
Abstract
Acute kidney injury (AKI) is common in the critically ill. Inadequate renal medullary tissue oxygenation has been linked to its pathogenesis. Moreover, renal medullary tissue hypoxia can be detected before biochemical evidence of AKI in large mammalian models of critical illness. This justifies medullary hypoxia as a pathophysiological biomarker for early detection of impending AKI, thereby providing an opportunity to avert its evolution. Evidence from both animal and human studies supports the view that non-invasively measured bladder urinary oxygen tension (PuO2) can provide a reliable estimate of renal medullary tissue oxygen tension (tPO2), which can only be measured invasively. Furthermore, therapies that modify medullary tPO2 produce corresponding changes in bladder PuO2. Clinical studies have shown that bladder PuO2 correlates with cardiac output, and that it increases in response to elevated cardiopulmonary bypass (CPB) flow and mean arterial pressure. Clinical observational studies in patients undergoing cardiac surgery involving CPB have shown that bladder PuO2 has prognostic value for subsequent AKI. Thus, continuous bladder PuO2 holds promise as a new clinical tool for monitoring the adequacy of renal medullary oxygenation, with its implications for the recognition and prevention of medullary hypoxia and thus AKI.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI) is a frequent complication in intensive care units, affecting 30–60% of critically ill patients [1]. The role of renal haemodynamics and oxygenation in the evolution of acute renal dysfunction has been summarised by Ricksten and colleagues [2], who have particularly focused on clinical states where the parlous balance between oxygen supply and demand might most likely explain the pathogenesis of AKI, so-called ischemic AKI. Such states include sepsis, cardiac and other major surgery, congestive heart failure, after liver transplantation [2] and in the context of renal transplantation [3]. In particular, it is the outer medulla, with lesser perfusion than the cortex but high oxygen consumption within the thick ascending limb of the loop of Henle that is particularly susceptible to hypoxia [4]. Oxygen availability in the renal medulla is also attenuated by counter-current exchange of oxygen within the vasa recta [4]. Additionally, the renal medullary circulation appears not to be as tightly autoregulated as the renal cortical circulation [5]. Thus, the medulla is at particular risk of inadequate perfusion and oxygenation, particularly under pathophysiological conditions [6, 7]. Renal tissue hypoxia may in fact be a final common pathway [8, 9] or a critical event [10, 11] in the development of multiple forms of AKI. Furthermore, renal tissue hypoxia has been implicated in the transition of AKI to chronic kidney disease [12, 13].
Serum creatinine is used to estimate glomerular filtration rate and diagnose patients with AKI. Other plasma and urinary biomarkers, including neutrophil gelatinase-associated lipocalin and cystatin C, have demonstrated promise in the early detection of AKI [14]. However, they are markers of renal injury that has already occurred. Thus, any diagnosis that depends on such biomarkers may lag hours behind the initiating insult. Using large mammalian experimental models, renal medullary hypoxia has been demonstrated in two major contributors to AKI: sepsis and cardiac surgery involving cardiopulmonary bypass (CPB) [15,16,17,18,19]. In these experiments, probes are inserted into the tissue of the renal medulla of sheep. When sepsis-induced AKI is induced by E. coli resulting in reduced creatinine clearance [15, 16] or oliguria [17], a fall in renal medullary tissue oxygenation (tPO2) is seen. A similar fall is demonstrated upon initiation of CPB [18, 19]. Moreover, in the ovine septic AKI model, medullary tissue hypoxia precedes the development of functional deficits by several hours [16]. Accordingly, developing minimally invasive techniques for reliably assessing renal medullary tPO2 may provide a means for early detection of risk of AKI and for guiding therapies in order to avoid exacerbating that risk. However, it is not clinically feasible to directly monitor renal medullary tPO2 in patients. Our research group has proposed that continuous measurement of bladder urinary oxygen tension (PuO2) could provide a non-invasive surrogate measure of renal medullary tPO2 and thus could be an appropriate pathophysiological biomarker of AKI [10]. The rationale for this proposition is supported by the following lines of evidence (summarised in [11]): that (i) the anatomy of the renal medulla facilitates diffusion of oxygen between vasa recta and the collecting ducts, so that (ii) PuO2 in the renal pelvis equilibrates with renal medullary tPO2, (iii) that oxygen diffusion across the epithelium of the ureter and bladder only partially confounds the relationship between medullary tPO2 and PuO2 and that (iv) bladder PuO2 provides reliable prognostic information. A selection of studies separated into animal and human data is presented below as support for these assertions.
Experimental studies in animals
Initial pioneering work between 1958 and 1960 focused on canine experiments, establishing that the urine in the renal pelvis and the tissue of the renal medulla were areas of low oxygen tension [20,21,22], with values lower than the oxygen tension within the renal vein. However, the polarographic methods for measurement of oxygen tension used at the time were subject to measurement error, with insufficient miniaturisation to always accurately determine site of measurement [23].
More recent work has focused on using different animal models (chiefly ovine [15,16,17, 19, 24,25,26,27], but also murine [28, 29] and leporine [30]) to examine this area, with the use of new technology such as fibre optic fluorescence lifetime oximetry probes to continuously measure oxygen tension within urine or renal tissue (Fig. 1). This has provided insights into changes that occur during development of septic AKI, as well as during clinically relevant resuscitation strategies such as fluids, vasopressors and diuretics.

