
Abstract
Background
Epigenetic biomarkers of aging (the “epigenetic clock”) have the potential to address puzzling findings surrounding mortality rates and incidence of cardio-metabolic disease such as: (1) women consistently exhibiting lower mortality than men despite having higher levels of morbidity; (2) racial/ethnic groups having different mortality rates even after adjusting for socioeconomic differences; (3) the black/white mortality cross-over effect in late adulthood; and (4) Hispanics in the United States having a longer life expectancy than Caucasians despite having a higher burden of traditional cardio-metabolic risk factors.
Results
We analyzed blood, saliva, and brain samples from seven different racial/ethnic groups. We assessed the intrinsic epigenetic age acceleration of blood (independent of blood cell counts) and the extrinsic epigenetic aging rates of blood (dependent on blood cell counts and tracks the age of the immune system). In blood, Hispanics and Tsimane Amerindians have lower intrinsic but higher extrinsic epigenetic aging rates than Caucasians. African-Americans have lower extrinsic epigenetic aging rates than Caucasians and Hispanics but no differences were found for the intrinsic measure. Men have higher epigenetic aging rates than women in blood, saliva, and brain tissue.
Conclusions
Epigenetic aging rates are significantly associated with sex, race/ethnicity, and to a lesser extent with CHD risk factors, but not with incident CHD outcomes. These results may help elucidate lower than expected mortality rates observed in Hispanics, older African-Americans, and women.
Similar content being viewed by others

Background
Many demographic and epidemiological studies explore the effects of chronological age, race/ethnicity, and sex on mortality rates and susceptibility to chronic disease [1–5], but it remains an open research question whether race/ethnicity and sex affect molecular markers of aging directly. To what extent clinical biomarkers of inflammation, dyslipidemia, and immune senescence relate to cellular markers of aging also remains an open question. One major challenge is the lack of agreement on how to define and measure biological aging rates [6]. Many biomarkers of aging have been proposed ranging from clinical markers (such as whole-body functional evaluations and gait speed) to molecular markers such as telomere length [7, 8]. Available biomarkers capture only particular aspects of aging. For example, African Americans have been shown to have longer telomere lengths than Caucasians [9], despite significantly higher levels of inflammation, lower average life expectancies, and higher disease incidence. To date, no studies have employed epigenetic measures to estimate and compare molecular aging rates among gender or racial/ethnic groups.
Measures incorporating DNA methylation levels have recently given rise to a new class of biomarkers that appear informative of aging given that age has a profound effect on DNA methylation levels in most human tissues and cell types [10–18]. Several recent studies have measured the epigenetic age of tissue samples by combining the DNA methylation levels of multiple dinucleotide markers, known as Cytosine phosphate Guanines or CpGs [19–21]. We recently developed the epigenetic clock (based on 353 CpGs) to measure the age, known as “DNA methylation age” or “epigenetic age,” of assorted human cell types (CD4+ T cells or neurons), tissues, and organs—including blood, brain, breast, kidney, liver, lung [20], and even prenatal brain samples [22]. The epigenetic clock is an attractive biomarker of aging because it applies to most human tissues and its accurate measurement of chronological age is unprecedented.
The following evidence shows that the epigenetic clock captures aspects of biological age. First, the epigenetic age of blood has been found to be predictive of all-cause mortality even after adjusting for chronological age and a variety of known risk factors [23–25]. Second, the blood of the offspring of Italian semi-supercentenarians (i.e. participants who reached an age of at least 105 years) has a lower epigenetic age than that of age-matched controls [26]. Third, the epigenetic age of blood relates to frailty [27] and cognitive/physical fitness in the elderly [28]. The utility of the epigenetic clock method has been demonstrated in applications surrounding obesity [29], Down’s syndrome [30], HIV infection [31], Parkinson’s disease [32], Alzheimer’s disease-related neuropathologies [33], lung cancer [34], and lifetime stress [35]. Here, we apply the epigenetic clock to explore relationships between epigenetic age and race/ethnicity, sex, risk factors of coronary heart disease (CHD), and the CHD outcome itself.
Results
Blood datasets and racial/ethnic groups
An overview of our DNA methylation datasets can be found in Table 1. We analyze multiple sources of DNA: mostly blood, saliva, and lymphoblastoid cell lines. In addition, brain datasets were used to compare men and women (Table 2). We considered the following racial/ethnic groups (Table 1): 1387 African Ancestry (African Americans and two groups from Central Africa), 2932 Caucasian (non-Hispanic whites), 657 Hispanic, 127 East Asians (mainly Han Chinese), and 59 Tsimane Amerindians.
Accuracy of the epigenetic clock
DNAm age, also referred to as epigenetic age, was calculated in human samples profiled with the Illumina Infinium 450 K platform using a previously described method [20]. As expected, we found DNAm age to have a strong linear relationship with chronological age in blood and saliva (correlations in the range of 0.65–0.93, Figs. 1, 2, 3, 4, and 5) and in lymphoblastoid cell lines (r = 0.59; Additional file 1). Based on a spline regression line, we defined a “universal” measure of epigenetic age acceleration, denoted “Age Accel.” in our figures, as the difference between the observed DNAm age value and the value predicted by a spline regression model in Caucasians. The term “universal” refers to the fact that this measure can be defined in a vast majority of tissues and cell types with the notable exception of sperm [20]. A positive value of the universal age acceleration measure indicates that DNA methylation age is higher than that predicted from the regression model for Caucasian participants of the same age. Our intrinsic and extrinsic age acceleration measures (see “Methods”) only apply to blood data. A measure of intrinsic epigenetic age acceleration (IEAA) measures cell-intrinsic epigenetic aging effects that are not confounded by extra-cellular differences in blood cell counts. The measure of IEAA is an incomplete measure of the age-related functional decline of the immune system because it does not track age-related changes in blood cell composition, such as the decrease of naïve CD8+ T cells and the increase in memory or exhausted CD8+ T cells [36–38]. The measure of extrinsic epigenetic age acceleration (EEAA) only applies to whole blood and aims to measure epigenetic aging in immune-related components. It keeps track of both intrinsic epigenetic changes and age-related changes in blood cell composition (see “Methods”). The estimated blood cell counts, which are used in these measures, correlate strongly with corresponding flow cytometric measurements from the MACS study (Additional file 2): r = 0.63 for CD8 + T cells, r = 0.77 for CD4+ T, r = 0.67 B cell, r = 0.68 naïve CD8+ T cell, r = 0.86 for naïve CD4+ T, and r = 0.49 for exhausted CD8+ T cells.

Intrinsic epigenetic age acceleration in Caucasians and Hispanics. a-d DNA methylation age (y-axis) versus chronological age (x-axis) in (a) Women’s Health Initiative, (b) blood data from PEG, (c) dataset 5, (d) saliva data from PEG. Dots corresponds to participants and are colored by ethnic group (gray = Caucasian, blue = Hispanic). The gray line depicts a spline regression line through Caucasians. We define two measures of age acceleration based on DNAm age. e-g The bar plots relate the universal measure of epigenetic age acceleration to race/ethnicity, which is defined as residual to the spline regression line through Caucasians, i.e. the vertical distance of a point from the line. By definition, the mean age acceleration in Caucasians is zero. h, m Results after combining the three blood datasets using Stouffer’s meta-analysis method. i Age acceleration residual versus ethnicity in the saliva data from PEG. j-m The y-axis reports the mean value of IEAA, which is defined as residual from a multivariate regression model that regresses DNAm age on age and several measures of blood cell counts. Each bar plot reports 1 standard error and the p value from a group comparison test (ANOVA). n Age acceleration in blood versus age acceleration in saliva for the subset of PEG participants for whom both data were available

Intrinsic epigenetic age acceleration in Tsimane, Hispanics, East Asians, and Caucasians. a-c DNA methylation age (y-axis) versus chronological age (x-axis) in (a) dataset 5, (b) dataset 6, (c) dataset 7. Dots corresponds to participants and are colored by race/ethnicity (green = African American, gray = Caucasian, blue = Hispanic, red = Tsimane, orange = East Asians). The gray line depicts a spline regression line through Caucasians. We define two measures of age acceleration based on DNAm age. d-f The bar plots relate the universal measure of epigenetic age acceleration to race/ethnicity, which is defined as residual to the spline regression line through Caucasians, i.e. the vertical distance of a point from the line. g-i The y-axis reports the mean value of IEAA, which is defined as residual from a multivariate regression model that regresses DNAm age on age and several measures of blood cell counts. Each bar plot reports 1 standard error and the p value from a group comparison test (ANOVA)

Intrinsic epigenetic age acceleration versus African or European Ancestry. a-c DNA methylation age (y-axis) versus chronological age (x-axis) in (a) Women’s Health Initiative, (b) Bogalusa study. Dots corresponds to participants and are colored by race/ethnicity (green = African Ancestry, gray = Caucasian). The gray line depicts a spline regression line through Caucasians. We define two measures of age acceleration based on DNAm age. c, d The bar plots relate the universal measure of epigenetic age acceleration to race/ethnicity, which is defined as residual to the spline regression line through Caucasians. e, h Results after combining the two blood datasets using Stouffer’s meta-analysis method. f, g The y-axis reports the mean value of IEAA, which is defined as residual from a multivariate regression model that regresses DNAm age on age and several measures of blood cell counts. Each bar plot reports 1 standard error and the p value from a group comparison test (ANOVA)

Extrinsic epigenetic age acceleration and blood cell counts across groups. EEAA versus race/ethnicity in (a, q) Women’s Health Initiative, (b) blood data from PEG, (c, k) dataset 5, (l) dataset 6, (o) dataset 7, (r) Bogalusa study. Flow cytometric, age adjusted estimates (e, t) naïve CD8+ T and (j, x) naïve CD4+ T cell counts in the WHI LLS. Age adjusted estimates of naïve CD4 + T cells based on DNA methylation data from (f, u) Women’s Health Initiative, (g) blood data from PEG, (h, m) dataset 5, (n) dataset 6, (p) dataset 7, (v) Bogalusa study. (d, i, s, w) Meta-analysis across the respective datasets based on Stouffer’s method

Analysis of African rainforest hunter-gatherers and farmers. a DNAm age versus age using 256 blood samples from [42]. The points are colored as follows: magenta = AGR (urban setting), turquoise = AGR (forest), brown = RHG (forest). Group status versus (b) universal age acceleration, (d) intrinsic age acceleration, (f) extrinsic age acceleration. Habitat versus (c) universal age acceleration, (e) intrinsic age acceleration, (g) extrinsic age acceleration. (h, i) are analogous to (a, b) but the y-axis is based on a DNAm age estimate that excluded CpG that were located near SNPs. In this robustness analysis, we removed CpG probes containing genetic variants at a frequency higher than 1 % in the populations studied
Hispanics have a lower intrinsic aging rate than Caucasians
We find that Hispanics have a consistently lower IEAA compared to Caucasians (p = 7.1 × 10–10, Fig. 1m). An important question is whether the observed differences in blood can also be observed in other tissues. Using a novel saliva dataset (dataset 4, saliva from PEG) we find that Hispanics have a lower epigenetic aging rate than Caucasians (p = 0.042, Fig. 1i). The fact that our findings in blood can also be validated in saliva is consistent with the strong correlation between epigenetic age acceleration measures of the two sources of DNA (r = 0.70, p = 1.4 × 10–12, Fig. 1n). The lower value of IEAA in Hispanics unlikely reflects country of birth or of residence (at age 35 years) given the robust findings across samples and our detailed analysis in the WHI, where we find that Hispanics born outside US, but living in the US, have a higher IEAA than Hispanics born and raised in the US (p = 0.025, Additional file 3B).
