and powerful tests for rare variants using Fisher’s method to combine evidence of association from two or more complementary tests. Genet Epidemiol. 2013;37:110–21.
Sun J, Zheng Y, Hsu L. A unified mixed-effects model for rare-variant association in sequencing studies. Genet Epidemiol. 2013;37:334–44.
Chen H, Dupuis J. Rare variant association analysis: beyond collapsing approaches. In: Zeggini E, Morris A, editors. Assessing rare variation in complex traits. 1st ed. New York: Springer-Verlag; 2015.
Chen Lin S, Hsu L, Gamazon Eric R, Cox Nancy J, Nicolae DL. An exponential combination procedure for set-based association tests in sequencing studies. Am J Hum Genet. 2012;91:977–86.
Zhou H, Sehl ME, Sinsheimer JS, Lange K. Association screening of common and rare genetic variants by penalized regression. Bioinformatics. 2010;26:2375–82.
Yi N, Zhi D. Bayesian analysis of rare variants in genetic association studies. Genet Epidemiol. 2011;35:57–69.
He L, Pitkäniemi J, Sarin A-P, Salomaa V, Sillanpää MJ, Ripatti S. Hierarchical Bayesian model for rare variant association analysis integrating genotype uncertainty in human sequence data. Genet Epidemiol. 2015;39:89–100.
Daye ZJ, Li H, Wei Z. A powerful test for multiple rare variants association studies that incorporates sequencing qualities. Nucleic Acids Res. 2012;40:e60.
Basu S, Pan W. Comparison of statistical tests for disease association with rare variants. Genet Epidemiol. 2011;35:606–19.
Dering C, Hemmelmann C, Pugh E, Ziegler A. Statistical analysis of rare sequence variants: an overview of collapsing methods. Genet Epidemiol. 2011;35:S12–7.
Derkach A, Lawless JF, Sun L. Pooled association tests for rare genetic variants: a review and some new results. Statist Sci. 2014;29:302–21.
Nicolae DL. Association tests for rare variants. Annu Rev Genomics Hum Genet. 2016;17:117–30.
Liu DJ, Peloso GM, Zhan X, Holmen OL, Zawistowski M, Feng S, et al. Meta-analysis of gene-level tests for rare variant association. Nat Genet. 2014;46:200–4.
Sanna S, Li B, Mulas A, Sidore C, Kang HM, Jackson AU, et al. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198.
Liu DJ, Leal SM. Replication strategies for rare variant complex trait association studies via next-generation sequencing. Am J Hum Genet. 2010;87:790–801.
Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44:886–9.
Mathieson I, McVean G. Differential confounding of rare and common variants in spatially structured populations. Nat Genet. 2012;44:243–6.
O’Connor TD, Kiezun A, Bamshad M, Rich SS, Smith JD, Turner E, et al. Fine-scale patterns of population stratification confound rare variant association tests. PLoS One. 2013;8:e65834.
Liu Q, Nicolae DL, Chen LS. Marbled inflation from population structure in gene-based association studies with rare variants. Genet Epidemiol. 2013;37:286–92.
Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95:5–23.
Zhang Y, Shen X, Pan W. Adjusting for population stratification in a fine scale with principal components and sequencing data. Genet Epidemiol. 2013;37:787–801.
Babron M-C, de Tayrac M, Rutledge DN, Zeggini E, Génin E. Rare and low frequency variant stratification in the UK population: description and impact on association tests. PLoS One. 2012;7:e46519.
Moltke I, Grarup N, Jorgensen ME, Bjerregaard P, Treebak JT, Fumagalli M, et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature. 2014;512:190–3.
Colonna V, Pistis G, Bomba L, Mona S, Matullo G, Boano R, et al. Small effective population size and genetic homogeneity in the Val Borbera isolate. Eur J Hum Genet. 2013;21:89–94.
Panoutsopoulou K, Hatzikotoulas K, Xifara DK, Colonna V, Farmaki A-E, Ritchie GRS, et al. Genetic characterization of Greek population isolates reveals strong genetic drift at missense and trait-associated variants. Nat Commun. 2014;5:5345.
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–8.
Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5.
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47:445–7.
Flannick J, Thorleifsson G, Beer NL, Jacobs SBR, Grarup N, Burtt NP, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46:357–63.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Saleheen D, Natarajan P, Zhao W, Rasheed A, Khetarpal S, Won HH, et al. Human knockouts in a cohort with a high rate of consanguinity. bioRxiv. 2015. doi: https://doi.org/10.1101/031518.
Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352:474–7.
Sulem P, Helgason H, Oddson A, Stefansson H, Gudjonsson SA, Zink F, et al. Identification of a large set of rare complete human knockouts. Nat Genet. 2015;47:448–52.
Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42:969–72.
Feng S, Pistis G, Zhang H, Zawistowski M, Mulas A, Zoledziewska M, et al. Methods for association analysis and meta-analysis of rare variants in families. Genet Epidemiol. 2015;39:227–38.