An example of a fibre optic probe (an “optode”) for fluorescence lifetime oximetry (NX-LAS-1/O/E; Oxford Optronix Ltd.; Abingdon, UK). Here, the optode has been advanced through a Foley bladder catheter to permit continuous monitoring of bladder urinary oxygen tension. Similar probes can be used to invasively measure tissue oxygen tension in various organs
Prognostic utility of PuO 2 in the early detection of AKI
In ovine sepsis, the initiation of renal medullary tissue hypoperfusion and hypoxia occurs within the first hour of Gram-negative infection followed by a progressive reduction over 24 h [15,16,17]. Such renal medullary microcirculatory abnormalities take place despite increased and/or preserved global renal blood flow, global renal oxygen delivery and cortical perfusion and cortical tPO2 in non-anesthetised septic sheep [15,16,17]. An early onset of renal medullary tissue hypoxia could potentially initiate and perpetuate a cycle of inflammation and oxidative/nitrosative stress that can lead to mitochondrial dysfunction, tubular cell injury and reduced kidney function [6, 31]. Reductions in renal medullary tPO2 were detected in ‘real time’ up to 8 to 24 h before any significant elevations in urinary neutrophil gelatinase-associated lipocalin and/or serum creatinine could be detected in septic sheep with AKI (Fig. 2C & D) [16]. Remarkably, the temporal profile and magnitude of the progressive decrease in renal medullary tPO2 in sepsis was closely reflected by continuously measured bladder PuO2 (Fig. 2A). Indeed, a positive significant correlation was detected during the development of AKI over 24 h (r = 0.7; P < 0.001; Fig. 2B) [17]. These findings strongly support the prognostic value of bladder PuO2 as an early physiological biomarker to evaluate risk of developing septic AKI.

The prognostic utility of bladder urinary oxygen tension in the early detection of acute kidney injury over 24 h in Gram-negative sepsis in non-anesthetised sheep. A The time course of changes in renal medullary tissue PO2 and bladder urinary PO2 and urine flow over 24 h of sepsis. Urine flow is presented as absolute values corrected for kilogram of body weight. B Scatterplot of the relationship between medullary tissue and urinary PO2 (different symbols represent individual sheep). The line of best fit, determined by ordinary least-product regression analysis, had an X intercept of 2.4 mmHg (95% confidence interval: 0.3–4.5) and a slope of 1.02 (95% confidence interval: 0.95–1.09) (P < 0.001). C Changes in renal medullary tissue and bladder urinary PO2 and D plasma creatinine and urinary neutrophil gelatinase-associated lipocalin (NGAL) over 24 h of Gram-negative sepsis-induced acute kidney injury. *P < 0.05 indicates significant differences from pre-morbid baseline (Time 0). Figures modified from [16, 17]
Potential utility of PuO 2 to guide clinical interventions
Renal medullary tPO2 is dominated by the energy requirements of the sodium–potassium ATPase pump required to drive tubular sodium reabsorption in the thick ascending limb of the loop of Henle. Thus, renal oxygen consumption is directly linked to the filtered load of sodium and thus glomerular filtration rate [32]. Consequently, the effects of goal-directed therapies such as fluid resuscitation, diuretics and vasopressors on renal medullary tPO2 reflect a complex interaction between their effects on renal and intra-renal perfusion and tubular function. For example, given that any therapy that increases glomerular filtration rate would be expected to increase renal oxygen consumption, this would, in turn, potentially promote renal tissue hypoxia. Conversely, furosemide treatment (which inhibits tubular sodium reabsorption) or fluid resuscitation was found to reverse medullary tissue hypoxia in ovine septic AKI back to healthy physiological levels [25, 26]. On the other hand, the actions of various vasoactive drugs to restore blood pressure were not universally favourable for renal medullary tissue perfusion and tPO2. Norepinephrine further worsened renal medullary tissue hypoperfusion and hypoxia, an undesirable effect; but this was not seen when vasopressin was used as primary vasopressor [17, 24]. Angiotensin II was found to induce minimal changes to medullary tPO2 in ovine septic AKI [16] but was also found to reduce medullary tPO2 in healthy sheep [24]. These varied effects are best understood by accounting for the myriad of factors that could affect medullary blood flow and medullary oxygen consumption, such as differential receptor occupancy of pre-glomerular (afferent) and post-glomerular (efferent) arterioles, changes in oxygen consumption due to alterations in sodium reabsorption accompanying changes in GFR or changes in microcirculatory shunting of oxygen within the counter-current exchange arrangement of vessels [24]. At a macro-circulatory level, when renal blood flow was increased in sheep undergoing CPB by increasing global perfusion (i.e. increasing systemic pump flow), increasing renal perfusion by vasopressor support with metaraminol or both [18, 33], medullary tPO2 also increased.