CHD risk factors bear little or no relationship with IEAA
We related our measures of age acceleration to risk factors related to CHD since the latter are significant predictors of mortality. In postmenopausal women from the Women’s Health Initiative (WHI), we found no evidence that IEAA is associated with disparities in education, high density lipoprotein (HDL) or low density lipoprotein (LDL) cholesterol, insulin, glucose, C-reactive protein (CRP), creatinine, alcohol consumption, smoking, diabetes status, or hypertension (see Table 3).
Tsimane have a lower intrinsic aging rate than Caucasians
The Tsimane are an indigenous population (~15,000 inhabitants) of forager-horticulturalists who reside in the remote lowlands of Bolivia. They reside mostly in open-air thatch huts, and actively fish, hunt, and cultivate plantains, rice, and manioc through slash-and-burn horticulture [39]. Tsimane provide a unique contribution to aging researchers and epidemiologists because they experience high rates of inflammation due to repeated bacterial, viral, and parasitic infections, yet show minimal risk factors for heart disease or type 2 diabetes as they age; they have minimal hypertension and obesity, low LDL cholesterol and no evidence of peripheral arterial disease [39–41]. Since Hispanics share genetic ancestry with peoples indigenous to the Americas, we hypothesized that a slower intrinsic aging rate might also be observable by analyzing Tsimane blood samples [39]. Among participants who are older than 35 years, Tsimane have the lowest intrinsic age acceleration (Fig. 2d, g). While Tsimane have a significantly lower IEAA than Caucasians after the age of 35 years (p = 0.0061), no significant difference could be observed in younger participants (Fig. 2e, h). In this analysis, the threshold of 35 years was chosen so that a sufficient number of young participants would be included in dataset 6. We found no significant difference in IEAA between older Hispanics and Tsimane, which might reflect the relatively low group sizes of n = 37 Tsimane versus n = 38 Hispanics.
IEAA is not associated with CHD in the WHI
Based on our findings above showing little or no relationship between IEAA and CVD risk factors at baseline, we hypothesized that IEAA would not predict future onset of CHD. A multivariate logistic regression model shows that IEAA is not significantly associated with an increased risk of incident CHD (Table 4). However, as expected, current smoking, prior history of diabetes, hypertension, high insulin and glucose levels, and lower HDL predicted an increased risk of CHD (Table 4).
Hispanics and Tsimane have a higher EEAA than Caucasians
According to our measure of EEAA, Hispanics have a significantly older extrinsic epigenetic age than Caucasians (meta-analysis p = 0.00012, Fig. 4a–d) and fewer naïve CD4+ T cells, based on cytometric data from the WHI LLS, the MACS study, and imputed blood cell counts (Fig. 4f–j, Additional file 2H, I). This pattern of fewer naïve CD4+ T cells is even more pronounced for Tsimane (Fig. 4m, n), who experience repeated acute infections and elevated, often chronic, inflammatory loads.
Epigenetic age analysis of East Asians
Because ancient Native American populations share common ancestral lineages with East Asians, we examined whether East Asians also differ from Caucasians in terms of epigenetic aging rates. We found no significant difference between Caucasians and East Asians in terms of IEAA (Fig. 2i), EEAA (Fig. 4o), or naïve CD4+ T cells (Fig. 4p). Similarly, we found no difference in lymphoblastoid cell lines (Additional file 1). However, these comparative analyses are limited by the relatively small number of samples and should be repeated in larger datasets.
Which risk factors for cardiometabolic disease are associated with EEAA?
Our multivariate model analysis in the WHI (Table 3) shows that EEAA tracks better than IEAA with risk factors for cardiometabolic disease; EEAA was positively associated (higher) with: triglyceride levels (multivariate model p = 0.04), CRP (p = 0.023), and creatinine (p = 0.008). EEAA was negatively associated (lower) with higher levels of education in all ethnic groups (p from 2.0 × 10–8 to 0.05, Additional file 4I–L). For each racial/ethnic group, we find that women who did not finish high school exhibit the highest levels of EEAA (leftmost bar in Additional file 4J–L).
Epigenetic aging rates of African Americans
In the following, we compare African Americans with European Americans in terms of IEAA and EEAA. Comparisons of African Americans with Caucasians in terms of IEAA yield contradictory findings across datasets that differ in age range: African American women have slightly lower IEAA than Caucasian women in the WHI (p = 0.017 Fig. 3f), but no significant difference can be observed for the younger participants of the Bogalusa study (Fig. 3g). Indeed, participants in the WHI (aged between 50 and 80 years) were older than those of the Bogalusa study (aged between 29 and 51 years). This failure to detect a significant racial/ethnic difference in IEAA in younger participants is consistent with our results from the comparison of younger Tsimane and Caucasians (Fig. 2h). A multivariate model analysis based on the Bogalusa study (comprising African Americans and Caucasians) confirms that IEAA does not differ between middle-aged African Americans and Caucasians but IEAA is higher among men (p = 0.025) and has a marginally significant association with hypertension (p = 0.064, Table 5). When relating individual variables to IEAA, we find significant associations for hypertension (p = 0.00035, Additional file 5D–F) but not for type II diabetes status or educational level.
Our findings for EEAA are highly consistent across the two studies and age groups: African Americans have lower EEAA than Caucasians in the WHI and in the Bogalusa study (p = 7.2 × 10–7, Fig. 4q, r, s). Our flow cytometric data from the WHI LLS show that African American women exhibit a higher abundance of naïve CD8+ T cells than Caucasian women (p = 1.7 × 10–9, Fig. 4t).
In multivariate regression analyses of EEAA, we find that African Americans have indications of a significantly younger immune system age than Caucasians (p = 0.0076) after controlling for gender, educational level, diabetes status, and hypertension. In the Bogalusa study, we find three significant predictors of EEAA: race/ethnicity, hypertension, and gender (p = 0.0093, Table 5). A marginal analysis in the Bogalusa study identifies a significant association between EEAA and hypertension (p = 8.0 × 10–5, Additional file 5G–I), type II diabetes status in Caucasians (p = 0.0085, Additional file 6H), but not in African Americans (Additional file 6I). Contrary to our findings in the WHI, no significant association can be observed between EEAA and educational level (Additional file 7).
African rainforest hunter-gatherers and farmers
To evaluate the effect of subsistence ecology and environment on epigenetic aging rates, we analyzed 256 blood samples from two different groups in Central Africa: rainforest hunter-gatherers (RHGs, traditionally known as “pygmies,” sampled from Baka and Batwa populations) and African populations that have adopted an agrarian lifestyle (AGRs, traditionally known as “Bantus,” sampled from the Nzebi, Fang, Bakiga, and Nzime populations) over the last 5000 years [42]. The ancestors of the RHGs and AGRs diverged ~60,000 years ago. These groups have historically occupied separate ecological habitats—the ancestors of RHGs in the equatorial rainforest while those of AGRs in drier, more open space savannahs and grasslands. Many RHG groups still live in the rainforest as mobile bands, whereas AGR populations now occupy primarily rural or urban deforested areas, though some AGR groups have settled in the rainforest over the last millennia.
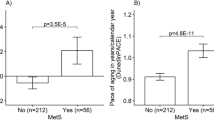
We considered three groups: (1) RHG (n = 102); (2) AGR living in the forest (n = 60); and (3) AGR living in an urban setting (n = 94). The forest habitat was significantly associated with an increase in AgeAccel (p = 2.4 × 10–8, Fig. 5c) and EEAA (p = 5.9 × 10–11, Fig. 5g), but no difference was found for IEAA (p = 0.11, Fig. 5e). Further, no significant difference could be observed between AGR and RHG when focusing on participants living in the rainforest, suggesting greater importance of environment over genetic differences. These results are not affected by differences in genetic variants between RHG and AGR as can be seen from a robustness analysis where we removed CpG probes containing genetic variants at a frequency higher than 1 % in the populations studied (Fig. 5h, i).
Sex effects in blood and saliva
We explored whether differences exist between men and women in epigenetic aging rates. According to measures of IEAA, men are older than women in two racial/ethnic groups: African Americans (Additional file 8A, B) and Caucasians (Additional file 9A, B, N, Z).
Overall, men have higher IEAA and EEAA than women even when controlling for education, diabetes, and hypertension (Table 5). Using saliva data from PEG, we find that Hispanic men age faster than Hispanic women (p = 0.021, Fig. 6j). According to EEAA, Caucasian men are epigenetically older than Caucasian women (Additional file 9C, O, ZA), but we do not observe a significant difference in other groups such as African Americans (Additional file 8C) or central African populations (Fig. 6p, q). The results for EEAA are also consistent with significant sex differences in blood cell counts suggesting more rapid immunosenescence in men. Men have fewer naïve CD4+ T cells than women in three racial/ethnic groups: Caucasians (p = 0.0015 in the Bogalusa study, p = 0.051 in PEG, p = 4.2 × 10–5 in dataset 5); Tsimane (p = 0.0088 in older Tsimane); and African Americans (p = 0.011 in the Bogalusa study).