Timpson NJ, Walter K, Min JL, Tachmazidou I, Malerba G, Shin S-Y, et al. A rare variant in APOC3 is associated with plasma triglyceride and VLDL levels in Europeans. Nat Commun. 2014;5:4871.
Taylor PN, Porcu E, Chew S, Campbell PJ, Traglia M, Brown SJ, et al. Whole-genome sequence-based analysis of thyroid function. Nat Commun. 2015;6:5681.
deCODE genetics. http://www.decode.com/. Accessed 30 Mar 2017.
Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature. 2013;497:517–20.
Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46:294–8.
Helgason H, Sulem P, Duvvari MR, Luo H, Thorleifsson G, Stefansson H, et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat Genet. 2013;45:1371–4.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–16.
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–9.
Nioi P, Sigurdsson A, Thorleifsson G, Helgason H, Agustsdottir AB, Norddahl GL, et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N Engl J Med. 2016;374:2131–41.
Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, et al. The Genome of the Netherlands: design, and project goals. Eur J Hum Genet. 2014;22:221–7.
Deelen P, Menelaou A, van Leeuwen EM, Kanterakis A, van Dijk F, Medina-Gomez C, et al. Improved imputation quality of low-frequency and rare variants in European samples using the ‘Genome of The Netherlands’. Eur J Hum Genet. 2014;22:1321–6.
van Leeuwen EM, Karssen LC, Deelen J, Isaacs A, Medina-Gomez C, Mbarek H, et al. Genome of the Netherlands population-specific imputations identify an ABCA6 variant associated with cholesterol levels. Nat Commun. 2015;6:6065.
Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327–32.
Danjou F, Zoledziewska M, Sidore C, Steri M, Busonero F, Maschio A, et al. Genome-wide association analyses based on whole-genome sequencing in Sardinia provide insights into regulation of hemoglobin levels. Nat Genet. 2015;47:1264–71.
Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80.
Morrison AC, Voorman A, Johnson AD, Liu X, Yu J, Li A, et al. Whole-genome sequence–based analysis of high-density lipoprotein cholesterol. Nat Genet. 2013;45:899–901.
ENGAGE (European network of genetic and genomic epidemiology). http://www.euengage.org/. Accessed 30 Mar 2017.
Surakka I, Horikoshi M, Mägi R, Sarin A-P, Mahajan A, Lagou V, et al. The impact of low-frequency and rare variants on lipid levels. Nat Genet. 2015;47:589–97.
Lange Leslie A, Hu Y, Zhang H, Xue C, Schmidt Ellen M, Tang Z-Z, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet. 2014;94:233–45.
Do R, Stitziel NO, Won H-H, Jørgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring. Nature. 2015;518:102–6.
Johnsen JM, Auer PL, Morrison AC, Jiao S, Wei P, Haessler J, et al. Common and rare von Willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: the NHLBI Exome Sequencing Project. Blood. 2013;122:590–7.
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–7.
Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, et al. Cancer Risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105:812–22.
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–82.
Kast K, Rhiem K, Wappenschmidt B, Hahnen E, Hauke J, Bluemcke B, et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet. 2016;53:465–71.
Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–33.
Määttä K, Rantapero T, Lindström A, Nykter M, Kankuri-Tammilehto M, Laasanen S-L, et al. Whole-exome sequencing of Finnish hereditary breast cancer families. Eur J Hum Genet. 2016;25:85–93.
Gracia-Aznarez FJ, Fernandez V, Pita G, Peterlongo P, Dominguez O, de la Hoya M, et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One. 2013;8:e55681.
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506.
Wood AR, Tuke MA, Nalls M, Hernandez D, Gibbs JR, Lin H, et al. Whole-genome sequencing to understand the genetic architecture of common gene expression and biomarker phenotypes. Hum Mol Genet. 2015;24:1504–12.
Thompson AJ, Fellay J, Patel K, Tillmann HL, Naggie S, Ge D, et al. Variants in the ITPA gene protect against ribavirin-induced hemolytic anemia and decrease the need for ribavirin dose reduction. Gastroenterology. 2010;139:1181–9. e1182.
Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–60.
Acknowledgements
Lorenzo Bomba acknowledges Open Targets for funding. We acknowledge Jennifer Asimit for critical review of the manuscript.
Funding
Nicole Soranzo’s research is supported by the Wellcome Trust (Grant Codes WT098051 and WT091310), the EU FP7 (EPIGENESYS Grant Code 257082 and BLUEPRINT Grant Code HEALTH-F5-2011-282510) and the National Institute for Health Research Blood and Transplant Research Unit (NIHR BTRU) in Donor Health and Genomics at the University of Cambridge in partnership with NHS Blood and Transplant (NHSBT). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health or NHSBT.
Authors’ contributions
All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
All rare variants (allele frequency <5%) that were discovered across different traits, together with chromosomal position (GRCh37), mapped gene, disease or traits, sample size, allele frequency, effect size, population and reference. (XLSX 39 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bomba, L., Walter, K. & Soranzo, N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol 18, 77 (2017). https://doi.org/10.1186/s13059-017-1212-4
Published:
DOI: https://doi.org/10.1186/s13059-017-1212-4