Despite the variable effects of various resuscitative measures on medullary tPO2, they all appear to mirror changes in bladder PuO2 in sheep (e.g. during resuscitation with fluids [25], norepinephrine [17] and angiotensin II [16]; as well as during administration of furosemide [26]). Similarly, when two pharmacological agents that reduced medullary tPO2 (vasopressin V1-receptor activation or blockade of nitric oxide synthesis) were administered to anesthetised rabbits, the changes in bladder PuO2, measured using the same technique as used in sheep, mimicked changes in medullary tPO2 [30]. Further evidence comes from a post-hoc analysis of experimental studies in septic sheep breathing room air, where it was demonstrated that bladder PuO2 varied linearly with medullary tPO2 under normoxic or slightly hypoxic conditions [34].
Together, these preclinical observations indicate that measurement of bladder PuO2 may be a clinically feasible technique to estimate renal medullary tPO2, and therefore vulnerability towards developing AKI, as well as a tool to gauge the impact of resuscitation strategies on medullary tPO2.
Clinical studies in humans
Although PuO2 in humans had been reported previously (e.g. [21, 35]), the utility of urine oximetry in humans was only properly investigated in a small series of studies by Leonhardt and colleagues between 1963 and 1965 [36,37,38,39]. This early exploration of its utility occurred concurrently alongside animal work in dogs, with early postulation that there must exist an equilibrium between the oxygen tension in the collecting ducts and the peritubular capillary blood given the permeable nature of the epithelium of the collecting ducts [35]. This series of studies was performed amongst patients undergoing urological procedures to enable measurement of both pelvic ureteric PuO2 and sometimes renal tissue PO2, together with patients with an indwelling urethral catheter for measurement of bladder PuO2. In such patients, Leonhardt and colleagues found good correlation between pelvic ureteric PuO2 and medullary tPO2 [36] as well as a relationship between pelvic ureteric PuO2 and bladder PuO2 [37]. Furthermore, pelvic ureteric PuO2 increased with hydration, although the response time was delayed in patients with reduced renal perfusion (e.g. sepsis or shock) [38], whilst hypertonic saline infusion reduced pelvic ureteric PuO2, presumably due to the increased medullary oxygen consumption associated with salt loading [39]. Both pelvic PuO2 and bladder PuO2 were reported to rise with increasing inspired fraction of oxygen, although the response was sluggish in disease states such as pyelonephritis or azotemia [38]. Additionally, the investigators reported that PuO2 was altered with agents that affect sympathetic tone, in a manner independent of urine flow [37]. Leonhardt and colleagues proposed that the oxygen tension in urine could be used as an indication of medullary oxygenation and that the response time of PuO2 to changes in FiO2 could be an index of medullary blood flow at periods of stable oxygen consumption, with a slower response time indicating poorer medullary blood flow, in a manner analogous to an indicator dilution technique for determining flow [36].
Continued reports of the use of PuO2 largely focused on measuring effects of various interventions on bladder PuO2, given the ease of sampling. These included demonstration of the reduction in PuO2 with sevoflurane and isoflurane [40], and the improvement in PuO2 with the renal vasodilatory effects of dopamine, prostaglandin E1 and fenoldopam [41, 42]. Dexmedetomidine was observed to decreased bladder PuO2 in critically ill patients, which was attributed to attenuation of cardiac output and thus renal blood flow and medullary perfusion [43]. Furosemide was reported to decrease ureteric PuO2 in otherwise well patients after relief of urologic stones [44], but was observed to increase bladder PuO2 in septic patients [45]. Whilst the latter response is consistent with reductions in medullary oxygen consumption observed in animal models [26] and humans [46, 47], furosemide has also been associated with a reduction in medullary blood flow in animals [48, 49], which could explain the former response.
Studies of the effects of non-pharmacological interventions on bladder PuO2 have included examination of the effects of red cell transfusion and the relationship between bladder PuO2 and cardiac output. In a small study involving eight patients, red cell transfusion was seen to increase bladder PuO2 as measured by a blood gas analyser [50]. Additionally, amongst 60 patients with a recent myocardial infarction or unstable angina, bladder PuO2 and cardiac output (as measurement by Swan-Ganz catheter) were positively correlated [51]. These imply that global oxygen delivery (DO2) is relevant for determining PuO2.
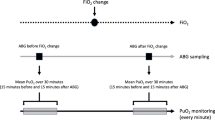
The association between DO2 and PuO2 or kidney outcomes has been further explored in patients undergoing CPB. During CPB, it is notable that increasing pump flow results in a more favourable renal oxygen supply relative to demand, with a lower oxygen extraction ratio [52], which is consistent with the observation that renal blood flow is directly proportional to systemic flow during CPB, implying a loss of renal autoregulatory capacity [53]. In a multicentre randomised controlled study that was terminated after interim analysis of 350 randomised patients (out of a target sample size of 700 patients), a goal-directed strategy aiming for DO2 > 280 ml/min/m2 was found to result in fewer patients developing Acute kidney Injury Network (AKIN) Stage 1 compared with usual care (relative risk 0.45, 95% confidence interval 0.25–0.83, P = 0.01) [54]. Furthermore, in a randomised controlled crossover trial amongst 20 patients, bladder PuO2 was successfully raised in response to higher CPB pump flow (and therefore systemic DO2) and mean arterial pressure (MAP) compared with normal CPB pump flow and MAP (achieving median [interquartile range] DO2 of 409 [374, 441] vs. 340 [297, 379] ml/min/m2 respectively) [55]. This demonstrated a successful dynamic manoeuvre to increase bladder PuO2 during CPB with increasing pump flow and MAP, with a response time of 17 min (Fig. 3). Notably, this higher systemic DO2 was achieved by targeting a CPB pump flow of 3.0 L/min/m2 and MAP of 80 mmHg compared with the control interval with target CPB pump flow of 2.4 L/min/m2 and MAP of 65 mmHg. This suggests that bladder PuO2 has the potential to guide management for optimising renal medullary tPO2 during cardiac surgery and that higher target CPB pump flow and MAP than traditionally used may be required for improving medullary oxygenation during bypass.