Sex effect on epigenetic age acceleration in blood and saliva. Panels of the first two rows (a-j) and last two rows (k-s) relate sex to intrinsic and extrinsic epigenetic age acceleration, respectively. Results are reported for blood tissue in all but one panel (j). The combined results across all blood studies can be found in panels (i) IEAA, (s) EEAA. Each bar plot reports 1 standard error and a Kruskal–Wallis test
Sex effects in brain tissue
We analyzed the effect of sex on the universal measure of age acceleration (Age Accel.) in six independent brain datasets (Table 2 and “Methods”). In total, we analyzed 2287 brain samples from 1370 participants. In our analysis, we distinguished the cerebellum from other brain regions because it is known to age more slowly than other brain regions according to the epigenetic clock [43]. While sex did not have a significant effect on the epigenetic age of the cerebellum (Fig. 7a), we found that other brain regions from men exhibit a significantly higher age acceleration than those from women (Fig. 7b, meta-analysis p = 3.1 × 10–5).

Effect of sex on the epigenetic age of brain tissue. Each panel depicts a forest plot resulting from the meta-analysis of sex effects. Each row in a forest plot shows the mean difference in epigenetic age between men and women and a 95 % confidence interval. To combine the coefficient estimates from the respective studies into a single estimate, we applied a fixed-effects model weighted by inverse variance, which is implemented in the metafor R package [89]. a Gender did not have a significant effect on the epigenetic age of the cerebellum, which is known to age more slowly than other brain regions according to the epigenetic clock [43]. b When excluding cerebellar samples from the analysis, we find that male brain regions exhibit a significantly higher age acceleration than female brain regions (mean difference = 0.82, meta-analysis p = 3.1 × 10–5). The difference remains significant even after adjusting for intra-subject correlations using a linear mixed effects model (mean difference = 0.77, p = 0.0034)
Studies of young participants
So far, our results have largely pertained to participants who are middle-aged or older (Table 1, column 6) as we only had access to two datasets involving newborns, infants, children, adolescents, and/or young adults. In dataset 6 (which involved participants between the ages of 2 and 35 years), we did not observe a significant difference epigenetic aging rates between Caucasians and Tsimane. In cord blood samples [44], we found no significant difference in the epigenetic ages of cord blood samples between African American and Caucasian newborns (p = 0.23).
Robustness analysis in the WHI
The epigenetic clock involves 47 CpGs whose broadly defined neighborhood includes a single nucleotide polymorphism (SNP) marker according to the probe annotation file from the Illumina 450 K array. Thus, genetic differences coupled with differences in hybridization efficiency could give rise to spurious differences between different racial/ethnic groups.
We addressed this concern in multiple ways. First, we re-analyzed the WHI data by removing the 47 CpGs (out of 353 epigenetic clock CpGs) from the analysis. The epigenetic clock software imputes the 47 missing CpGs using a constant value (the mean value observed in the original training set). Using the resulting modified epigenetic clock, we validate our findings of racial/ethnic differences in terms of IEAA and EEAA (Additional file 8A–C). However, this type of robustness analysis is limited because the removal of a subset of DNA methylation probes, potentially influenced by proximal genetic variation, is not as good a control as directly having matched genetic data. Second, we used a completely independent epigenetic biomarker based on a published signature of age-related CpGs from Teschendorff et al. [13]. Again, these results corroborate our findings (Additional file 8D, E). Third, we validated our findings using the original blood-based aging measure by Hannum [19] (Additional file 8F, G). Fourth, we highlight that both the Horvath and Hannum age estimators were developed based on training data from mixed populations. The training data underlying the Horvath clock involved four racial/ethnic groups (mainly Caucasians, Hispanics, African Americans, and to a lesser extent East Asians). The Hannum clock was trained on Caucasians and Hispanics. While race/ethnicity can lead to a significant offset between DNAm age and chronological age (which is interpreted as age acceleration), these two variables are highly correlated in all racial/ethnic groups.
Discussion
Our main findings are that: (1) Hispanics and Tsimane have a lower intrinsic but a higher extrinsic aging rate than Caucasians; (2) African Americans have a lower extrinsic epigenetic aging rate than Caucasians and Hispanics; (3) levels of education are associated with a decreased level of EEAA in each race/ethnic group (Additional file 4); (4) neither intrinsic nor extrinsic aging rates of blood tissue are predictive of incident CHD in the WHI even though EEAA is weakly associated with several cardiometabolic risk factors of CHD (such as hypertension, triglycerides, and CRP); (5) men exhibit higher epigenetic aging rates than women in blood, saliva, and brain samples, and (6) the rain forest habitat is significantly associated with extrinsic age acceleration but not with intrinsic age acceleration in African populations. Although precise understanding of the significance of epigenetic aging measures awaits further elaboration, our principal findings may provide additional context towards resolving several controversial, epidemiological paradoxes, including the Hispanic paradox, black–white mortality cross-over, the Tsimane inflammation paradox, and the sex morbidity–mortality paradox.
Hispanic paradox
The lower level of IEAA in Hispanics echo the finding that Hispanics in the US have a lower overall risk of mortality than Caucasians despite having a disadvantaged risk profile [45–48]. Our findings stratified by country of birth suggest that the lower intrinsic aging rate of Hispanics does not reflect biases arising through immigration such as a “healthy immigrant effect” (Additional file 3). Our finding regarding higher levels of EEAA in Hispanics parallels the findings that Hispanics have higher levels of metabolic/inflammatory risk profiles [49] and that Hispanics have a lower relative CD4+ T cell percentage than Caucasians [50]. Several articles have explored the question of why the immune system of Hispanics might differ from that of Caucasians [51–53].
Black–white mortality cross-over
In the US, the black–white mortality cross-over refers to the reported pattern of lower mortality after the age of 85 years among black men and women, compared to whites, despite their higher observed mortality rates at younger ages [54–57]. Although we find no differences in IEAA between African Americans and Caucasians at younger ages, older African American adults from the Bogalusa study had lower IEAA than their Caucasian counterparts. This finding might reflect selective survival of more robust individuals or other aspects of health and systemic risk given its independence from common risk factors for cardiovascular disease and type II diabetes mellitus. Our finding regarding the lower EEAA of African Americans, compared to Caucasians, is consistent with the longer leukocyte telomere lengths of African Americans relative to those of Caucasians [3, 9]. Lastly, our flow cytometric data show that African Americans have a larger number of naïve CD8+ T cells than Caucasians (Fig. 4t).
Tsimane inflammation paradox
Our results regarding the low intrinsic aging rate in Tsimane may help address another paradox (which we refer to as the Tsimane inflammation paradox), wherein high levels of inflammation and infection, and low HDL levels, are not associated with accelerated cardiovascular aging [39]. The finding that Tsimane have decreased levels of IEAA has parallels to the following clinical/epidemiological observations: even older Tsimane show little evidence of chronic diseases common in high-income countries, like diabetes, atherosclerosis, asthma, and other autoimmune disorders [39]. High levels of physical activity are maintained well into late adulthood [58].
The finding that Tsimane have increased levels of EEAA has parallels to the following observation: a lifetime of diverse pathogen stresses, elevated inflammation and extensive immune activation, seems to lead to more rapid depletion of naïve CD4+ T cells and greater expression of exhausted T cells, i.e. more rapid immunosenescence [39, 40, 59]. Infectious disease and high chronic inflammatory load contribute to the low life expectancy of Tsimane, 43.5 years at birth during the period 1950–1989, and 54.1 years during 1990–2002 [40, 60].
Sex morbidity–mortality paradox
The sex morbidity–mortality paradox was first described in the 1970s and refers to the observation that women possess a lower age-adjusted mortality rate compared to men despite a higher suffering from a higher burden of co-morbid conditions [61, 62]. Most explanations focus on differences in lifestyle behaviors or healthcare utilization. However, marked sex differences in health and disability remain after controlling for differences in work-related behavior, smoking, obesity, and other behaviors [63]. Whereas other explanations attest to sex differences in a variety of biomarkers, our epigenetic aging markers show robust and consistent male-biased vulnerability in multiple tissues (blood, brain, and saliva) in all racial groups. Similar sex differences in blood-based epigenetic aging rates have also been reported in minors and teenagers [64].
Strengths and limitations
Our study has several strengths including the analysis of 18 DNA methylation datasets (Tables 1 and 2), large sample sizes (almost 6000 samples), multiple tissues (blood, saliva, brain), access to unique populations (Tsimane Amerindians; rainforest hunter-gatherers and farmers), two flow cytometric studies, and robust epigenetic biomarkers of aging. Our analysis of race/ethnicity also spanned seven different racial/ethnic groups (African American, Caucasian, Hispanic, Tsimane, East Asian, RHGs, and AGRs from Central Africa). Another strength is that our analysis of race/ethnicity involved two sources of DNA: blood and saliva. Limitations include the use of some datasets that are cross-sectional as opposed to longitudinal datasets and the fact that both IEAA and EEAA rely on imputed blood cell counts based on DNA methylation levels. Fortunately, the imputed blood cell counts are quite accurate (Additional file 2). Our results reported here concerning ethnic/racial differences in blood cell counts are supported both by our two flow cytometric datasets and by the literature. However, these measured data are not fully reflective of the breakdown of blood cell types, representing only T and B cells.
Conclusion
Our exploratory study demonstrates that epigenetic aging rates differ between different racial/ethnic groups and between men and women. Further, intrinsic epigenetic aging rates tend to have insignificant associations with well-studied risk factors of CHD whereas extrinsic aging rates tend to have significant (but weak) associations with several pro-inflammatory risk factors. While racial/ethnic differences have previously been observed in DNA methylation levels [44], we are the first to directly compare epigenetic aging rates across different racial/ethnic groups. Our derived intrinsic and extrinsic epigenetic aging rates in blood offer an independent glimpse into biological aging that incorporates genetics and the environment and provides potential insight into a number of epidemiological paradoxes. The application of genome-wide DNAm-based epigenetic analysis to understand race/ethnic and sex disparities in biological aging is novel and offers an important perspective that complements existing approaches based on other biomarkers. Future studies will need to confirm our findings with longitudinal designs and to extend the epigenetic age analysis to other tissues and organs.