Urine oxygen tension (PuO2) across time during Intervention H (target pump flow of 3.0 L/min/m2 and mean arterial pressure (MAP) of 80 mmHg) and Intervention N (target pump flow of 2.4 L/min/m2 and MAP of 65 mmHg). Each box represents the interquartile range (IQR) for the 20-secondly median values across patients, with lines out to \(\pm\) 1.5 \(\times\) IQR. Dots represent outliers. Mixed model for repeated measures showed statistical significance for group and time interaction (P < 0.001). Differences in PuO2 between the intervention groups, adjusted for repeat measurements, showed statistically significance after approximately 17 min (marked *). Reproduced from Hu et al. [59] with permission
Whilst it is possible that the concomitant increase in renal perfusion or cortical perfusion with increased systemic DO2 is at least somewhat contributory to PuO2 (or improved kidney outcomes), the preceding discussions describing the association of PuO2 with medullary tPO2 in both animal and human studies suggest that it is medullary perfusion (and thereby oxygenation) that is the key variable that is altered. This is particularly relevant because the medullary circulation is poorly autoregulated and is determined by factors that are distinct from cortical or global renal perfusion [5].
Further support for the association between renal medullary tissue hypoxia and AKI comes from observational studies involving patients undergoing cardiac surgery requiring CPB that examined the association of bladder PuO2 and AKI [56,57,58,59]. In these studies, a reduction in PuO2 was observed upon initiation of CPB, and a slower rate of rise of PuO2 after weaning from bypass [56] or a lower mean PuO2 in the post bypass period [58] or a lower nadir and longer duration of low PuO2 values [57] were associated with AKI. Notably, there were no statistically significant differences in cardiac index between groups that did or did not develop AKI. However, measurement of PuO2 using a blood gas analyser six hours after admission to ICU had modest discrimination in detecting patients who later developed AKI [59].
Limitations
Whilst bladder PuO2 is more convenient and less invasive for approximating medullary tPO2 than ureteric measurement, potential confounders abound. These include oxygen from the presence of blood in urine, which would be expected to contaminate samples with arterialised oxygen tension [60]. This is nevertheless easy to identify. More subtle sources of bias in bladder PuO2 measures include the presence of substances that can alter oxygen within the bladder, such as the microbiota [61] or ascorbic acid [21]. It is also well established that oxygen consumption can be observed in urine in a static state [35, 62]. Therefore, it becomes imperative that PuO2 should be obtained in the fasted state [11] and that continuous rather than static measurements of bladder PuO2 be used, ensuring that urine flow does not stop.
Bladder PuO2 measurements can also be altered by the diffusion of oxygen between urine and the wall of the bladder and ureter, or exposure of urine to atmospheric oxygen [34]. The uncertainty created by these confounding factors can be minimised in two ways: (i) by measuring bladder PuO2 in a way that minimises the opportunity for diffusion of oxygen from these sources, and (ii) through use of computational models to estimate the magnitude of this diffusion and correct for it.
Bladder PuO2 can be measured continuously using a fibre optic probe inserted into the bladder catheter (Fig. 1) so that its sensing tip lies within the bladder [45, 55, 57], or in the urine line external to the bladder, either using a polarographic electrode [56] or fibre optic probe [58]. Sampling external to the bladder has potential advantages in avoidance of risks associated with insertion of a probe within the bladder and potentially allowing re-use of sensors. However, it also risks the possibility of further confounding from diffusion of oxygen between the urine and the wall of the Foley catheter. Consistent with this possibility, in studies in humans, urinary PO2 measured in the urine line external to the bladder (38–107 mmHg) [56, 58] was higher than that measured within the bladder (26–66 mmHg) [57]. However, no direct comparison is available to determine the extent of this confounding.
Urinary PO2 can also potentially be measured by collecting urine from a Foley catheter for analysis in a blood gas machine [50, 51, 59]. However, when measured in this way there is the potential for oxygen to diffuse from the atmosphere (PO2 159 mmHg at sea level) to the urine sample. Consistent with this proposition, reported mean levels of urinary PO2 in humans using this approach [50, 59, 63,64,65] were markedly higher (80–153 mmHg) than those reported from studies in which urinary PO2 was measured in the bladder [57] (26–66 mmHg) or in the urine line external to the bladder [56, 58] (38–107 mmHg).
The rate of oxygen diffusion between the urine and the walls of the ureter or bladder is driven by the gradient in PO2 between interfacing compartments. Bladder tPO2 has been shown to correlate with renal blood flow in animal studies [66, 67] although confounders to this relationship exist at the lower end of the autoregulatory limit [68], and the relationship is uncertain in the context of sepsis [69,70,71,72], and vasopressors [73, 74]. Whilst bladder tPO2 could contaminate bladder PuO2 measurement, it appears that the oxygen diffusion between bladder tPO2 and bladder PuO2 contributes little to final bladder PuO2 value, which is chiefly influenced by the PuO2 of urine entering the bladder from the ureter [30]. This ureteric PuO2 value is in turn determined by the time available for oxygen diffusion between ureteric wall and the urine (a function of the transit time of each bolus of urine and the bolus volume) [75]. Consequently, the major confounders of the relationship between renal medullary (or pelvic ureteric) PuO2 and bladder PuO2 appear to be the patient’s systemic oxygenation and their rate of urine flow. Thus, as urine flow becomes greater, the error in the estimate of medullary PO2 generated by measurement of urinary PO2 becomes less. In contrast, as urine flow becomes less and a patient becomes progressively hyperoxemic or hypoxemic, the error becomes greater [75]. Therefore, in a clinical setting, estimating medullary oxygenation directly from measurement of urinary PO2 is probably most useful when patients have a normal or high urine flow. But even when such conditions are not entirely met, computational models can account for variations in urine flow and systemic oxygenation. The most sophisticated model developed to date predicts the existence of a family of linear relationships between bladder PuO2 and pelvic ureteric PuO2 for any given set of input conditions of urine flow and systemic arterial PO2 in human patients (Fig. 4) [75]. Thus, it should be technically possible to predict renal medullary tPO2 in real time based on continuous measurement of bladder PuO2, provided accurate and real-time measurements of urine flow and arterial PO2 are available.

Contour plot of urine flow, bladder PuO2 and pelvic PuO2 under modelling conditions of A normoxia (PaO2 = 90 mmHg) and B hyperoxia (PaO2 = 300 mmHg) and other assumptions as per Lee et al. [75]. Figure reproduced with permission
Finally, the implications of disease states of the kidney for measurements of bladder PuO2 remain to be fully elucidated, as recent clinical studies have been performed largely on patients without established kidney disease [55,56,57,58]. Furthermore, an elevated ureteric PuO2 has been noted in established hydronephrosis and could also occur with renal cysts, due to medullary thinning and the influence of renal cortical oxygenation on measurements [38]. Nevertheless, it remains possible that trends in bladder PuO2 could still be useful as a reflection of the adequacy of medullary oxygenation.
Conclusion
AKI is common in critical care units, and attenuating its severity or even preventing its development is a key therapeutic target. There is increasing evidence that the inadequacy of renal medullary oxygenation is linked with its development. Furthermore, medullary tissue hypoxia appears to precede other evidence of AKI. Continuous measurements of bladder PuO2 promise to provide a non-invasive method for estimating medullary tPO2 and have prognostic value. Furthermore, it is a physiological variable that can be manipulated by pharmacological and other non-pharmacological interventions, making it an ideal candidate for targeted therapies to maintain or restore its value and thereby protect the kidney from injury. Sources of error or confounding variables remain a challenge, although many of these can be overcome by appropriate sampling and computational modelling. Harnessing the value of continuous bladder PuO2 monitoring appears promising in critical care environments.
Availability of data and materials
All data described in this manuscript are derived from published articles and have been appropriately referenced throughout.
References
Pickkers P, Darmon M, Hoste E, Joannidis M, Legrand M, Ostermann M, et al. Acute kidney injury in the critically ill: an updated review on pathophysiology and management. Intensive Care Med. 2021;47(8):835–50.
Ricksten SE, Bragadottir G, Lannemyr L, Redfors B, Skytte J. Renal hemodynamics, function, and oxygenation in critically ill patients and after major surgery. Kidney360. 2021;2(5):894–904.
Rosenberger C, Eckardt KU. Oxygenation of the transplanted kidney. Semin Nephrol. 2019;39(6):554–66.
Brezis M, Rosen S. Hypoxia of the renal medulla—its implications for disease. N Engl J Med. 1995;332(10):647–55.
Pallone TL, Edwards A, Mattson DL. Renal medullary circulation. Compr Physiol. 2012;2(1):97–140.
Lankadeva YR, Okazaki N, Evans RG, Bellomo R, May CN. renal medullary hypoxia: A new therapeutic target for septic acute kidney injury? Semin Nephrol. 2019;39(6):543–53.
Lankadeva YR, May CN, Bellomo R, Evans RG. Role of perioperative hypotension in postoperative acute kidney injury: a narrative review. Br J Anaesth. 2022;128(6):931–48.
Le Dorze M, Legrand M, Payen D, Ince C. The role of the microcirculation in acute kidney injury. Curr Opin Crit Care. 2009;15(6):503–8.
Heyman SN, Evans RG, Rosen S, Rosenberger C. Cellular adaptive changes in AKI: mitigating renal hypoxic injury. Nephrol Dial Transplant. 2012;27(5):1721–8.
Ow CPC, Ngo JP, Ullah MM, Hilliard LM, Evans RG. Renal hypoxia in kidney disease: Cause or consequence? Acta Physiol (Oxf). 2018;222(4): e12999.