Methods
We differentiate groups according to “race/ethnicity,” mindful about existing controversies over rigid racial definitions. Our use of these terms reflects self-identified group membership based on macro-categories commonly employed in censuses, human genetics, demography, and epidemiology. The term race/ethnicity thus combines elements of genetic ancestry, population history, and culture.
DNA methylation age and epigenetic clock
All of the described epigenetic measures of aging and age acceleration are implemented in our freely available software. The epigenetic clock is defined as a prediction method of age based on the DNAm levels of 353 CpGs. Predicted age, referred to as DNAm age, correlates with chronological age in sorted cell types (CD4+ T cells, monocytes, B cells, glial cells, neurons), tissues, and organs, including: whole blood, brain, breast, kidney, liver, lung, saliva [20]. Mathematical details and software tutorials for the epigenetic clock can be found in the Additional files of [20]. An online age calculator can be found at our webpage (https://dnamage.genetics.ucla.edu).
Intrinsic versus extrinsic measures of epigenetic age acceleration in blood
Empirical studies show that DNAm has a relatively weak correlation with various measures of white blood cell counts [31], which probably reflects the fact that dozens of different tissue and blood cell types were used to define DNAm age. However, we find it useful to explicitly define another measure of age acceleration that is completely independent of blood cell counts as described in the following. We distinguish intrinsic from extrinsic measures of epigenetic age acceleration in whole blood according to their relationship with blood cell counts. A measure of intrinsic epigenetic age acceleration (IEAA) measures “pure” epigenetic aging effects that are not confounded by differences in blood cell counts. Our measure of IEAA is defined as the residual resulting from a multivariate regression model of DNAm age on chronological age and various blood immune cell counts (naïve CD8+ T cells, exhausted CD8+ T cells, plasma B cells, CD4+ T cells, natural killer cells, monocytes, and granulocytes). The measure of IEAA is an incomplete measure of the age-related functional decline of the immune system because it does not track age-related changes in blood cell composition, such as the decrease of naïve CD8+ T cells and the increase in memory or exhausted CD8+ T cells [36–38].
We defined a measure of EEAA that only applies to whole blood and aims to measure epigenetic aging in immune-related components in two steps. First, we formed a weighted average of the epigenetic age measure from Hannum et al. [19] and three estimated measures of blood cells for cell types that are known to change with age: naïve (CD45RA + CCR7+) cytotoxic T cells; exhausted (CD28-CD45RA-) cytotoxic T cells; and plasma B cells using the approach by Klemera Doubal [65]. Second, we defined the measure of EEAA as the residual resulting from a univariate model that regressed the weighted average on chronological age. By definition, our measure of EEAA has a positive correlation with the amount of exhausted CD8+ T cells and plasmablast cells and a negative correlation with the amount of naïve CD8+ T cells. Blood cell counts were estimated based on DNA methylation data. EEAA tracks both age-related changes in blood cell composition and intrinsic epigenetic changes. In most blood datasets, EEAA has a moderate correlation (r = 0.5) with IEAA. We note that, by definition, none of our three measures of epigenetic age acceleration are associated with the chronological age of the participant at the time of blood draw.
Relationship to mortality prediction
Although the epigenetic clock method was only published in 2013, there is already a rich body of literature that shows that it relates to biological age. Using four human cohort studies, we previously demonstrated that both the Horvath and Hannum epigenetic clocks are predictive of all-cause mortality [23]. Published results in Marioni et al. [23] show that DNAm age adjusted for blood cell counts (i.e. IEAA) is prognostic of mortality in four cohort studies. We recently expanded our original analysis by analyzing 13 different cohorts (including three racial/ethnic groups) and by evaluating the prognostic utility of both IEAA and EEAA. All considered measures of epigenetic age acceleration were predictive of age at death in univariate Cox models (pAgeAccel = 1.9 × 10–11, pIEAA = 8.2 × 10–9, pEEAA = 7.5 × 10–43) and multivariate Cox models adjusting for risk factors and pre-existing disease status (pAgeAccel = 5.4 × 10–5, pIEAA = 5.0 × 10–4, pEEAA = 3.4 × 10–19) where the latter adjusted for chronological age, body mass index, education, alcohol, smoking pack years, recreational physical activity, and prior history of disease (diabetes, cancer, hypertension). These results will be published elsewhere. Further, the offspring of centenarians age more slowly than age matched controls according to Age Accel and IEAA [26] which strongly suggests that these measures relate to heritable components of biological age. Two independent research groups have shown that epigenetic age acceleration predicts mortality [24, 25].
Description of the blood datasets listed in Table 1
All data presented in this article have been made publicly available as indicated in the column “Available” of Table 1.
Dataset 1: Women’s Health Initiative (WHI)
Participants included a subsample of participants of the WHI study, a national study that began in 1993 which enrolled postmenopausal women between the ages of 50 and 79 years into either one of two three randomized clinical trials [66]. None of these women had CHD at baseline but about half of these women had developed CHD by 2010. Women were selected from one of two WHI large subcohorts that had previously undergone genome-wide genotyping as well as profiling for seven cardiovascular disease related biomarkers including total cholesterol, HDL, LDL, triglycerides, CRP, creatinine, insulin, and glucose through two core WHI ancillary studies [67]. The first cohort is the WHI SNP Health Association Resource (SHARe) cohort of minorities that includes >8000 African American women and >3500 Hispanic women. These women were genotyped through WHI core study M5-SHARe (www.whi.org/researchers/data/WHIStudies/StudySites/M5) and underwent biomarker profile through WHI Core study W54-SHARe (…data/WHIStudies/StudySites/W54). The second cohort consists of a combination of European Americans from the two Hormonal Therapy trials selected for GWAS and biomarkers in core studies W58 (…/data /WHIStudies/StudySites/W58) and W63 (…/data/WHIStudies/StudySites/W63). From these two cohorts, two sample sets were formed. The first (sample set 1) is a sample set of 637 CHD cases and 631 non-CHD cases as of 30 September 2010. The second sample set (sample set 2) is a non-overlapping sample of 432 cases of CHD and 472 non-cases as of 17 September 2012. The ethnic groups differed in terms of the age distribution in the sense that Caucasian women tended to be older. Therefore, we randomly removed 80 % of the Caucasian women who were older than 65 years when it came to the direct comparisons reported in our figures. This resulted in a total sample size of 1462 women, comprising 673 African Americans, 353 Caucasians, and 433 Hispanics. There was no significant difference in age between the three ethnic groups. However, we kept all of the samples in our analysis of clinical characteristics, such as future CHD status and baseline characteristics such as education, hypertension, diabetes, and smoking, in order to ensure that sufficient sample sizes were available for these analyses. Our results are highly robust with respect to using the smaller or larger versions of the datasets. All results are qualitatively the same for the two versions of the datasets. We acknowledge a potential for selection bias using the above-described sampling scheme in WHI but suspect if such bias is present it is minimal. First, some selection bias is introduced by restricting our methylation profiling at baseline to women with GWAS and biomarker data from baseline as well, given the requirement that these participants must have signed the WHI supplemental consent for broad sharing of genetic data in 2005. However, we believe that selection bias at this stage is minimized by the inclusion of participants who died between the time of the start of the WHI study and the time of supplemental consent in 2005, which resulted in the exclusion of only ~6–8 % of all WHI participants. Nevertheless, participants unable or unwilling to sign consent in 2005 may not represent a random subset of all participants who survived to 2005. Second, some selection bias may also occur if similar gross differences exist in the characteristics of participants who consented to be followed in the two WHI extension studies beginning in 2005 and 2010 compared to non-participants at each stage. We believe these selection biases if present have minimal effects on our effect estimates. Data are available from the page https://www.whi.org/researchers/Stories/June%202015%20WHI%20Investigators'%20Datasets%20Released.aspx, see the link https://www.whi.org/researchers/data/Documents/WHI%20Data%20Preparation%20and%20Use.pdf.
Dataset 2: Bogalusa
We analyzed the blood DNA methylation levels of 968 participants (680 Caucasians, 288 African Americans; age range = 28–51.3 years) from the Bogalusa Heart study [68] who were examined in Bogalusa, Louisiana during 2006–2010 for cardiovascular risk factors. All participants in this study gave informed consent at each examination. Study protocols were approved by the Institutional Review Board (IRB reference no. 12-395283) of the Tulane University Health Sciences Center. DNA was extracted from 1106 whole blood samples using the PureLink Pro 96 Genomic DNA Kit (LifeTechnology, CA, USA) following the manufacturer’s instructions. The Infinium HumanMethylation450 BeadChip (Methy450K) was used for whole genome DNA methylation analysis.
All the samples were processed at the Microarray Core Facility, University of Texas Southwestern Medical Center at Dallas, Texas. For DNA methylation analysis, 750 ng genomic DNA from each participant was bisulphite converted using the EZ-96 DNA Methylation Kit (Zymo Research, CA, USA) and the efficiency of the bisulphite conversion was confirmed by built-in controls on the Methy450K array. The methylation profile of each individual was measured by processing 4 μL of bisulphite-converted DNA, at a concentration of 50 ng/μL, on a Methy450K array. The bisulphite-converted DNA was amplified, fragmented, and hybridized to the array. The arrays were scanned on an Illumina HiScan scanner and the raw methylation data were extracted using Illumina’s Genome Studio methylation module. Data cleaning procedures were undertaken using R package “minfi” [69], generating quality control report, finding sample outliers, cell counts estimation, and annotation accessing. The R package wateRmelon [70] was used for β-value normalization and quality control. For correction of systematic technical biases in the 450 K assay, β-value normalization was performed by the “dasen” function, in which type I and type II intensities and methylated and unmethylated intensities will be quantile normalized separately after backgrounds equalization of type I and type II. The R package ChAMP [71] was used for batch effect analysis and correction with “champ.SVD” and “champ.runCombat” functions. The clinical variables and participant characteristics are defined in the captions of the respective Additional files.