Evans RG, Smith JA, Wright C, Gardiner BS, Smith DW, Cochrane AD. Urinary oxygen tension: a clinical window on the health of the renal medulla? Am J Physiol Regul Integr Comp Physiol. 2014;306(1):R45-50.
Tanaka S, Tanaka T, Nangaku M. Hypoxia as a key player in the AKI-to-CKD transition. Am J Physiol Renal Physiol. 2014;307(11):F1187–95.
Ullah MM, Basile DP. Role of renal hypoxia in the progression from acute kidney injury to chronic kidney disease. Semin Nephrol. 2019;39(6):567–80.
Haase M, Bellomo R, Haase-Fielitz A. Novel biomarkers, oxidative stress, and the role of labile iron toxicity in cardiopulmonary bypass-associated acute kidney injury. J Am Coll Cardiol. 2010;55(19):2024–33.
Calzavacca P, Evans RG, Bailey M, Bellomo R, May CN. Cortical and medullary tissue perfusion and oxygenation in experimental septic acute kidney injury. Crit Care Med. 2015;43(10):e431–9.
Lankadeva Y, Kosaka J, Evans R, Bellomo R, May C. Urinary oxygenation as a surrogate marker of medullary oxygenation during angiotensin II therapy in septic acute kidney injury. Crit Care Med. 2018;46:e41–8.
Lankadeva YR, Kosaka J, Evans RG, Bailey SR, Bellomo R, May CN. Intrarenal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int. 2016;90(1):100–8.
Lankadeva YR, Cochrane AD, Marino B, Iguchi N, Hood SG, Bellomo R, et al. Strategies that improve renal medullary oxygenation during experimental cardiopulmonary bypass may mitigate postoperative acute kidney injury. Kidney Int. 2019;95(6):1338–46.
Lankadeva YR, May CN, Cochrane AD, Marino B, Hood SG, McCall PR, et al. Influence of blood haemoglobin concentration on renal haemodynamics and oxygenation during experimental cardiopulmonary bypass in sheep. Acta Physiol (Oxf). 2021;231(3): e13583.
Aukland K, Krog J. Renal oxygen tension. Nature. 1960;188:671.
Rennie DW, Reeves RB, Pappenheimer JR. Oxygen pressure in urine and its relation to intrarenal blood flow. Am J Physiol. 1958;195(1):120–32.
Reeves RB, Rennie DW, Pappenheimer JR. Oxygen tension of urine and its significance. Fed Proc. 1957;16(3):693–6.
Ulfendahl HR. Intrarenal oxygen tension. Acta Soc Med Ups. 1962;67:95–106.
Calzavacca P, Evans RG, Bailey M, Bellomo R, May CN. Variable responses of regional renal oxygenation and perfusion to vasoactive agents in awake sheep. Am J Physiol Regul Integr Comp Physiol. 2015;309(10):R1226–33.
Lankadeva Y, Kosaka J, Iguchi N, Evans R, Booth L, Bellomo R, et al. Effects of fluid bolus therapy on renal perfusion, oxygenation, and function in early experimental septic kidney injury. Crit Care Med. 2019;47(1):e36–43.
Iguchi N, Lankadeva YR, Mori TA, Osawa EA, Cutuli SL, Evans RG, et al. Furosemide reverses medullary tissue hypoxia in ovine septic acute kidney injury. Am J Physiol Regul Integr Comp Physiol. 2019;317(2):R232–9.
Evans RG, Iguchi N, Cochrane AD, Marino B, Hood SG, Bellomo R, et al. Renal hemodynamics and oxygenation during experimental cardiopulmonary bypass in sheep under total intravenous anesthesia. Am J Physiol Regul Integr Comp Physiol. 2020;318(2):R206–13.
Liss P, Nygren A, Revsbech NP, Ulfendahl HR. Measurements of oxygen tension in the rat kidney after contrast media using an oxygen microelectrode with a guard cathode. Adv Exp Med Biol. 1997;411:569–76.
Johannes T, Mik EG, Nohé B, Unertl KE, Ince C. Acute decrease in renal microvascular PO2 during acute normovolemic hemodilution. Am J Physiol Renal Physiol. 2007;292(2):F796-803.
Sgouralis I, Kett MM, Ow CP, Abdelkader A, Layton AT, Gardiner BS, et al. Bladder urine oxygen tension for assessing renal medullary oxygenation in rabbits: experimental and modeling studies. Am J Physiol Regul Integr Comp Physiol. 2016;311(3):R532–44.
Ow CPC, Trask-Marino A, Betrie AH, Evans RG, May CN, Lankadeva YR. Targeting oxidative stress in septic acute kidney injury: from theory to practice. J Clin Med. 2021;10(17):3798.
Evans RG, Gardiner BS, Smith DW, O’Connor PM. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Renal Physiol. 2008;295(5):F1259–70.
Lankadeva YR, Evans RG, Cochrane AD, Marino B, Hood SG, McCall PR, et al. Reversal of renal tissue hypoxia during experimental cardiopulmonary bypass in sheep by increased pump flow and arterial pressure. Acta Physiol (Oxf). 2021;231(4): e13596.
Ngo JP, Lankadeva YR, Zhu MZL, Martin A, Kanki M, Cochrane AD, et al. Factors that confound the prediction of renal medullary oxygenation and risk of acute kidney injury from measurement of bladder urine oxygen tension. Acta Physiol (Oxf). 2019;227:e13294.
Hong SK, Boylan JW, Tannenberg AM, Rahn H. Total and partial gas tensions of human bladder urine. J Appl Physiol. 1960;15:115–20.
Leonhardt KO, Landes RR, McCauley RT. Anatomy and physiology of intrarenal oxygen tension: preliminary study of the effects of anesthetics. Anesthesiology. 1965;26(5):648–58.
Leonhardt KO, Landes RR. Oxygen tension of the urine and renal structures. Preliminary report of clinical findings. N Engl J Med. 1963;269:115–21.
Leonhardt KO, Landes RR. Urinary oxygen pressure in renal parenchymal and vascular disease. Effects of breathing oxygen. JAMA. 1965;194(4):345–50.
Landes RR, Leonhardt KO, Duruman N. A clinical study of the oxygen tension of the urine and renal structures. II. J Urol. 1964;92:171–8.
Terashima M, Komatsu T, Watanabe K, Isosu T, Ohtsuki M, Okuaki A, et al. Changes in urine oxygen tension (PuO2) during anesthesia with isoflurane and sevoflurane. Masui. 1994;43(4):467–71.
Watanabe K, Terashima M, Isosu T, Komatsu T, Ohtsuki M, Okuaki A. The effect of dopamine and prostaglandin E1 on urine oxygen tension (PuO2). Masui. 1995;44(7):950–5.
Morelli A, Rocco M, Conti G, Orecchioni A, De Blasi RA, Coluzzi F, et al. Monitoring renal oxygen supply in critically-ill patients using urinary oxygen tension. Anesth Analg. 2003;97(6):1764–8.
Plummer MP, Lankadeva YR, Finnis ME, Harrois A, Harding C, Peiris RM, et al. Urinary and renal oxygenation during dexmedetomidine infusion in critically ill adults with mechanistic insights from an ovine model. J Crit Care. 2021;64:74–81.
Tolley PM, Purcell A, Bolsin SN. Effect of iv furosemide on pelvic urinary oxygen tension in humans. Br J Anaesth. 1999;83(2):328–9.
Osawa EA, Cutuli SL, Bitker L, Canet E, Cioccari L, Iguchi N, et al. Effect of furosemide on urinary oxygenation in patients with septic shock. Blood Purif. 2019;48(4):336–45.
Haddock B, Larsson HBW, Francis S, Andersen UB. Human renal response to furosemide: simultaneous oxygenation and perfusion measurements in cortex and medulla. Acta Physiol (Oxf). 2019;227(1): e13292.
Redfors B, Swärd K, Sellgren J, Ricksten SE. Effects of mannitol alone and mannitol plus furosemide on renal oxygen consumption, blood flow and glomerular filtration after cardiac surgery. Intensive Care Med. 2009;35(1):115–22.
Kirchner KA. Role of medullary plasma flow in the attenuated furosemide response in indomethacin-treated rats. J Pharmacol Exp Ther. 1989;249(3):757–61.
Dobrowolski L, Badzyńska B, Grzelec-Mojzesowicz M, Sadowski J. Renal vascular effects of frusemide in the rat: influence of salt loading and the role of angiotensin II. Exp Physiol. 2001;86(5):611–6.
Valente A, Sorrentino L, La Torre G, Draisci G. Post-transfusional variation in urinary oxygen tension in surgical patients. Clin Exp Pharmacol Physiol. 2008;35(9):1109–12.
Kitashiro S, Sugiura T, Takayama Y, Tamura T, Izuoka T, Inada M, et al. Clinical significance of the urinary oxygen tension in patients with ischemic heart disease. Cardiology. 1997;88(6):540–3.
Lannemyr L, Bragadottir G, Hjärpe A, Redfors B, Ricksten SE. Impact of cardiopulmonary bypass flow on renal oxygenation in patients undergoing cardiac operations. Ann Thorac Surg. 2019;107(2):505–11.
Andersson LG, Bratteby LE, Ekroth R, Hallhagen S, Joachimsson PO, van der Linden J, et al. Renal function during cardiopulmonary bypass: influence of pump flow and systemic blood pressure. Eur J Cardiothorac Surg. 1994;8(11):597–602.
Ranucci M, Johnson I, Willcox T, Baker RA, Boer C, Baumann A, et al. Goal-directed perfusion to reduce acute kidney injury: a randomized trial. J Thorac Cardiovasc Surg. 2018;156(5):1918-27.e2.
Hu R, Yanase F, McCall P, Evans R, Raman J, Bellomo R. The Effects of targeted changes in systemic blood flow and mean arterial pressure on urine oximetry during cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2022;36(9):3551–60.
Kainuma M, Yamada M, Miyake T. Continuous urine oxygen tension monitoring in patients undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 1996;10(5):603–8.