The are available from https://biolincc.nhlbi.nih.gov/studies/bhs/.
Dataset 3: blood from Hispanics and Caucasians of PEG
The Parkinson’s disease, Environment, and Genes (PEG) case-control study aims to identify environmental risk factors (e.g. neurotoxic pesticide exposures) for Parkinson’s disease.
The PEG study is a large population-based study of Parkinson’s disease of mostly rural and township residents of California’s central valley [72]. Here we only used diseased participants from wave 1 (PEG1). Since all participants of dataset 3 had Parkinson’s disease, disease status could not confound associations with epigenetic aging. Medication status was not associated with epigenetic age acceleration. The data are available from Gene Expression Omnibus.
Dataset 4: saliva samples from PEG
This novel dataset comes from the PEG study (described above). Since PD disease status did not relate to epigenetic age acceleration in these data, we ignored it in the analysis. However, our findings are unchanged after incorporating PD status in a multivariate model. About half of the samples overlapped with those of dataset 3, which is why we could correlate epigenetic age acceleration between blood and saliva.
Datasets 5 and 6: blood from Tsimane, Hispanics, and Caucasians
Datasets 5 and 6, which were collected and generated in the same way, only differ in terms of the chronological ages. All participants in dataset 5 are older than 35 years while those in dataset 6 are younger or equal to 35 years. The dataset involved three different ethnic groups: Tsimane Amerindians, Hispanics living in the US, and Caucasians living in the US. Fasting whole-blood samples were collected from Tsimane via venipuncture in field villages in the vicinity of San Borja, Bolivia as a part of the annual biomedical data collection for a longitudinal project on aging during 2004–2009 (Tsimane Health and Life History Project). Manual complete blood counts were conducted using a hemocytometer, erythrocyte sedimentation rate was calculated following the Westergren method, and hemoglobin was analyzed with a QBC Autoread Plus Dry Hematology System (Drucker Diagnostics, Port Matilda, PA, USA). Specimens were stored in liquid nitrogen until transfer to the US on dry ice, where they were stored at –80 °C. All participants provided written and informed consent; study protocols and procedures were approved at the individual, village, and Tsimane government level, as well as by the University of California, Santa Barbara and University of New Mexico Institutional Review Boards (IRB Reference numbers 14-0604 and 07-157, respectively). Specimens were shipped on dry ice to the University of Southern California for extraction. The same core facility provided blood samples that were collected at the same time and stored in the same condition as Hispanic participants living in the US. The DNA samples from all participants (Caucasians, Hispanics, Tsimane) were randomized across the Illumina chips to avoid confounding due to chip effects. For our age prediction analysis, we used background corrected beta values resulting from Genome Studio.
Hispanics for datasets 5 + 6: Participant recruitment: Participation in the BetaGene study was restricted to Mexican Americans from families of a proband with gestational diabetes mellitus (GDM) diagnosed within the previous 5 years. Probands were identified from the patient populations at Los Angeles County/USC Medical Center, OB/GYN clinics at local hospitals, and the Kaiser Permanente health plan membership in Southern California. Probands qualified for participation if they: (1) were of Mexican ancestry (defined as both parents and ≥3/4 of grandparents Mexican or of Mexican descent); (2) had a confirmed diagnosis of GDM within the previous 5 years; (3) had glucose levels associated with poor pancreatic β-cell function and a high risk of diabetes when not pregnant; and (4) had no evidence of β-cell autoimmunity by GAD-65 antibody testing. Recruitment targeted two general family structures using siblings and/or first cousins of GDM probands, all with fasting glucose levels <126 mg/dl (7 mM): (1) at least two siblings and three first cousins from a single nuclear family; or (2) at least five siblings available for study. Using information from the proband to determine preliminary eligibility, siblings and first cousins were invited to participate in screening and, if eligible, detailed phenotyping (below) and collection of DNA. Available parents and connecting uncles and aunts were asked to provide DNA and had a fasting glucose determination. In addition, women of Mexican ancestry who have gone through pregnancy without GDM, as evidenced by a plasma or serum glucose level <120 mg/dl after a 50 g oral glucose screen for GDM, were also collected. Recruitment criteria for control probands were similar to that of the GDM probands, but were also selected to be age, BMI, and parity-matched to the GDM probands. Unrelated samples for the present methylation analysis were selected randomly from all BetaGene participants. The BetaGene protocol (HS-06-00045) has been approved by the Institutional Review Boards of the USC Keck School of Medicine.
Dataset 7: blood from East Asians and Caucasians
Here we downloaded the publicly available DNA methylation data from GSE53740 [73]. Since we found that progressive supranuclear palsy (PSP) had a significant effect on epigenetic age acceleration, we removed PSP samples from the analysis. Further, we focused on comparing East Asians to Caucasians since other racial/ethnic groups were represented by fewer than 10 samples.
Dataset 8: blood from African populations
We used blood methylation data from [42]. We studied peripheral whole-blood DNA from a total of 256 samples (for which the chronological age at the time of blood draw was available).
As detailed in Fagny et al. [42], the samples come from seven populations located across the Central African belt. These populations can be divided into two main groups: RHG populations, historically known as “pygmies,” who have traditionally relied on the equatorial forest for subsistence and who live close to, or within, the forest; and AGR populations, living either in rural/urban deforested regions or in forested habitats in which they practice slash-and-burn agriculture. Informed consent was obtained from all participants and from both parents of any participants under the age of 18 years. Ethical approval for this study was obtained from the institutional review boards of Institut Pasteur, France (RBM 2008-06 and 2011-54/IRB/3).
Dataset 9: cord blood samples from African Americans and Caucasians
These 216 cord blood samples from 92 African American and 70 Caucasian participants come from a study that described racial differences in DNA methylation levels [44].
Datasets 10 and 11
Saliva samples from Caucasians and Hispanics. The data were generated by splitting the data from [74] by sex, which reflected the use of these data in the development of the epigenetic clock software [20]. Note that these data were generated on the older Illumina platform (27 K array). Some of the data were used as training data in the development of the epigenetic clock, which might bias the results. By contrast, the novel saliva data from PEG (dataset 4) provide an unbiased analysis.
Dataset 12: lymphoblastoid cell lines from Han Chinese, African Americans, and Caucasians
We clustered the samples based on the interarray correlation. Since 51 samples were very distinct from the remaining samples, they were removed as potential outliers. Disease status did not affect the estimates of DNAm age, which is why we ignored it.
Description of brain datasets
We collected brain datasets from six independent studies to assess gender effect on epigenetic age acceleration. We focused on Caucasian samples since there were insufficient numbers of other racial/ethnic groups.
-
Study 1: brain DNA methylation data from a study of Alzheimer’s disease study from [75], GEO accession GSE59685. DNA methylation profiles of the cerebellum, entorhinal cortex, prefrontal cortex, and superior temporal gyrus were available from 117 individuals. We ignored disease status since it was not associated with age acceleration.
-
Study 2: brain DNA methylation data from neurologically normal participants from [76], GEO accession GSE15745. DNA methylation data of the cerebellum, frontal cortex, pons, and temporal cortex regions from up to 148 neurologically normal participants of European ancestry [76].
-
Study 3: cerebellar DNA methylation data from [77], GEO GSE38873. DNA methylation data from the cerebellum of 147 participants from a case-control study (121 cases/32 controls) of psychiatric disorders. Since disease status did not affect DNAm age, we ignored it.
-
Study 4: prefrontal cortex samples from [78], GEO GSE61431. We analyzed 37 Caucasian participants (European ancestry).
-
Study 5: frontal cortex and cerebellum from neurologically normal Caucasian participants from [79]. The DNA methylation data and corresponding SNP data can be found in dbGAP, http://www.ncbi.nlm.nih.gov/gap (accession: phs000249.v2.p1). We only analyzed 209 Caucasian participants who met our stringent quality control criteria. We excluded several putative outliers from the original dataset including three individuals who were genotyped on a different platform, six participants who were outliers according to a genetic analysis (PC plot), and 13 participants who had the wrong gender according to the gender prediction algorithm of the epigenetic clock software.
-
Study 6: dorsolateral prefrontal cortex samples from 718 Caucasian participants from the Religious Order Study (ROS) and the Memory and Aging Project (MAP). The DNA methylation data are available at the following webpage https://www.synapse.org/#!Synapse:syn3168763. We focused on brain samples of Caucasian participants from these two prospective cohort studies of aging that include brain donation at the time of death [80]. Additional details on the DNA methylation data can be found in [81]. We were not able to evaluate the effect of race/ethnicity on epigenetic age acceleration since the dataset contained only 12 Hispanic samples (which did not differ significantly from Caucasians in terms of epigenetic age). Further, we found no association between disease status and epigenetic age acceleration, which is why we ignored disease status in our analysis.
Preprocessing of Illumina Infinium 450 K arrays
In brief, bisulfite conversion using the Zymo EZ DNA Methylation Kit (ZymoResearch, Orange, CA, USA) as well as subsequent hybridization of the HumanMethylation450k Bead Chip (Illumina, San Diego, CA, USA), and scanning (iScan, Illumina) were performed according to the manufacturers’ protocols by applying standard settings. DNA methylation levels (β values) were determined by calculating the ratio of intensities between methylated (signal A) and unmethylated (signal B) sites. Specifically, the β value was calculated from the intensity of the methylated (M corresponding to signal A) and unmethylated (U corresponding to signal B) sites, as the ratio of fluorescent signals β = Max(M,0)/[Max(M,0) + Max(U,0) + 100]. Thus, β values range from 0 (completely unmethylated) to 1 (completely methylated) [82]. The epigenetic clock software implements a data normalization step that repurposes the BMIQ normalization method from Teschendorff [83] so that it automatically references each sample to a gold standard based on type II probes as detailed in [20].
Estimating blood cell counts based on DNA methylation levels
We estimate blood cell proportions using two different software tools. Houseman’s estimation method [84], which is based on DNA methylation signatures from purified leukocyte samples, was used to estimate the proportions of cytotoxic (CD8+) T cells, helper (CD4+) T, natural killer, B cells, and granulocytes. The software does not allow us to identify the type of granulocytes in blood (neutrophil, eosinophil, or basophil) but we note that neutrophils tend to be the most abundant granulocyte (~60 % of all blood cells compared with 0.5–2.5 % for eosinophils and basophils). The advanced analysis option of the epigenetic clock software [20] was used to estimate the percentage of exhausted CD8+ T cells (defined as CD28-CD45RA-) and the number (count) of naïve CD8+ T cells (defined as (CD45RA + CCR7+) as described in [31].
Flow cytometric data from the Long Life Study of the WHI
While our DNA methylation data from the WHI were assessed at baseline, the flow cytometric data were measured 14.6 years after baseline. Between March 2012 and May 2013, a subset of WHI participants were enrolled in the Long Life Study (LLS) and additional biospecimens, physiometric, and questionnaire data were collected. All surviving Hormone Trial participants followed through 2010 and all African American and Hispanic/Latino participants from the SNP Health Association Resource (WHI-SHARe) sub-cohort were included if CVD biomarker from WHI baseline exam and genome-wide genotyping (GWAS) data were available and if they were at least 63 years old by 1 January 2012. Women who were either unable to provide informed consent (e.g. dementia) or those residing in an institution (e.g. skilled nursing facility) were excluded. Of a total of 14,081 eligible WHI participants, 9242 women consented to participate, 7875 were enrolled, and 7481 underwent successful blood draws. Blood was collected at locations across the US using a standardized protocol between March 2012 and May 2013 (Examination Management Services, Inc.) Fresh peripheral blood samples were packaged in Styrofoam with cold packs and were sent overnight to a central testing facility in Seattle.
A random sample of 600 residual fresh peripheral blood specimens (single tube, following CBC analysis) was transported to the University of Washington Medical Center’s (UWMC’s) flow cytometry laboratory and high-sensitivity, multi-parameter flow cytometry was performed utilizing a modified four-laser, multi-color Becton-Dickinson (BD; San Jose, CA, USA) LSRII flow cytometer. All of the flow cytometry studies were performed within 72 h of sample collection between June 2012 and February 2013. A single tube was used to evaluate T lymphocyte subsets: CD45 (KO), CD8 (BV), CD45RA (F), CCR7 (PE), CD5 (ECD), CD56 (PC5), CD3 (APC-H7), CD4 (A594), CD28 (APC), CD27 (PC7). A second tube evaluated B lymphocyte subsets: CD45 (APC-H7), CD20 (V450), kappa (F), lambda (PE), CD23 (ECD), CD5 (PC5.5), CD19 (BV650), CD38 (A594), CD10 (APC), CD27 (PC7), CD3 (APC-A700). Categories of circulating cells were quantified using a predefined population-based gating strategy based on established gating strategies for both T lymphocyte [85] and B lymphocyte [86] subsets.
Flow cytometric data from the MACS cohort
As part of Additional file 2, we validated imputed blood cell counts using flow cytometric data and DNA methylation data collected from men of the Multi-Center AIDS Cohort Study (MACS). The data were generated as described in [87]. Briefly, human peripheral blood mononuclear cell (PBMC) samples were isolated from fresh blood samples and either stained for flow cytometry analysis or used for genomic DNA isolation. DNA was isolated from 1 × 106 PBMC using Qiagen DNeasy blood and tissue mini spin columns. Quality of DNA samples was assessed using Nanodrop measurements and accurate DNA concentrations were measured using a Qubit assay kit (Life Technology). Cryopreserved PBMC obtained from the repository were thawed and assayed for viability using trypan blue. The mean viability of the samples was 88 %. Samples were stained for 30 min at 4 °C with the following antibody combinations of fluorescently conjugated monoclonal antibodies using the manufacturers recommended amounts for 1 million cells: tube 1: CD57 FITC (clone HNK-1), CD28 phycoerythrin (PE, L293), CD3 peridinin chlorophyll protein (PerCP,SK7), CD45RA phycoerythrin cyanine dye Cy7 tandem (PE-Cy7, L48), CCR7 Alexa Fluor 647 (AF647, 150503), CD8 allophycocyanin H7- tandem (APC-H7, SK1) and CD4 horizon V450 (V450, RPA-T4); tube 2: HLA-DR FITC (L243), CD38 PE (HB7), CD3 PercP, CD45RO PE-Cy7 (UCHL-1), CD95-APC(DXZ), CD8 APC-H7, and CD4 V450); tube 3: CD38 FITC (HB7), IgD PE (1A6–2), CD3 PerCP, CD10 PE-Cy7 (HI10a), CD27 APC (eBioscience, clone 0323, San Diego, CA), CD19 APC-H7 (SJ25C1) and CD20 V450 (L27). Antibodies were purchased from BD Biosciences, San Jose, CA (BD) except as noted. Stained samples were washed twice with staining buffer and run immediately on an LSR2 cytometer equipped with a UV laser (BD, San Jose, CA, USA) for the detection of 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) which was used as a viability marker at a final concentration of 0.1 ug/mL. Lineage gated isotype controls to measure non-specific binding were run and used CD3, CD4, and CD8 for T-cells or CD19 for B-cells. Fluorescence minus one controls (FMO) were also utilized to assist gating and cursor setting. A range of 20,000–100,000 lymphocytes were acquired and analyzed per sample using the FACSDiva software package (BD, San Jose, CA, USA).
References
Levine M, Crimmins E. Evidence of accelerated aging among African Americans and its implications for mortality. Soc Sci Med. 2014;118:27–32.
Diez Roux AV, Ranjit N, Jenny NS, Shea S, Cushman M, Fitzpatrick A, Seeman T. Race/ethnicity and telomere length in the Multi-Ethnic Study of Atherosclerosis. Aging Cell. 2009;8:251–7.
Rewak M, Buka S, Prescott J, De Vivo I, Loucks EB, Kawachi I, Non AL, Kubzansky LD. Race-related health disparities and biological aging: does rate of telomere shortening differ across blacks and whites? Biol Psychol. 2014;99:92–9.
Crimmins EM, Kim JK, Seeman TE. Poverty and biological risk: the earlier “aging” of the poor. J Gerontol A Biol Sci Med Sci. 2009;64:286–92.
Arbeev KG, Butov AA, Manton KG, Sannikov IA, Yashin AI. Disability trends in gender and race groups of early retirement ages in the USA. Soz Praventivmed. 2004;49:142–51.
Ingram DK, Nakamura E, Smucny D, Roth GS, Lane MA. Strategy for identifying biomarkers of aging in long-lived species. Exp Gerontol. 2001;36:1025–34.
Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol. 1978;120:33–53.
Lin J, Epel E, Cheon J, Kroenke C, Sinclair E, Bigos M, et al. Analyses and comparisons of telomerase activity and telomere length in human T and B cells: insights for epidemiology of telomere maintenance. J Immunol Methods. 2010;352:71–80.
Hunt SC, Chen W, Gardner JP, Kimura M, Srinivasan SR, Eckfeldt JH, Berenson GS, Aviv A. Leukocyte telomeres are longer in African Americans than in whites: the National Heart, Lung, and Blood Institute Family Heart Study and the Bogalusa Heart Study. Aging Cell. 2008;7:451–8.
Christensen B, Houseman E, Marsit C, Zheng S, Wrensch M, Wiemels J, Nelson H, Karagas M, Padbury J, Bueno R et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602.
Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–9.
Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6.
Vivithanaporn P, Heo G, Gamble J, Krentz H, Hoke A, Gill M, Leistung C. Neurologic disease burden in treated HIV/AIDS predicts survival. Neurology. 2010;75:1150–8.
Horvath S, Zhang Y, Langfelder P, Kahn R, Boks M, van Eijk K, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97.
Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22:623–32.
Johansson A, Enroth S, Gyllensten U. Continuous aging of the human DNA methylome throughout the human lifespan. PLoS One. 2013;8:e67378.
Day K, Waite L, Thalacker-Mercer A, West A, Bamman M, Brooks J, Myers R, Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14:R102.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Lin Q, Weidner CI, Costa IG, Marioni RE, Ferreira MRP, Deary IJ. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging. 2016;8:394–401.
Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CC, O’Donovan MC, Bray NJ, Mill J. Methylomic trajectories across human fetal brain development. Genome Res. 2015;25:338–52.
Marioni R, Shah S, McRae A, Chen B, Colicino E, Harris S, Gibson J, Henders A, Redmond P, Cox S et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25.
Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016;15:149–54.
Perna L, Zhang Y, Mons U, Holleczek B, Saum K-U, Brenner H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics. 2016;8:1–7.
Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, Mari D, Arosio B, Monti D, Passarino G, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY). 2015;7:1159–70.
Breitling LP, Saum KU, Perna L, Schöttker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016;8:21.
Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 1936;2015:44.
Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, Heits N, Bell JT, Tsai P-C, Spector TD et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A. 2014;111:15538–43.
Horvath S, Garagnani P, Bacalini M, Pirazzini C, Salvioli S, Gentilini D, DiBlasio A, Giuliani C, Tung S, Vinters H, Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015;14:491–5.
Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015;212:1563–73.
Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015;7:1130–42.
Levine M, Lu A, Bennett D, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015;7:1198–211.
Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY). 2015;7:690–700.
Zannas A, Arloth J, Carrillo-Roa T, Iurato S, Roh S, Ressler K, Nemeroff C, Smith A, Bradley B, Heim C, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015;16:266.
Fagnoni FF, Vescovini R, Mazzola M, Bologna G, Nigro E, Lavagetto G, Franceschi C, Passeri M, Sansoni P. Expansion of cytotoxic CD8+ CD28- T cells in healthy ageing people, including centenarians. Immunology. 1996;88:501–7.
Fagnoni FF, Vescovini R, Passeri G, Bologna G, Pedrazzoni M, Lavagetto G, Casti A, Franceschi C, Passeri M, Sansoni P. Shortage of circulating naive CD8+ T cells provides new insights on immunodeficiency in aging. Blood. 2000;95:2860–8.
Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol. 2007;211:144–56.
Gurven M, Kaplan H, Winking J, Eid Rodriguez D, Vasunilashorn S, Kim JK, Finch C, Crimmins E. Inflammation and infection do not promote arterial aging and cardiovascular disease risk factors among lean horticulturalists. PLoS One. 2009;4:e6590.