Zhu MZL, Martin A, Cochrane AD, Smith JA, Thrift AG, Harrop GK, et al. Urinary hypoxia: an intraoperative marker of risk of cardiac surgery-associated acute kidney injury. Nephrol Dial Transpl. 2018;33(12):2191–201.
Silverton NA, Lofgren LR, Hall IE, Stoddard GJ, Melendez NP, Van Tienderen M, et al. Noninvasive urine oxygen monitoring and the risk of acute kidney injury in cardiac surgery. Anesthesiology. 2021;135(3):406–18.
Kato T, Kawasaki Y, Koyama K. Intermittent urine oxygen tension monitoring for predicting acute kidney injury after cardiovascular surgery: a preliminary prospective observational study. Cureus. 2021;13(7): e16135.
Washington JA 2nd, Holland JM. Urine oxygen tension: effects of osmotic and saline diuresis and of ethacrynic acid. Am J Physiol. 1966;210(2):243–50.
Shannon MB, Limeira R, Johansen D, Gao X, Lin H, Dong Q, et al. Bladder urinary oxygen tension is correlated with urinary microbiota composition. Int Urogynecol J. 2019;30(8):1261–7.
Brodwall EK, Skrede S, Murio JR. Ureteral oxygen tension in a human material. Invest Urol. 1964;2:226–34.
Patel RP, Joshi H, Patel H, Kothari J, Pandya H, Solanki A. Use of urinary PO2 for early detection of renal dysfunction in cardiac surgical patients. Int J Med Sci Public Health. 2016;5:847–51.
Ikemoto N, Katayama H, Ishida N, Michida M, Yoshida Y, Ochiai Y, et al. Assessment of urine partial oxygen preesure to predict postoperative acute kidney injury in major surgical patients. Kawasaki Med J. 2021;47:77–84.
Tosun M, Ulugol H, Aksu U, Toraman F. Can partial oxygen pressure of urine be an indicator for tissue perfusion? Turk J Anaesthesiol Reanim. 2019;47(3):187–91.
Singer M, Millar C, Stidwill R, Unwin R. Bladder epithelial oxygen tension–a new means of monitoring regional perfusion? Preliminary study in a model of exsanguination/fluid repletion. Intensive Care Med. 1996;22(4):324–8.
Lang JD Jr, Evans DJ, deFigueiredo LP, Hays S, Mathru M, Kramer GC. A novel approach to monitor tissue perfusion: bladder mucosal PCO2, PO2, and pHi during ischemia and reperfusion. J Crit Care. 1999;14(2):93–8.
Anele UA, Ratz PH, Colhoun AF, Roberts S, Musselman R, Vince RA, et al. Potential vascular mechanisms in an ex vivo functional pig bladder model. Neurourol Urodyn. 2018;37(8):2425–33.
Rosser DM, Stidwill RP, Jacobson D, Singer M. Oxygen tension in the bladder epithelium rises in both high and low cardiac output endotoxemic sepsis. J Appl Physiol (1995). 1995;79(6):1878–82.
Rosser DM, Stidwill RP, Jacobson D, Singer M. Cardiorespiratory and tissue oxygen dose response to rat endotoxemia. Am J Physiol. 1996;271(3 Pt 2):H891–5.
Dyson A, Stidwill R, Taylor V, Singer M. Tissue oxygen monitoring in rodent models of shock. Am J Physiol Heart Circ Physiol. 2007;293(1):H526–33.
Dyson A, Rudiger A, Singer M. Temporal changes in tissue cardiorespiratory function during faecal peritonitis. Intensive Care Med. 2011;37(7):1192–200.
Rosser DM, Stidwill RP, Millar CG, Singer M. The effect of norepinephrine and dobutamine on bladder epithelial oxygen tension. Chest. 1995;108(5):1368–72.
Dyson A, Simon F, Seifritz A, Zimmerling O, Matallo J, Calzia E, et al. Bladder tissue oxygen tension monitoring in pigs subjected to a range of cardiorespiratory and pharmacological challenges. Intensive Care Med. 2012;38(11):1868–76.
Lee C-J, Gardiner BS, Evans RG, Smith DW. Predicting oxygen tension along the ureter. Am J Physiol-Ren Physiol. 2021;321(4):F527–47.
Acknowledgements
Not applicable
Funding
YRL and RGE were jointly supported by project grants from the National Health and Medical Research Council of Australia (NHMRC) [GNT1122455, GNT1185777] and the National Heart Foundation of Australia [VG101377; VG104674]. RGE was supported by project grants from the NHMRC [GNT1188514, GNT1050672] and the National Heart Foundation Australia [101853 and 102282]. YRL was supported by a Future Leader Fellowship from the National Heart Foundation of Australia [NHF105666]. RH was supported by the Austin Medical Research Foundation.
Ethics declarations
Ethics approval and consent to participate
Not applicable. No new unpublished human or laboratory studies are described in this manuscript.
Consent for publication
Not applicable.
Competing interests
RGE reports receipt of consulting fees from Medtronic Inc.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, R.T., Lankadeva, Y.R., Yanase, F. et al. Continuous bladder urinary oxygen tension as a new tool to monitor medullary oxygenation in the critically ill. Crit Care 26, 389 (2022). https://doi.org/10.1186/s13054-022-04230-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-022-04230-7