Gurven M, Kaplan H, Winking J, Finch C, Crimmins EM. Aging and inflammation in two epidemiological worlds. J Gerontol A Biol Sci Med Sci. 2008;63:196–9.
Gurven M, Blackwell AD, Rodriguez DE, Stieglitz J, Kaplan H. Does blood pressure inevitably rise with age?: longitudinal evidence among forager-horticulturalists. Hypertension. 2012;60:25–33.
Fagny M, Patin E, MacIsaac JL, Rotival M, Flutre T, Jones MJ, Siddle KJ, Quach H, Harmant C, McEwen LM, et al. The epigenomic landscape of African rainforest hunter-gatherers and farmers. Nat Commun. 2015;6:10047.
Horvath S, Mah V, Lu AT, Woo JS, Choi OW, Jasinska AJ, Riancho JA, Tung S, Coles NS, Braun J et al. The cerebellum ages slowly according to the epigenetic clock. Aging (Albany NY). 2015;7:294–306.
Adkins RM, Krushkal J, Tylavsky FA, Thomas F. Racial differences in gene-specific DNA methylation levels are present at birth. Birth Defects Res A Clin Mol Teratol. 2011;91:728–36.
Markides KS, Coreil J. The health of Hispanics in the southwestern United States: an epidemiologic paradox. Public Health Rep. 1986;101:253–65.
Sorlie PD, Backlund E, Johnson NJ, Rogot E. Mortality by Hispanic status in the United States. JAMA. 1993;270:2464–8.
Arias E, Eschbach K, Schauman WS, Backlund EL, Sorlie PD. The Hispanic mortality advantage and ethnic misclassification on US death certificates. Am J Public Health. 2010;100 Suppl 1:S171–7.
Ruiz JM, Steffen P, Smith TB. Hispanic mortality paradox: a systematic review and meta-analysis of the longitudinal literature. Am J Public Health. 2013;103:e52–60.
Crimmins EM, Kim JK, Alley DE, Karlamangla A, Seeman T. Hispanic paradox in biological risk profiles. Am J Public Health. 2007;97:1305–10.
Tracy RP, Doyle MF, Olson NC, Huber SA, Jenny NS, Sallam R, Psaty BM, Kronmal RA. T-helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: the Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2013;2:e000117.
Contreras G, Alaez C, Murguia A, Garcia D, Flores H, Gorodezky C. Distribution of the killer cell immunoglobulin-like receptors in Mexican Mestizos. Tissue Antigens. 2007;69 Suppl 1:125–9.
Dowd JB, Aiello AE, Alley DE. Socioeconomic disparities in the seroprevalence of cytomegalovirus infection in the US population: NHANES III. Epidemiol Infect. 2009;137:58–65.
Dowd JB, Zajacova A, Aiello A. Early origins of health disparities: Burden of infection, health, and socioeconomic status in U.S. children. Soc Sci Med. 2009;68:699–707.
Johnson NE. The racial crossover in comorbidity, disability, and mortality. Demography. 2000;37:267–83.
Corti MC, Guralnik JM, Ferrucci L, Izmirlian G, Leveille SG, Pahor M, Cohen HJ, Pieper C, Havlik RJ. Evidence for a black-white crossover in all-cause and coronary heart disease mortality in an older population: the North Carolina EPESE. Am J Public Health. 1999;89:308–14.
Yao L, Robert SA. Examining the racial crossover in mortality between African American and white older adults: a multilevel survival analysis of race, individual socioeconomic status, and neighborhood socioeconomic context. J Aging Res. 2011;2011:8.
Fenelon A. An examination of black/white differences in the rate of age-related mortality increase. Demogr Res. 2013;29:441–72.
Gurven M, Jaeggi AV, Kaplan H, Cummings D. Physical activity and modernization among Bolivian Amerindians. PLoS One. 2013;8:e55679.
Blackwell AD, Trumble BC, Maldonado Suarez I, Stieglitz J, Beheim B, Snodgrass JJ, et al. Immune function in Amazonian horticulturalists. Ann Hum Biol. 2016;43:382–96.
Gurven M, Kaplan H, Supa AZ. Mortality experience of Tsimane Amerindians of Bolivia: regional variation and temporal trends. Am J Hum Biol. 2007;19:376–98.
Case A, Paxson C. Sex differences in morbidity and mortality. Demography. 2005;42:189–214.
Oksuzyan A, Juel K, Vaupel JW, Christensen K. Men: good health and high mortality. Sex differences in health and aging. Aging Clin Exp Res. 2008;20:91–102.
Crimmins EM, Kim JK, Langa KM, Weir DR. Assessment of cognition using surveys and neuropsychological assessment: the Health and Retirement Study and the Aging, Demographics, and Memory Study. J Gerontol B Psychol Sci Soc Sci. 2011;66 Suppl 1:i162–71.
Simpkin AJ, Hemani G, Suderman M, Gaunt TR, Lyttleton O, McArdle WL, et al. Prenatal and early life influences on epigenetic age in children: a study of mother-offspring pairs from two cohort studies. Hum Mol Genet. 2016;25:191–201.
Klemera P, Doubal S. A new approach to the concept and computation of biological age. Mech Ageing Dev. 2006;127:240–8.
[No authors listed]. Design of the Women’s Health Initiative clinical trial and observational study. The Women’s Health Initiative Study Group. Control Clin Trials. 1998;19:61–109.
Curb JD, McTiernan A, Heckbert SR, Kooperberg C, Stanford J, Nevitt M, Johnson KC, Proulx-Burns L, Pastore L, Criqui M, Daugherty S. Outcomes ascertainment and adjudication methods in the Women’s Health Initiative. Ann Epidemiol. 2003;13:S122–8.
Freedman DS, Newman 3rd WP, Tracy RE, Voors AE, Srinivasan SR, Webber LS, Restrepo C, Strong JP, Berenson GS. Black-white differences in aortic fatty streaks in adolescence and early adulthood: the Bogalusa Heart Study. Circulation. 1988;77:856–64.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293.
Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450 k Chip Analysis Methylation Pipeline. Bioinformatics. 2014;30:428–30.
Costello S, Cockburn M, Bronstein J, Zhang X, Ritz B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the Central Valley of California. Am J Epidemiol. 2009;169:919–26.
Li Y, Chen JA, Sears RL, Gao F, Klein ED, Karydas A, Geschwind MD, Rosen HJ, Boxer AL, Guo W, et al. An epigenetic signature in peripheral blood associated with the haplotype on 17q21.31, a risk factor for neurodegenerative tauopathy. PLoS Genet. 2014;10:e1004211.
Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PLoS One. 2010;5:e10028.
Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, Troakes C, Al-Sarraj S, Burrage J, Macdonald R, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17:1164–70.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai S-L, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952.
Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, Craig DW, Redman M, Gershon ES, Liu C. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86:411–9.
Pidsley R, Viana J, Hannon E, Spiers H, Troakes C, Al-Saraj S, Mechawar N, Turecki G, Schalkwyk L, Bray N, Mill J. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 2014;15:483.
Hernandez D, Nalls M, Gibbs J, Arepalli S, van der Brug M, Chong S, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–72.
Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the religious orders study. Curr Alzheimer Res. 2012;9:628–45.
De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–63.
Dunning M, Barbosa-Morais N, Lynch A, Tavare S, Ritchie M. Statistical issues in the analysis of Illumina data. BMC Bioinformatics. 2008;9:85.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96.
Houseman E, Accomando W, Koestler D, Christensen B, Marsit C, Nelson H, Wiencke J, Kelsey K. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73:975–83.
Perez-Andres M, Paiva B, Nieto WG, Caraux A, Schmitz A, Almeida J, Vogt RF, Jr., Marti GE, Rawstron AC, Van Zelm MC, et al. Human peripheral blood B-cell compartments: a crossroad in B-cell traffic. Cytometry B Clin Cytom. 2010;78 Suppl 1:S47–60.
Rickabaugh TM, Baxter RM, Sehl M, Sinsheimer JS, Hultin PM, Hultin LE, Quach A, Martínez-Maza O, Horvath S, Vilain E, Jamieson BD. Acceleration of age-associated methylation patterns in HIV-1-infected adults. PLoS One. 2015;10(3):e0119201. doi:10.1371/journal.pone.0119201. eCollection 2015.
Heyn H, Moran S, Hernando-Herraez I, Sayols S, Gomez A, Sandoval J, Monk D, Hata K, Marques Bonet T, Wang L, Esteller M. DNA methylation contributes to natural human variation. Genome Res. 2013;23(9):1363–72.
Viechtbauer W. Conducting meta-analyses in R with the metafor Package. J Stat Software. 2010;36:1–48.
Acknowledgements
We would like to acknowledge the following WHI investigators. Program Office (National Heart, Lung, and Blood Institute, Bethesda, MD, USA): Jacques Rossouw, Shari Ludlam, Dale Burwen, Joan McGowan, Leslie Ford, and Nancy Geller. Clinical Coordinating Center (Fred Hutchinson Cancer Research Center, Seattle, WA, USA): Garnet Anderson, Ross Prentice, Andrea LaCroix, and Charles Kooperberg. Investigators and Academic Centers: JoAnn E. Manson (Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA); Barbara V. Howard (MedStar Health Research Institute/Howard University, Washington, DC, USA); Marcia L. Stefanick (Stanford Prevention Research Center, Stanford, CA, USA); Rebecca Jackson (The Ohio State University, Columbus, OH, USA); Cynthia A. Thomson (University of Arizona, Tucson/Phoenix, AZ, USA); Jean Wactawski-Wende (University at Buffalo, Buffalo, NY, USA); Marian Limacher (University of Florida, Gainesville/Jacksonville, FL, USA); Robert Wallace (University of Iowa, Iowa City/Davenport, IA, USA); Lewis Kuller (University of Pittsburgh, Pittsburgh, PA, USA); Sally Shumaker (Wake Forest University School of Medicine, Winston-Salem, NC, USA). Women’s Health Initiative Memory Study (Wake Forest University School of Medicine, Winston-Salem, NC): Sally Shumaker.
Funding
This study was supported by NIH/NHLBI 60442456 BAA23 (Assimes, Absher, Horvath), National Institutes of Health NIH/NIA 1U34AG051425-01 (Horvath). The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C. The PEG data were supported by NIEHS RO1ES10544 (Ritz) and NIEHS R21 ES024356 (Horvath, Ritz). Gurven and Trumble were funded by NIH/NIA R01AG024119 and R56AG02411. The Religious Order study and Rush Memory and Aging Project (brain dataset 6) were funded by P30AG10161, R01AG17917, RF1AG15819, and R01AG36042.
One of our flow datasets was collected by the Multicenter AIDS Cohort Study (MACS) at UCLA (Principal Investigators, Roger Detels and Otoniel Martinez-Maza), U01-AI35040. The MACS is funded primarily by the National Institute of Allergy and Infectious Diseases (NIAID) with additional co-funding from the National Cancer Institute (NCI P30 CA016042), the National Institute on Drug Abuse (NIDA 5P30 AI028697), the National Institute of Mental Health (NIMH), the National Institute on Aging (NIA Grant 1RO1-AG-030327 by BDJ), and UL1-TR000424 (JHU CTSA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or donors to the David Geffen School of Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
Our DNA methylation data are publicly available through gene expression omnibus (GEO) accession numbers: GSE72775, GSE78874, GSE72773, and GSE72777. Further, the WHI and Bogalusa datasets are available through dbGAP (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000200.v10.p3 and https://biolincc.nhlbi.nih.gov/studies/bhs/).
African Populations: The genotyping data generated in this study have been deposited in the European Genome-Phenome Archive under accession codes EGAS00001000605, EGAS00001000908 and EGAS00001001066. The DNA methylation data generated in this study have been deposited in the European Genome-Phenome Archive under accession code EGAS00001001066.
The GSE numbers for the brain datasets are as follows: GSE59685, GSE15745, GEO GSE38873, and GEO GSE61431. Brain data 5 can be found at http://www.ncbi.nlm.nih.gov/gap (accession: phs000249.v2.p1) and brain data 6 at https://www.synapse.org/#!Synapse:syn3168763.
Authors’ contributions
SH conceived of the study, developed the methods, analyzed the data, and wrote the first draft of the article. MG, BT, HK, and HA contributed the DNA from the Tsimane Amerindians and interpreted the findings. ML, BR, and BC helped to interpret the data and edited the article. BR and SH contributed the PEG DNA methylation data. AL analyzed the brain datasets. DS, SL, and WC contributed the DNA methylation data from the Bogalusa Heart Study. SH, PT, DA, and TA contributed the DNA methylation data from the WHI. KE and AR contributed flow cytometric data from the WHI LLS. BJ and TR contributed flow data from the MACS. LQM, MF and MSK contributed DNAm data from African hunter gatherers. All authors helped interpret the data and edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
This study was reviewed by the UCLA institutional review board (IRB#13-000671 and IRB#14-000061) as well as the University of California Santa Barbara and University of New Mexico Institutional Review Boards (IRB Reference numbers 14-0604 and 07-157 respectively).
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Lymphoblastoid cell lines from Han Chinese, Caucasians, and African Americans. A Gray line corresponds to a natural spline regression through Caucasian samples. Age acceleration was defined as residual with respect to this line. B Marginally significant evidence that African American’s are younger than other ethnic groups. (PDF 33 kb)
Additional file 2:
Accuracy of imputed blood cell counts. Here we used an independent dataset, which was not used to develop estimators of blood cell counts based on DNA methylation data, to evaluate the accuracy of the imputed blood cell counts. For each participant, both flow cytometric measures and Illumina Inf450 data were available from 96 participants as described in [88]. A-G The scatter plots depict the predicted abundance of blood cell count (based on DNA methylation levels) versus the corresponding observed flow cytometric measurement (y-axis). Each panel reports a robust correlation coefficient (biweight midcorrelation) and a corresponding p value. The Houseman method was used to impute (A) CD8+ T cells, (B) CD4+ T, (C) B cells. The epigenetic clock software was used for imputing (D) naïve CD8+ T cells, (E) naïve CD4 + T cells, (F) plasma blasts, and (G) exhausted CD8+ T cells. H, I Another flow cytometric dataset was used to test for ethnic differences in naïve CD4+ T cells. The y-axis shows the log transformed flow cytometric measurement of naïve CD4+ T cells (adjusted for age). Specifically, the y-axis reports the residual resulting from regressing log(naïve CD4+ T cell abundance) on chronological age. H Findings for HIV– participants (198 Caucasians versus 34 Hispanics). I Findings for HIV+ participants (101 Caucasians, 58 Hispanics). Stouffer’s meta-analysis across the two strata (HIV+ and HIV– strata) shows that Hispanics have significantly fewer naïve CD4+ T cells (Stouffer’s p = 0.030, Stouffer’s Z = (1.75 + 1.31)/sqrt(2)). (PDF 130 kb)
Additional file 3:
Epigenetic age acceleration in Hispanics versus country of residence in the WHI. Each column corresponds to different measure of age acceleration: (A, D) age acceleration residual, (B, E) IEAA (C, F) EEAA. (A-C, first row) results for “country of birth” (x-axis). (D-F, second row) results for “country of residence” at age 35 years, which was defined by combining two variables country of birth and “living in the US at age 35.” The left-most bar corresponds to Hispanic women who were born outside the US and lived outside the US at age 35 years, the middle bar corresponds to Hispanic women who were born outside the US but lived already in the US at the age of 35 years; the right-most bar reports results for women who were born in the US and lived in the US at age 35 years. Incidentally, all of these postmenopausal Hispanic women lived in the US at the age of the blood draw. As a caveat, we mention the relatively small group sizes (small gray numbers underneath the bars). (PDF 3 kb)
Additional file 4:
Educational level versus age acceleration in the WHI. Each row relates educational level (x-axis) to three respective measures of epigenetic age acceleration: (A-D) Age Accel., (E-H) IEAA, and (I-L) EEAA. The columns correspond to different groups of women from the WHI. The first, second, third, and fourth columns report findings for (A, E, I) all women, (B, F, J) Caucasians, (C, G, K) African Americans, and (D, H, L) Hispanics, respectively. Each bar plot reports the mean values, 1 standard error, and the p value from a non-parametric group comparison test (Kruskal–Wallis). Education was assessed using the form “Demographics and Study Membership.” We find that education predicts future EEAA. (PDF 6 kb)
Additional file 5:
Hypertension status versus age acceleration in the Bogalusa study. Each row relates hypertension status (x-axis) to three respective measures of epigenetic age acceleration: (A-C) Age Accel., (D-F) IEAA, and (G-I) EEAA. The columns correspond to different groups. The first, second, and third columns report findings for (A, D, G) all participants, (B, E, H) Caucasians, (C, F, I) African Americans, respectively. Each bar plot reports the mean values, 1 standard error, and the p value from a non-parametric group comparison test (Kruskal–Wallis). Hypertension status was defined as meeting any of the three conditions: (1) blood pressure > =140/90; (2) taking medication; or (3) having been diagnosed as having hypertension. (PDF 4 kb)
Additional file 6:
Type II diabetes status versus age acceleration in the Bogalusa study. Each row relates type II diabetes status (x-axis) to three respective measures of epigenetic age acceleration: (A-C) Age Accel., (D-F) IEAA, and (G-I) EEAA. The columns correspond to different groups. The first, second, and third columns report findings for (A, D, G) all participants, (B, E, H) Caucasians, (C, F, I) African Americans, respectively. Each bar plot reports the mean values, 1 standard error, and the p value from a non-parametric group comparison test (Kruskal–Wallis). Type 2 diabetes status was defined as fasting glucose > =126 mg/dl or taking diabetes medication. (PDF 3 kb)
Additional file 7:
Educational level versus age acceleration in the Bogalusa study. Each row relates educational level (x-axis) to three respective measures of epigenetic age acceleration: (A-C) Age Accel., (D-F) IEAA, and (G-I) EEAA. The columns correspond to different groups. The first, second, and third columns report findings for (A, D, G) all participants, (B, E, H) Caucasians, (C, F, I) African Americans, respectively. Each bar plot reports the mean values, 1 standard error, and the p value from a non-parametric group comparison test (Kruskal–Wallis). Education was grouped as follows: group 1 = grades 1–7; group 2 = grades 8–9; group 3 = grades 10–12; group 4 = vocational/tech training; group 5 = college; group 6 = postgraduate. (PDF 5 kb)
Additional file 8:
Robustness analysis with respect to other epigenetic biomarkers of aging in the WHI. A-C Results for the Horvath method when 47 out of 353 CpGs were removed from the epigenetic clock (because they are in the vicinity of a SNP). Since none of the remaining clock CpGs are near a SNP, the resulting age acceleration is not trivially related to race/ethnicity. A DNA methylation age versus chronological age. B Ethnicity versus age acceleration (defined as residual resulting from regressing DNAm age on chronological age). C Intrinsic epigenetic age acceleration versus ethnicity. D, E Alternative epigenetic biomarker of aging based on 589 age-related CpGs from Teschendorff [13]. The biomarker was defined using the following steps. First, the DNA methylation levels of each CpGs were standardized (to mean zero and variance 1). Second, a weighted average was formed by multiplying each CpG by the T test statistic from the chronological age relationship based on the table from the original reference. Third, the weighted average was regressed on chronological age to arrive at a residual. The resulting residual is referred to as extrinsic measure of age acceleration since it was not adjusted for blood cell counts. Fourth, the resulting measure was regressed on estimated blood cell counts (analogous to those used for IEAA) in order to arrive an intrinsic measure of age acceleration. F, G Epigenetic measures of age acceleration using the Hannum method 71 CpGs [19]. D, F Results for intrinsic measures, i.e. measures of age acceleration that adjust both for blood cell counts and chronological age. E, G reports extrinsic measures, i.e. no adjustment for imputed blood cell counts. Each bar plot depicts 1 standard error and reports the results from a Kruskal–Wallis test. (PDF 55 kb)
Additional file 9:
Demographic and physiologic characteristics of women from the WHI. Case-control status refers to CHD. Two designs were used to select samples: case/control and case-cohort. (DOC 48 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Horvath, S., Gurven, M., Levine, M.E. et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol 17, 171 (2016). https://doi.org/10.1186/s13059-016-1030-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-016-1030-0